
Abstract
Huntington’s disease (HD) or Huntington’s chorea is the most common inherited, dominantly transmitted, neurodegenerative disorder. It is caused by increased CAG repeats number in the gene coding for huntingtin (Htt) and characterized by motor, behaviour and psychiatric symptoms, ultimately leading to death. HD patients also exhibit alterations in glucose and energetic metabolism, which result in pronounced weight loss despite sustained calorie intake. Glucose metabolism decreases in the striatum of all the subjects with mutated Htt, but affects symptom presentation only when it drops below a specific threshold. Recent evidence points at defects in glucose uptake by the brain, and especially by neurons, as a relevant component of central glucose hypometabolism in HD patients. Here we review the main features of glucose metabolism and transport in the brain in physiological conditions and how these processes are impaired in HD, and discuss the potential ability of strategies aimed at increasing intracellular energy levels to counteract neurological and motor degeneration in HD patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Brain glucose metabolism
The brain relies predominantly on glucose as a substrate for its metabolism, at variance with other body tissues that can utilize other energy sources, mostly fatty acids. Brain requirements for glucose are quite high: the adult brain weighs ~2% of the whole body, but consumes ~20% of total body glucose in the resting state (Mergenthaler et al. 2013). Glucose is used by the brain to both provide the energy for cellular functions (e.g., generation of synaptic and action potentials, maintenance of ion gradients, etc.) and as a carbon source for the biosynthesis of a variety of molecules (e.g., neurotransmitters) (Siegel et al. 1999).
In brain cells glucose is phosphorylated by hexokinase (HK) to glucose-6-phosphate (G6P) (Patel et al. 2014) and metabolized, as in other cells, through one of the following pathways.
-
1.
Glycolysis. It takes place in the cytoplasm under anaerobic conditions, and leads to formation, for each glucose molecule, of two molecules of pyruvate, two molecules of adenosine triphosphate (ATP) and two molecules of NADPH. NADPH is a reducing agent, important for the management of oxidative stress, which in animals is mostly produced by the pentose phosphate pathway (see below). ATP stores chemical energy in a form ready to be released for cell needs. Pyruvate molecules can be further metabolized in two ways. Most pyruvate enters the mitochondria and is converted to acetyl-CoA, which undergoes the tricarboxylic acid (TCA) cycle and oxidative phosphorylation. Pyruvate aerobic metabolism produces the highest amount of cellular ATP (~30 molecules). A small amount of pyruvate is reduced to lactate, at the expense of NADPH, in the cytosol. This reaction is reversible; therefore lactate can be oxidized to pyruvate, producing NADPH, and join the mitochondrial aerobic metabolism. The fate of brain lactate is still controversial, and different models have been proposed to take into account the available experimental evidence: (a) “astrocyte-to-neuron lactate shuttle” (ANLS) (Pellerin and Magistretti 2012); (b) “neuron-to-astrocyte lactate shuttle” (NALS) (Mangia et al. 2011); and (c) “lactate release” (Dienel 2012). According to these models, during brain activation in response to a specific stimulus, lactate is: (a) released by astrocytes and taken up by neurons (ANLS); (b) released by neurons and taken up by astrocytes (NALS); or (c) released from the brain to the blood, respectively. In any case, the contribution of lactate to the overall glucose metabolism in the brain is reputed to be very small (Mangia et al. 2011; Dienel 2012).
-
2.
Pentose phosphate pathway. It generates NADPH and metabolites that re-enters the glycolytic pathway downstream of G6P.
-
3.
Glycogenesis. It occurs almost exclusively in astrocytes and leads to the formation of glycogen, a polymer comprising glucose molecules. This may serve as energy reserve in the brain, as in the periphery. However, the amount of brain glycogen is too limited to maintain brain function in the absence of permanent glucose supply from the blood. Indeed, brain reliance on glucose is so pronounced that even a transient decline in glucose metabolism causes serious disruption of brain functions; if blood glucose flux is interrupted, unconsciousness and coma occur in less than 10 s (Clarke and Sokoloff 1999).
Because of the strong brain dependence on blood glucose, impairment of glucose supply to brain cells can dramatically affect brain function and lead to a variety of neurological syndromes.
Glucose transport in the brain
In spite of the brain reliance on glucose as a food source, glucose ability to diffuse from the blood into the brain is restricted (Vannucci et al. 1997). This restriction is mainly due to the presence of the blood–brain barrier (BBB), which is considered to be the most selective of the biological barriers that separate blood from the interstitial fluids that surround body tissues (Keaney and Campbell 2015). The BBB is a specialized anatomical-physiological barrier made of microvascular endothelial cells connected by tight junction complexes, which effectively prevent passage of molecules between cells (Wilhelm et al. 2011). While lipophilic substances and gases, like oxygen, can reach the brain by freely diffusing through the membranes of endothelial cells, water-soluble substances, like glucose, can only cross the BBB by taking advantage of specific membrane transporters. Transporter molecules that allow glucose to move from blood to brain belong to the glucose transporter (GLUT) protein family (Olson and Pessin 1996). All mammalian cells contain one or more members of this family. The human genome encodes 14 different GLUT proteins that facilitate the passage of sugars (e.g., glucose, fructose) across cell and intracellular membranes. The most highly expressed GLUTs in the brain are GLUT1 and GLUT3.
GLUT1 is encoded by the SLC2A1 gene and expressed mostly in erythrocytes and endothelial cells in adults, as well as at high levels in all foetal tissues. The high molecular weight (i.e., highly glycosylated) form is densely expressed by endothelial cells of the BBB and transports glucose from microvasculature blood to brain interstitial fluid (Jurcovicova 2014). Glucose is then taken up into astrocytes by a lower molecular weight (i.e., less glycosylated) GLUT1 form, and into neurons by GLUT3. Heterozygous mutations in the SLC2A1 gene resulting in decreased glucose presence in the cerebrospinal fluid (hypoglycorrhachia) cause the GLUT1 deficiency syndrome (GLUT1-DS) (Seidner et al. 1998), a neurological disorder characterized by variable phenotypes and severity of symptoms. These include: mental retardation (from mild to severe cognitive impairment); microcephaly; movement disorders (from mild motor dysfunction to severe neurological disability); early onset absence epilepsy. Epileptic and movement symptoms can be effectively reduced by a ketogenic (i.e., high fat, low carbohydrate, adequate protein) diet, which provides the body with an alternative energy source to glucose. In the presence of low carbohydrates, the liver metabolizes fatty acids to ketone bodies (i.e., β-hydroxybutyrate, acetoacetate and acetone), which are able to cross the BBB. Ketone bodies can be taken up by other tissues such as heart, skeletal muscle and brain, and converted to acetyl-CoA, which enters the TCA cycle to be oxidized for energy production (Deng et al. 2014).
GLUT3 is encoded by the SLC2A3 gene and highly and specifically expressed in neurons. It has both higher glucose affinity and transport capacity than GLUT1, and its expression increases concomitantly with the maturation of synaptic connections (Simpson et al. 2008).
Both GLUT1 and GLUT3 are overexpressed in several types of solid tumours. Since they generate ATP mostly by anaerobic glycolysis in the cytoplasm (the Warburg effect), their need for glucose is dramatically increased with respect to normal cells, which generate the majority of ATP molecules via aerobic metabolism in the mitochondria (see above). As a consequence, overexpression of GLUT1 and/or GLUT3 may be exploited for the diagnosis or therapy of solid tumours (Deng et al. 2015).
GLUT4 is found in considerable amounts in the olfactory bulb, hippocampus and cerebellum, and its expression levels vary according to the levels of circulating insulin (see below) (Vannucci et al. 1997, 1998a).
Other members of the GLUT family have been detected in relatively small amounts in specific brain regions; e.g., GLUT5 (fructose transporter) is mostly found in the microglia and microvessels; GLUT2 in astrocytes (Vannucci et al. 1997; Joost and Thorens 2001; Choeiri et al. 2002).
Mechanism of glucose transport by GLUT1 and GLUT3
GLUT1 and GLUT3 are uniporter carriers, i.e., integral membrane proteins that bind one molecule of substrate at a time and transport it across the membrane. Transport occurs down the substrate concentration gradient, thereby allowing a constant supply of blood sugar to cell metabolism (Olson and Pessin 1996). Since chemical energy deriving from ATP hydrolysis is not required, this process is named “facilitated diffusion” or “passive transport”. Glucose transport by GLUT1 and GLUT3 is generally agreed to occur by the so-called “alternating access mechanism”, according to which the substrate binding site is made alternatively accessible from either side of the membrane by conformational changes of the receptor (Jardetzky 1966). This hypothesis has recently gained strong support by the determination of the three-dimensional (3D) atomic-level structures of GLUT1 and GLUT3 in different conformations by X-ray crystallography (Deng et al. 2014, 2015). All of these structures are now freely available from the Protein Data Bank (PDB).
Both GLUT1 and GLUT3 assume the conserved fold shared by both the closely related sugar porter family members, i.e., the sugar_tr family in the Pfam database (Finn et al. 2016), and the more distant homologues belonging to the major facilitator superfamily (MFS) (Quistgaard et al. 2016). The conserved fold is made of two domains, N- and C-terminal, comprising six trans-membrane spanning α-helices each. In members of the sugar porter family the N- and C-terminal domains are connected by a third domain, comprising five short intracellular α-helices. This intracellular helical (ICH) domain has not been detected in other MSF transporters until now, and may be a unique feature of the sugar porter family. Comparative analyses of the 3D structures of GLUT1 and GLUT3, as well as of homologous proteins belonging to the sugar porter family and MSF transporter superfamily, have allowed both overall conformational changes taking place during the sugar transport cycle, and specific residues and interactions responsible for these changes, to be elucidated (Deng et al. 2015). In particular, the following conformations have been proposed to take part in the transport cycle.
Outward-open: it has roughly the shape of a “V” letter, with a cavity opened towards the extracellular side of the membrane; it allows extracellular glucose to reach and bind to the internal substrate binding site (see Fig. 1, left panels).

Three-dimensional structure of GLUT1 and GLUT3. a Ribbon representation of GLUT1 (Deng et al. 2014) and GLUT3 (Deng et al. 2015) structures determined by X-ray crystallography in different conformational states. The N-terminal, C-terminal and ICH domains are coloured blue, green and orange, respectively. Ligand atoms are shown as spheres proportional to their van der Waals radii and coloured red. Left Outward-open GLUT3 in complex with maltose (PDB code: 4ZWC, resolution = 2.60 Å). Centre Outward-partially occluded GLUT3 in complex with glucose (PDB code: 4ZW9, resolution = 1.50 Å). Right Inward-open GLUT1 in complex with n-nonyl-β-d-glucopyranoside (PDB code: 4PYP, resolution = 3.17 Å). b Extracellular views of GLUT3 and cytoplasmic view of GLUT1 structures shown in panel a. Left In outward-open GLUT3 the substrate binding site can be accessed by extracellular glucose. Centre In outward-partially occluded GLUT3 the substrate binding site is not accessible. Right In inward-open GLUT1 the cavity allowing substrate exit into the cytoplasm is widely open (Colour figure online)
Outward-partially occluded: similar to the previous one but for the closure of the substrate entrance cavity by small conformational changes, which effectively trap glucose within the binding site (Fig. 1, central panels).
Occluded-substrate bound: the bound glucose is inaccessible from either side of the membrane.
Inward-open: it is also “V”-shaped, but the cavity is opened towards the cytoplasmic side of the membrane, allowing glucose to be released from the substrate binding site (Fig. 1, right panels).
Inward-partially occluded: similar to the inward-open, but for the fact that the cytoplasmic side is not completely open, therefore glucose cannot bind again to the receptor.
Occluded-free: the empty substrate binding site is not accessible from either side of the membrane.
The structure of GLUT1 has been determined in the inward-open conformation in complex with either a glucose analogue (Fig. 1, right panels) (Deng et al. 2015) or different inhibitors (Kapoor et al. 2016). The structure of GLUT3 has been determined in two distinct conformations: outward-open (Fig. 1, left panels), in complex with maltose, and outward-partially occluded (Fig. 1, central panels) in complex with either glucose or maltose (Deng et al. 2015). The conformations observed in GLUT1 and GLUT3 are shared by other members of the sugar porter family (i.e., mammalian fructose transporter GLUT5 (Nomura et al. 2015); fungal phosphate transporter (Pedersen et al. 2013); bacterial Glucose/H+ or d-xylose/H+ symporters (Sun et al. 2012; Quistgaard et al. 2013; Wisedchaisri et al. 2014; Iancu et al. 2013)) or of the MSF transporter superfamily (e.g., FucP, GlpT and LacY) (Quistgaard et al. 2016), whose structures have been determined either in the free state or in complex with their natural substrate or substrate analogues. This high degree of structural conservation indicates that the substrate transport mechanism is highly conserved throughout evolution, from bacterial to human homologues. The inward-partially occluded and occluded-substrate bound conformations have been observed in one of the structures of d-xylose/H+ symporter (XylE) from E. coli (Quistgaard et al. 2013), which belongs to the sugar porter family, and in the MSF transporter superfamily member EmrD (Quistgaard et al. 2016), respectively. The occluded-substrate free conformation is, at present, not supported by experimentally determined 3D structures of proteins homologous to GLUT1 and GLUT3.
The structural elements playing the main role in determining the conformational changes taking place during the transport cycle affect mostly the C-terminal domain, and in particular transmembrane (TM) helices TM7 and TM10 (Fig. 2); conversely, the N-terminal domain remains mostly unmodified, and moves as a rigid body relative to the C-terminal domain. While all the 12 TMs comprised in the N- and C-terminal domains are visible in all the GLUT1 and GLUT3 structures (Fig. 1a), the five helices of the ICH domain are fully visible only in GLUT3 outward-open and outward-partially closed conformations, where the ICH domain acts as a gate that occludes the tunnel between the substrate binding site and the cytoplasmic side of the membrane (Fig. 1a, right and central panels). In inward-open GLUT1 structure, a part of the fifth α-helix of the ICH domain is not visible, indicating that it is highly flexible (Fig. 1b, right panel). As a result, the tunnel towards the cytoplasmic side of the membrane is open.

Comparison of GLUT1 in the inward-open conformation and GLUT3 in the outward-open and outward-partially occluded conformations. Ribbon representation of GLUT1 (PDB code: 4PYP) and GLUT3 (PDB codes: 4ZWC and 4ZW9) structures determined by X-ray crystallography (Deng et al. 2014, 2015). a GLUT3 structures are coloured grey except for α-helix TM7, which is orange in outward-partially occluded and blue in outward-open conformations. The TM7 α-helix has a straight conformation (blue) and surrounds the substrate entry cavity in outward-open GLUT3. In outward-partially occluded GLUT3, TM7 is bent (orange), preventing substrate exit towards the extracellular side of the membrane. b Residues Asn286 and Tyr290 of TM7 point away from the outward-open cavity in outward-open GLUT3. Glucose passage to the extracellular side of the membrane is prevented by the hydrogen bond interaction with Asn286, and steric hindrance by the bulky Tyr290 side-chain in outward-partially occluded GLUT3. c GLUT1 and GLUT3 are coloured as in Figure 1 and panels a and b of Figure 2, respectively. Trp386 in TM10 prevents glucose exit towards the cytoplasm in outward-partially occluded GLUT3, but a conformational change of TM10 shifts the homologous Trp388 away from the substrate exit cavity in inward-open GLUT1, allowing substrate release (Colour figure online)
Regulation of GLUT1 and GLUT3 levels
GLUT1 and GLUT3 expression and activity regulation are complex, and vary in different cell types and depending on environmental conditions. Short-term increases in glucose transport can be achieved by stimulating the activity of cell surface GLUTs and/or translocation of intracellular GLUTs to the plasma membrane. Long-term glucose transport enhancements require synthesis of new GLUT proteins. GLUTs regulation has been shown to involve several factors, such as: hypoxia and hypoxia-inducible factor 1 (HIF-1α); glucose deprivation; insulin and insulin-like growth factor I (IGF-I); and human antigen R (HuR).
HIF-1α and hypoxia increase GLUT1 and GLUT3 levels
HIF-1 is considered to be the master transcriptional regulator of cellular and developmental response to a decrease in available oxygen concentration within cells (hypoxia). It is highly conserved in most oxygen-breathing species and it has an essential role in human metabolism (Formenti et al. 2010).
From a structural point of view HIF-1 is a heterodimer, formed by one α and one β subunit. In normal oxygen conditions, HIF-1α is constitutively expressed at low levels in most cells, and rapidly degraded via the ubiquitin protease pathway. In hypoxic conditions HIF-1α expression is upregulated, degradation is prevented and HIF-1α accumulates to associate with constitutively expressed HIF-1β.
HIF-1 exerts transcriptional activity on tens of genes whose expression promotes survival in hypoxic conditions, including: vascular endothelial growth factor and erythropoietin, which promote vascularization and erythropoiesis and, therefore, increase oxygen delivery to hypoxic regions (Semenza 2001; Vannucci et al. 1998b); glycolysis enzymes, which allow ATP to be synthetized in the absence of oxygen; and glucose transporters GLUT1 and GLUT3, which increase glucose uptake within cells.
A number of studies have investigated the effects of oxygen deprivation on GLUT transcription and translation in the central nervous system. GLUT1 and GLUT3 gene expression has been shown to be upregulated in rat brain in vivo, by hypoxia–ischemia induced by carotid artery ligation (Vannucci et al. 1998b) or by treatment with the hypoxia-mimetic cobalt chloride, which prevents HIF-1α degradation under normoxic conditions (Badr et al. 1999). Accordingly, in primary cultures of rat neurons, GLUT1 and GLUT3 mRNA levels increased in hypoxic conditions. Interestingly, the increase in GLUT1 transcript levels determined by joint hypoxia and glucose deprivation was significantly higher than that obtained by glucose deprivation alone (Bruckner et al. 1999).
HuR regulates GLUT1 gene expression
Human antigen R (HuR) or ELAV-like protein 1 is a member of the ELAVL family of RNA binding proteins. These contain several RNA recognition motifs and bind selectively to adenylate-uridylate-rich elements (AREs) in 3′ untranslated regions of mRNAs. HuR contains a nucleus–cytoplasm shuttling sequence. In the nucleus, HuR binds mRNAs and the resulting complex is exported to the cytoplasm (Peng et al. 1998). Since AREs are mRNA degradation signals, HuR binding in the cytoplasm stabilizes mRNA transcripts. Therefore, HuR regulates mRNA translational efficiency by acting as a molecular chaperone (Cherry et al. 2006).
The interaction between HuR and GLUT1 mRNA has been studied in preadipocytes and adipocytes. In these studies HuR regulated GLUT1 gene expression at a post-transcriptional level in a differentiation specific manner. In fact, while both HuR and GLUT1 mRNA are present in both stages of differentiation, the complex is detected only in adipocytes, where HuR localization in the cytoplasm is increased with respect to preadipocytes, where HuR is mostly nuclear. HuR regulatory effect on GLUT1 in adipocytes was ascribed to different mechanisms: on the one hand, stabilization of GLUT1 mRNA by binding to AREs and inducing translocation from the nucleus to the cytosol; on the other, initiation of translation and movement of GLUT1 mRNA to the polyribosomes (Gantt et al. 2006; Vander Heiden et al. 2001). More recently, HuR has been shown to be essential for pancreatic cancer cells survival under acute glucose deprivation. This condition stimulated HuR translocation from the nucleus to the cytoplasm, where HuR binds several mRNAs coding for enzymes involved in glucose metabolism, but not GLUT1 mRNA. These results may be ascribed to metabolic differences between cancer and normal cells, but may also imply that HuR binding to mRNA substrates is tissue-specific (Burkhart et al. 2013).
Insulin-dependent GLUT1 expression and translocation
Insulin is a protein hormone that promotes glucose absorption from the blood into almost all cell types, and most extensively in the liver, skeletal muscle and adipose tissue. Insulin binding to insulin receptors (IRs) determines the high affinity GLUT4 receptor to be transported from intracellular vesicles, where it is stored, to the membrane of adipose and muscle cells, resulting in an increased rate of glucose uptake. Within cells, glucose is used in anabolic reactions to build large molecules such as carbohydrates, lipids and proteins. Insulin hypoglycemic activity is essential to maintain blood glucose concentration within a narrow range. When glucose levels increase above the physiological threshold, large amounts of glucose are taken up via the low affinity receptor GLUT2 by the β-cells within the islets of Langerhans in the pancreas, which respond by secreting insulin. If glucose levels drop below physiological levels, the α-cells of the islets of Langerhans release glucagon, a peptide hormone with hyperglycemic activity: it stimulates liver cells to produce glucose, via glycogenolysis and gluconeogenesis, and release it into the blood.
Insulin has been considered for a long time to be a peripheral hormone, but its role in brain glucose metabolism is now well established. Insulin has been reported to modulate thermoregulation, synaptic transmission, neuronal growth and differentiation, neuroprotection, learning, memory, longevity, aging, age-associated neurodegenerative disorders, and fertility (Duarte et al. 2012; Laron 2009).
Both insulin and IRs are present at high levels in the human brain. Most of the insulin present in adults is produced by pancreatic β-cells and transported to the brain. However, a number of evidences indicate that both insulin and IRs are synthesized by neurons as well (Duarte et al. 2012). IRs are highly expressed in neurons (neuron-specific isoform) and less in glial cells (peripheral-like isoform, also expressed by BBB cells). Insulin stimulates glucose uptake in cultured glial cells and neurotransmitters release in neurons; therefore, glucose uptake by neurons may be the consequence of neuronal activation. Insulin-induced glucose uptake has been proposed to result from the recruitment of the partially insulin-sensitive GLUT1 to the membrane of brain cells, since GLUT3 is not sensitive to the hormone (Bingham et al. 2002). This hypothesis is supported by the observation that in three different osteosarcoma cell lines insulin induced GLUT1 translocation to the plasma membrane, while not affecting GLUT1 expression levels (Cifuentes et al. 2011). Insulin has also been reported to increase GLUT1 expression via activation of the anti-apoptotic PI3K/Akt/glycogen synthase kinase-3β (GSK-3β) pathway, one of the two major signalling pathways activated by this hormone (Duarte et al. 2012); in turn, increased GLUT1 expression has been correlated with Akt-mediated enhancement of mitochondrial membrane potential (Huang et al. 2005). In addition to stimulating glucose uptake, insulin can provide energy to neurons by stimulating glucose release from glycogen stores in astrocytes via the partially insulin-sensitive GLUT1 to the extracellular fluid; from here, neurons would uptake glucose via the insulin-insensitive GLUT3 (Duarte et al. 2012).
IGF-I and glucose
Both GLUT1 and GLUT3 mRNAs were shown to increase as a consequence of IGF-I activation and glucose deprivation in bovine adrenomedullary chromaffin cells. IGF-I activity was entirely mediated by synthesis of new GLUT proteins, and in particular GLUT3. Conversely, only about half of the glucose uptake resulting from glucose deprivation could be accounted for by new GLUT protein synthesis, the rest having been ascribed to post-transcriptional mechanisms such as reduced internalization rate of pre-existing GLUTs (Fladeby et al. 2003).
Impairment of energetic metabolism in Huntington’s disease
Huntington’s disease (HD) is an autosomal dominant and progressive neurodegenerative disease. It is characterized by progressive neuron dysfunction and death, most extensively in the brain (striatum, white matter and cortex), and is associated with involuntary movements, cognitive and behavioural symptoms and ultimately death (Roos 2010).
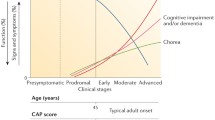
HD is caused by the elongation of the CAG trinucleotide repeat in the HTT gene, which encodes a polyglutamine (polyQ) stretch in the huntingtin (Htt) protein. In normal individuals, the CAG region of HTT contains 11–35 trinucleotide repeats; a repeats number higher than 35 indicates a high probability of developing HD. The expanded CAG repeat region tends to be unstable in both somatic and germline cells, particularly male, and frequently expands, rather than contract, during intergenerational transmission. The disease is typically diagnosed in adults around the age of 40, on average, and no treatment is currently available to reverse the neurodegeneration induced by the mutated Htt (mHtt).
In addition to the neurological symptoms, HD patients face pronounced weight loss, despite sustained caloric intake. This was a first indication that alterations in energetic metabolism played a role in HD pathogenesis; strong evidence of early glucose hypometabolism in the brain of HD patients is now available (Squitieri and Ciarmiello 2010). HD subjects exhibit reduced glycolysis and lactate levels (Powers et al. 2007) and alterations in mitochondrial function. While the activity and expression levels of TCA cycle enzymes are increased in both the post-mortem cortex of human patients, and cortex and striatum of a mouse HD model with >175 glutamine repeats (Q175) (Naseri et al. 2015), oxidative phosphorylation is impaired (Milakovic and Johnson 2005), and ATP is depleted in brain and peripheral tissues of HD patients (Chaturvedi et al. 2009). Other mitochondrial dysfunctions include: defective calcium handling (Lim et al. 2008) and mitochondria trafficking (Li et al. 2010); oxidative damage (Browne et al. 1999); and dysregulation of the peroxisome proliferator-activated receptor gamma co-activator-1-alpha (PGC-1α), which is the master regulator of mitochondrial biogenesis (Cui et al. 2006). Overall, metabolic reductions occur early in HD, and may precede the clinical onset of HD symptoms by many years.
HD causes defective glucose transport in the brain
Several evidences indicate that energetic metabolism impairment in HD patients is contributed by defective brain glucose uptake, in particular by neurons (Powers et al. 2007; Lalic et al. 2008; Russo et al. 2013; Adanyeguh et al. 2015). In mouse models, embryonic cortical neurons have been shown to take up significantly less glucose than WT neurons. The quantity of GLUT1 and GLUT3 present in membranes of post-mortem samples of caudate and cortex brain regions has been reported to be significantly reduced in advanced HD stages patients with respect to non-HD controls: in grade 3 HD caudate the amount of GLUT1 and GLUT3 was 3- and 4-fold lower than in non-HD controls, respectively, whereas at earlier stages (grade 1), there was no significant difference in GLUT1 and GLUT3 membrane concentration compared with non-diseased controls (Gamberino and Brennan 1994). Recent data also highlighted the presence of abnormalities in the BBB (i.e., leakage) and blood vessels (Drouin-Ouellet et al. 2015).
Reduced surface expression of GLUT3 has also been demonstrated to occur in the striatum and cortex of HD 140Q mice brain slices immunolabeled with an antibody recognizing an extracellular GLUT3 epitope, compared with WT mice (Li et al. 2012). The effects of GLUT3 gene expression levels and glucose uptake activity by neurons on HD progression have been observed also in Drosophila HD models. In particular, expression of GLUT3 in Drosophila neurons was shown to be sufficient to suppress most of the neurological phenotypes induced by mHtt, since it determined a striking improvement in fly survival, restored locomotor activity and rescued neurodegeneration (Besson et al. 2015).
Glucose hypometabolism and reduced GLUT3 presence in neuron membranes in HD are caused, at least in part, by deficient Rab11 activity (Li et al. 2012). Rab11, a member of the Ras superfamily of small GTPases (Rab: Ras-related in brain), regulates trafficking of numerous receptors and transporters to cell membranes, including transferrin receptor, neuronal glutamate/cysteine transporter EAAC1 and GLUT3 (McClory et al. 2014). Enhancements in Rab11 activity may increase glucose uptake by HD neurons. Alterations in insulin secretion and sensitivity have also been detected in normoglycemic HD patients (Russo et al. 2013; Lalic et al. 2008).
Several alterations have been shown to take place in HD mutation carriers before symptom presentation. Reduced uptake of glucose or 18-fluoro-desossi-glucose and progressive white matter loss, especially in the caudate nuclei has been demonstrated to occur by positron emission tomography (PET-scan) and MRI in adults (Ciarmiello et al. 2006; Squitieri and Ciarmiello 2010). Pre-manifest HD subjects also showed a metabolic decrease in striatum, frontal, and temporal lobes, albeit to a different extent, and thalamic hypermetabolism, as a compensation reaction to progressive decrease of glucose uptake in the caudate (Feigin et al. 2007). In all HD subjects the reduction in glucose metabolism is particularly enhanced and progressive in the caudate nucleus (Ciarmiello et al. 2006; Feigin et al. 2007), likely affecting symptom presentation only when glucose metabolism decreases below a certain threshold (Ciarmiello et al. 2012). These findings contribute to explain why brain functions of pre-manifest HD subjects remain indistinguishable from those of the general population for many years, and are expected to improve predictions of HD onset age (Feigin et al. 2007; Ciarmiello et al. 2012).
Future perspectives
Large evidence indicating that glucose metabolism is impaired in HD is now available. Of the many factors and mechanisms that have been discovered to affect energetic and glucose metabolism, glucose transport into brain cells has been only marginally analysed so far. Recent studies have unveiled the three-dimensional structure of glucose transporters that are responsible for glucose uptake inside endothelial cells and astrocytes (GLUT1) or neurons (GLUT3), at atomic level. Additionally, multiple factors controlling GLUT1 and GLUT3 expression and cellular localization have been identified. The amount of data now available about structure–function relationships among members of the GLUT family, accumulated knowledge about the factors and mechanisms affecting glucose transport inside cells, and evidences about deficiency of these receptors in membranes of HD patients and animal models, point to this pathway as an attractive target for new treatments against HD and, potentially, other incurable neurodegenerative disorders related to glucose hypometabolism.
To ameliorate the neurodegenerative process in HD, several strategies aimed at increasing intracellular energy levels have been explored.
Creatine is an endogenous nitrogenous organic acid whose phosphorylated form is used by the enzyme creatine kinase to the resynthesize ATP from ADP in a reversible reaction. Creatine is actively taken up by skeletal muscle, where the majority of the compound is stored, brain and other highly energy consuming tissues. For these reasons, it is widely used as a supplement to build muscle mass. Creatine supplementation to HD mouse model was shown to significantly improve body weight and motor performance, delay the onset of diabetes and slow down the development of brain atrophy, loss of striatal neurons and formation of Htt-positive aggregate. Unfortunately, creatine neuroprotective effects have not been replicated in clinical trials so far. However, it has been proposed that to reliably assess the actual therapeutic efficacy of this compound clinical trials should be carried out on larger cohorts of patients and for longer periods of time (Klopstock et al. 2011).
A different strategy consists in providing HD subjects with energy sources alternative to glucose. As mentioned above, a ketogenic diet causes the body to transform common dietary fat to ketone bodies. These cross the BBB and are transformed by brain cells to acetyl-CoA, which is used in the TCA cycle to produce energy. This treatment effectively reduces seizures in disorders associated with impaired glucose metabolism, such as GLUT1-DS, but it does not control other symptoms. Additionally, a high fat diet involves potential health risks and is not easy to maintain, leading to frequent dropouts (Kovac et al. 2013). An alternative to the ketogenic diet is provided by treatment with anaplerotic substances, i.e., compounds able to generate metabolic pathway intermediates. As mentioned above, glucose has anabolic, as well as energy production functions. In mitochondria, pyruvate molecules generated by glycolysis can be carboxylated to oxaloacetate, one of the intermediates of the TCA cycle. This is an anaplerotic reaction, since it produces a metabolic pathway intermediate. As a result of glucose deficit, not only energetic metabolism, but also glucose anaplerotic functions may be impaired in the brain of HD or GLUT1-DS patients. The most commonly studied anaplerotic compound is triheptanoin, a triglyceride comprising three heptanoate fatty acids. At variance with most dietary fats, which comprise fatty acids with even number of carbon atoms and are metabolized to three- and four-carbon atom ketone bodies, fatty acids with odd number of carbon atoms are metabolized to ketone bodies with five carbon atoms, which are also able to cross the BBB. Within tissues, three- and four-atom carbon ketone bodies can be transformed to acetyl-CoA only, whereas five-carbon atom ketones produce both acetyl-CoA and propionyl-CoA. This is an efficient anaplerotic substrate for the TCA cycle, since it can be converted, by a series of reactions, to succinyl-CoA, one of the intermediates of the TCA cycle. Because of the ability to generate the anaplerotic propionyl-CoA, in addition to the energy-providing acetyl-CoA, triheptanoin is expected to be more effective than the ketogenic diet as a glucose replacement strategy (Adanyeguh et al. 2015). Indeed, triheptanoin treatment has been shown to improve energy metabolism in HD patients (Mochel et al. 2010; Adanyeguh et al. 2015). In a small-scale study on 10 early stage HD patients vs. controls, treatment with triheptanoin improved brain metabolic profile and motor functions (Adanyeguh et al. 2015). Triheptanoin has also been shown to have beneficial effects in a clinical study on eight children and adult GLUT1-DS patients with non-epileptic paroxysmal manifestations (Mochel et al. 2015). Importantly, since triheptanoin was well tolerated by both adults and children, it can be tested in young HD patients, who present the rarest, so far trial-excluded, form of HD.
Taken together, the evidences reported in this review open the road to new research fields to recapitulate the mechanisms related to the transportation of glucose and other substances into brain cells through the BBB and identify possible therapeutic strategies to contrast this devastating disease.
References
Adanyeguh IM, Rinaldi D, Henry P-G et al (2015) Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 84:490–495. doi:10.1212/WNL.0000000000001214
Badr GA, Zhang JZ, Tang J et al (1999) Glut1 and Glut3 expression, but not capillary density, is increased by cobalt chloride in rat cerebrum and retina. Mol Brain Res 64:24–33. doi:10.1016/S0169-328X(98)00301-5
Besson MT, Alegría K, Garrido-Gerter P et al (2015) Enhanced neuronal glucose transporter expression reveals metabolic choice in a HD Drosophila model. PLoS One 10:1–23. doi:10.1371/journal.pone.0118765
Bingham EM, Hopkins D, Smith D et al (2002) The role of insulin in human brain glucose metabolism: an 18fluoro-deoxyglucose positron emission tomography study. Diabetes 51:3384–3390. doi:10.2337/diabetes.51.12.3384
Browne SE, Ferrante RJ, Beal MF (1999) Oxidative stress in Huntington’s disease. Brain Pathol 9:147–163
Bruckner BA, Ammini CV, Otal MP et al (1999) Regulation of brain glucose transporters by glucose and oxygen deprivation. Metabolism 48:422–431. doi:10.1016/S0026-0495(99)90098-7
Burkhart RA, Pineda DM, Chand SN et al (2013) HuR is a post-transcriptional regulator of core metabolic enzymes in pancreatic cancer. RNA Biol 10:1312–1323. doi:10.4161/rna.25274
Chaturvedi RK, Adhihetty P, Shukla S et al (2009) Impaired PGC-1α function in muscle in Huntington’s disease. Hum Mol Genet 18:3048–3065. doi:10.1093/hmg/ddp243
Cherry J, Karschner V, Jones H, Pekala PH (2006) HuR, an RNA-binding protein, involved in the control of cellular differentiation. In Vivo 20:17–23
Choeiri C, Staines W, Messier C (2002) Immunohistochemical localization and quantification of glucose transporters in the mouse brain. Neuroscience 111:19–34. doi:10.1016/S0306-4522(01)00619-4
Ciarmiello A, Cannella M, Lastoria S et al (2006) Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington’s disease. J Nucl Med 47:215–222
Ciarmiello A, Giovacchini G, Orobello S et al (2012) 18F-FDG PET uptake in the pre-Huntington disease caudate affects the time-to-onset independently of CAG expansion size. Eur J Nucl Med Mol Imaging 39:1030–1036. doi:10.1007/s00259-012-2114-z
Cifuentes M, García MA, Arrabal PM et al (2011) Insulin regulates GLUT1-mediated glucose transport in MG-63 human osteosarcoma cells. J Cell Physiol 226:1425–1432. doi:10.1002/jcp.22668
Clarke D, Sokoloff L (1999) Circulation and energy metabolism of the brain. In: Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD (eds) Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th edn. Lippincott-Raven, Philadelphia, pp 637–639
Cui L, Jeong H, Borovecki F et al (2006) Transcriptional repression of PGC-1α by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127:59–69. doi:10.1016/j.cell.2006.09.015
Deng D, Xu C, Sun P et al (2014) Crystal structure of the human glucose transporter GLUT1. Nature 510:121–125. doi:10.1038/nature13306
Deng D, Sun P, Yan C et al (2015) Molecular basis of ligand recognition and transport by glucose transporters. Nature 526:391–396. doi:10.1038/nature14655
Dienel GA (2012) Fueling and imaging brain activation. ASN Neuro 4:267–321. doi:10.1042/AN20120021
Drouin-Ouellet J, Sawiak SJ, Cisbani G et al (2015) Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: potential implications for its pathophysiology. Ann Neurol 78:160–177. doi:10.1002/ana.24406
Duarte AI, Moreira PI, Oliveira CR (2012) Insulin in central nervous system: more than just a peripheral hormone. J Aging Res. doi:10.1155/2012/384017
Feigin A, Tang C, Ma Y et al (2007) Thalamic metabolism and symptom onset in preclinical Huntington’s disease. Brain 130:2858–2867. doi:10.1093/brain/awm217
Finn RD, Coggill P, Eberhardt RY et al (2016) The Pfam protein families database: towards a more sustainable future. Nucl Acids Res 44:D279–D285. doi:10.1093/nar/gkv1344
Fladeby C, Skar R, Serck-Hanssen G (2003) Distinct regulation of glucose transport and GLUT1/GLUT3 transporters by glucose deprivation and IGF-I in chromaffin cells. Biochim Biophys Acta Mol Cell Res 1593:201–208. doi:10.1016/S0167-4889(02)00390-7
Formenti F, Constantin-teodosiu D, Emmanuel Y et al (2010) Regulation of human metabolism by hypoxia-inducible factor. Proc Natl Acad Sci USA 107:12722–12727. doi:10.1073/pnas.1002339107/-/DCSupplemental.www.pnas.org/cgi/doi/10.1073/pnas.1002339107
Gamberino WC, Brennan WA Jr (1994) Glucose transporter isoform expression in Huntington’s disease brain. J Neurochem 63:1392–1397
Gantt KR, Cherry J, Richardson M et al (2006) The regulation of glucose transporter (GLUT1) expression by the RNA binding protein HuR. J Cell Biochem 99:565–574. doi:10.1002/jcb.20950
Huang TJ, Verkhratsky A, Fernyhough P (2005) Insulin enhances mitochondrial inner membrane potential and increases ATP levels through phosphoinositide 3-kinase in adult sensory neurons. Mol Cell Neurosci 28:42–54. doi:10.1016/j.mcn.2004.08.009
Iancu CV, Zamoon J, Bum S et al (2013) Crystal structure of a glucose/H+ symporter and its mechanism of action. Proc Natl Acad Sci USA 110:17862–17867. doi:10.1073/pnas.1311485110
Jardetzky O (1966) Simple allosteric model for membrane pumps. Nature 211:969–970. doi:10.1038/211969a0
Joost HG, Thorens B (2001) The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members (review). Mol Membr Biol 18:247–256. doi:10.1080/09687680110090
Jurcovicova J (2014) Glucose transport in brain—effect of inflammation. Endocr Regul 48:35–48. doi:10.4149/endo_2014_01_35
Kapoor K, Finer-Moore JS, Pedersen BP et al (2016) Mechanism of inhibition of human glucose transporter GLUT1 is conserved between cytochalasin B and phenylalanine amides. Proc Natl Acad Sci 113:4711–4716. doi:10.1073/pnas.1603735113
Keaney J, Campbell M (2015) The dynamic blood–brain barrier. FEBS J 282:4067–4079. doi:10.1111/febs.13412
Klopstock T, Elstner M, Bender A (2011) Creatine in mouse models of neurodegeneration and aging. Amino Acids 40:1297–1303. doi:10.1007/s00726-011-0850-1
Kovac S, Abramov AY, Walker MC (2013) Energy depletion in seizures: anaplerosis as a strategy for future therapies. Neuropharmacology 69:96–104. doi:10.1016/j.neuropharm.2012.05.012
Lalic NM, Maric J, Svetel M et al (2008) Glucose homeostasis in Huntington disease: abnormalities in insulin sensitivity and early-phase insulin secretion. Arch Neurol 65:476–480. doi:10.1001/archneur.65.4.476
Laron Z (2009) Insulin and the brain. Arch Physiol Biochem 115:112–116. doi:10.1016/S0140-6736(66)90770-7
Li XJ, Orr AL, Li S (2010) Impaired mitochondrial trafficking in Huntington’s disease. Biochim Biophys Acta Mol Basis Dis 1802:62–65
Li X, Valencia A, McClory H et al (2012) Deficient Rab11 activity underlies glucose hypometabolism in primary neurons of Huntington’s disease mice. Biochem Biophys Res Commun 421:727–730. doi:10.1016/j.bbrc.2012.04.070
Lim D, Fedrizzi L, Tartari M et al (2008) Calcium homeostasis and mitochondrial dysfunction in striatal neurons of Huntington disease. J Biol Chem 283:5780–5789. doi:10.1074/jbc.M704704200
Mangia S, DiNuzzo M, Giove F et al (2011) Response to ‘comment on recent modeling studies of astrocyte-neuron metabolic interactions’: much ado about nothing. J Cereb Blood Flow Metab 31:1346–1353. doi:10.1038/jcbfm.2011.29
McClory H, Williams D, Sapp E et al (2014) Glucose transporter 3 is a rab11-dependent trafficking cargo and its transport to the cell surface is reduced in neurons of CAG140 Huntington’s disease mice. Acta Neuropathol Commun 2:179. doi:10.1186/s40478-014-0178-7
Mergenthaler P, Lindauer U, Dienel GA, Meisel A (2013) Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci 36:587–597. doi:10.1016/j.tins.2013.07.001
Milakovic T, Johnson GVW (2005) Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J Biol Chem 280:30773–30782. doi:10.1074/jbc.M504749200
Mochel F, Duteil S, Marelli C et al (2010) Dietary anaplerotic therapy improves peripheral tissue energy metabolism in patients with Huntington’s disease. Eur J Hum Genet 18:1057–1060. doi:10.1038/ejhg.2010.72
Mochel F, Hainque E, Gras D et al (2015) Triheptanoin dramatically reduces paroxysmal motor disorder in patients with GLUT1 deficiency. J Neurol Neurosurg Psychiatry. doi:10.1136/jnnp-2015-311475
Naseri NN, Xu H, Bonica J et al (2015) Abnormalities in the tricarboxylic acid cycle in Huntington disease and in a Huntington disease mouse model. J Neuropathol Exp Neurol 74:527–537. doi:10.1097/nen.0000000000000197
Nomura N, Verdon G, Kang HJ et al (2015) Structure and mechanism of the mammalian fructose transporter GLUT5. Nature 526:397–401. doi:10.1038/nature14909
Olson AL, Pessin JE (1996) Structure, function, and regulation of the mammalian facilitative glucose transporter gene family. Annu Rev Nutr 16:235–256. doi:10.1146/annurev.nu.16.070196.001315
Patel AB, Lai JC, Chowdhury GM et al (2014) Direct evidence for activity-dependent glucose phosphorylation in neurons with implications for the astrocyte-to-neuron lactate shuttle. Proc Natl Acad Sci USA 111:5385–5390. doi:10.1073/pnas.1403576111
Pedersen BP, Kumar H, Waight AB et al (2013) Crystal structure of a eukaryotic phosphate transporter. Nature 496:533–536. doi:10.1038/nature12042
Pellerin L, Magistretti PJ (2012) Sweet sixteen for ANLS. J Cereb Blood Flow Metab 32:1152–1166. doi:10.1038/jcbfm.2011.149
Peng SSY, Chen CYA, Xu N, Bin Shyu A (1998) RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J 17:3461–3470. doi:10.1093/emboj/17.12.3461
Powers WJ, Videen TO, Markham J et al (2007) Selective defect of in vivo glycolysis in early Huntington’s disease striatum. Proc Natl Acad Sci 104:2945–2949. doi:10.1073/pnas.0609833104
Quistgaard EM, Löw C, Moberg P et al (2013) Structural basis for substrate transport in the GLUT-homology family of monosaccharide transporters. Nat Struct Mol Biol 20:766–768. doi:10.1038/nsmb.2569
Quistgaard EM, Löw C, Guettou F, Nordlund P (2016) Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat Rev Mol Cell Biol 17:123–132. doi:10.1038/nrm.2015.25
Roos RAC (2010) Huntington’s disease: a clinical review. Orphanet J Rare Dis 5:40. doi:10.1186/1750-1172-5-40
Russo CV, Salvatore E, Saccà F et al (2013) Insulin sensitivity and early-phase insulin secretion in normoglycemic Huntington’s disease patients. J Huntingt Dis 2:501–507. doi:10.3233/JHD-130078
Seidner G, Alvarez MG, Yeh J-I et al (1998) GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat Genet 18:188–191. doi:10.1038/ng0298-188
Semenza GL (2001) Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med 7:345–350. doi:10.1016/S1471-4914(01)02090-1
Siegel A, Roeling TA, Gregg TR, Kruk MR (1999) Neuropharmacology of brain-stimulation-evoked aggression. Neurosci Biobehav Rev 23:359–389. doi:10.1016/S0149-7634(98)00040-2
Simpson IA, Dwyer D, Malide D et al (2008) The facilitative glucose transporter GLUT3: 20 years of distinction. Am J Physiol Endocrinol Metab 295:E242–E253. doi:10.1152/ajpendo.90388.2008
Squitieri F, Ciarmiello A (2010) Editorial: key role of nuclear medicine in seeking biomarkers of Huntington’s disease. Eur J Nucl Med Mol Imaging 37:1124–1127. doi:10.1007/s00259-010-1439-8
Sun L, Zeng X, Yan C et al (2012) Crystal structure of a bacterial homologue of glucose transporters GLUT1–4. Nature 490:361–366. doi:10.1038/nature11524
Vander Heiden MG, Plas DR, Rathmell JC et al (2001) Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol Cell Biol 21:5899–5912. doi:10.1128/MCB.21.17.5899-5912.2001
Vannucci SJ, Maher F, Simpson IA (1997) Glucose transporter proteins in brain: delivery of glucose to neurons and glia. Glia 21:2–21. doi:10.1002/(SICI)1098-1136(199709)21:1<2:AID-GLIA2>3.0.CO;2-C
Vannucci SJ, Koehler-Stec EM, Li K et al (1998a) GLUT4 glucose transporter expression in rodent brain: effect of diabetes. Brain Res 797:1–11. doi:10.1016/S0006-8993(98)00103-6
Vannucci SJ, Reinhart R, Maher F et al (1998b) Alterations in GLUT1 and GLUT3 glucose transporter gene expression following unilateral hypoxia-ischemia in the immature rat brain. Brain Res Dev Brain Res 107:255–264. doi:10.1016/S0165-3806(98)00021-2
Wilhelm I, Fazakas C, Krizbai IA (2011) In vitro models of the blood–brain barrier. Acta Neurobiol Exp (Wars) 71:113–128
Wisedchaisri G, Park M-S, Iadanza MG et al (2014) Proton-coupled sugar transport in the prototypical major facilitator superfamily protein XylE. Nat Commun 5:4521. doi:10.1038/ncomms5521
Acknowledgements
We thank the Foundation “Lega Italiana Ricerca Huntington e malattie correlate” (http://www.lirh.it) for supporting research on HD (FS) and taking care of families.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics statement
This research did not involve human participants and/or animals.
Rights and permissions
About this article

Cite this article
Morea, V., Bidollari, E., Colotti, G. et al. Glucose transportation in the brain and its impairment in Huntington disease: one more shade of the energetic metabolism failure?. Amino Acids 49, 1147–1157 (2017). https://doi.org/10.1007/s00726-017-2417-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-017-2417-2