
Abstract
Recent genome-wide association studies have revealed the HLA region on chromosome 6p21 as a susceptibility locus for hepatitis B virus (HBV) infection, a finding subsequently replicated in independent samples. However, only limited single nucleotide polymorphisms (SNPs) were analyzed in most of these studies, and it remains to be determined which SNPs contribute to the detected association. After genotyping 140 SNPs within this genomic region in a total of 1657 HBV-positive patients and 1456 HBV-negative controls, we conducted a series of genetic epidemiological and bioinformatics analysis, including individual SNP-based association analysis, haplotype-based association analysis, and conditional analysis. We identified 76 SNPs and 5 LD blocks in HLA-DP/DQ clusters that are significantly associated with HBV infection, with the smallest P value being 3.88 × 10−18 for rs9277535 in HLA-DPB1. With conditional analysis, we further revealed that the genes contributing to the effects of variants in HLA-DP/DQ on infection are independent of each other, and the LD block 5 in the 3′-UTR region of HLA-DPB1 had a predominant effect in the association of HLA-DP with HBV infection. We also found that the SNPs in the 3′-UTR region of HLA-DPB1 were significant between the subgroups of inactive HBV carrier, chronic hepatitis B, or hepatic cirrhosis from the case group and the spontaneous HBV-clearance subgroup from the control group. Finally, we did further association analysis of SNPs in this region with different subgroups from the case group, which revealed no association of these SNPs with the progression of HBV-related diseases. In sum, we showed, for the first time, that the HLA-DP/DQ clusters contribute independently to HBV infection, and the 3′-UTR region of HLA-DPB1 represents an important functional region involved in HBV infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatitis B virus (HBV) infection is one of the world’s most common and serious infectious diseases. According to the recent World Health Organization report, released in July 2014, there are approximately 240 million people in the world known to have chronic HBV infection. After developing serious diseases such as hepatic cirrhosis (HC) and hepatocellular carcinoma (HCC), about 780,000 infected people die each year. China, currently recognized as a low and high HBV endemicity co-existing region, still has 7.18 % of the general population positive for hepatitis B surface antigen (HBsAg) and more than 50,000 people die annually from the end stage of the disease (Cui and Jia 2013; Sun and Hou 2010).
Because infection and progression of HBV-related diseases vary, genetic factors are considered to be as important as the viral and environmental factors. As early as the 1970s, a familial aggregation study of hepatitis B infection implied that genetics plays an important role in HBV infection (Szmuness et al. 1975). In 1989, a twin study showed that host genetic background has an impact on infection outcome (Lin et al. 1989). Since then, many candidate gene-based association studies have been conducted to determine the genetic mechanism underlying HBV infection. Genes in the HLA region on chromosome 6 (such as HLA-DP and HLA-DQ, which are expressed as surface antigens and determine the immune response to virus infection) were associated, not only with HBV infection and progression, but also with the response to HBV vaccination and interferon treatment of hepatitis B (Desombere et al. 1998; Ahn et al. 2000; Jiang et al. 2003; Zang et al. 2004; Xi-Lin et al. 2006).
In 2009, significant associations between chronic hepatitis B (CHB) and single nucleotide polymorphisms (SNPs) rs3077 and rs9277535 in the HLA-DP gene cluster were reported in 786 Japanese individuals having CHB and 2201 controls as a result of a two-stage genome-wide association study (GWAS) (Kamatani et al. 2009). A second GWAS using 2667 Japanese patients with persistent HBV infection and 6496 controls revealed an independent effect of CHB susceptibility of SNPs rs2856718 and rs7453920 in the HLA-DQ gene cluster, which is located adjacent to HLA-DP (Mbarek et al. 2011a). Those positive associations have been replicated in several studies with populations of Chinese, Korean, and Saudi Arabian origin (Al-Qahtani et al. 2014; Nishida et al. 2012; Li et al. 2011; Hu et al. 2012). In addition, a study of a Caucasian population revealed a significant association of rs3077 in HLA-DPA1, but not rs9277535 in HLA-DPB1, with HBV infection (Vermehren et al. 2012).
Moreover, there exists a significant association of SNPs rs2856718 and rs9275319 in HLA-DQ as well as rs3077 in HLA-DP with the progression of CHB to HCC (Hu et al. 2012; Jiang et al. 2013; Chen et al. 2013). In contrast, no significant association was revealed between SNPs rs3077 and rs9277542 in HLA-DP in a Korean sample and rs2856718, rs7453920 and rs9275572 in HLA-DQ, and rs3077 and rs9277535 in HLA-DP in the Saudi Arabian sample of patients with HC or HCC (Al-Qahtani et al. 2014; Nishida et al. 2012). Also, for the progression of HBV carriers to CHB, no association of SNPs rs2395309 and rs9277535 in HLA-DP/DQ was revealed in studies of Caucasian and Chinese subjects (Vermehren et al. 2012; Li et al. 2011). Thus, more studies are greatly needed to determine whether the variants in this broad and important genomic region contribute to HBV infection and HBV-related diseases (Jiang et al. 2014).
Considering that previously reported studies examined only limited SNPs within this broad region containing the gene-rich HLA complex (180 protein-coding genes in a span of 4 Mb) (Strachan and Read 2010), in this study, we conducted a comprehensive fine mapping analysis of this region, especially in the HLA-DP/DQ gene clusters, with the primary goals of identifying: (1) which subset of SNPs in this region affect the susceptibility to HBV infection; (2) whether these involvements are dependent or independent; and (3) whether the variants in this regions are associated with the progression of HBV-related diseases.
Subjects and methods
Subject recruitment and demographic characteristics
A total of 1657 patients were recruited from the Outpatient Department of Infectious Diseases, First Affiliated Hospital of Zhejiang University, between February 2011 and August 2012. Within the same period, based on the age and sex proportion of the case group, 1456 control subjects were recruited from among healthy volunteers during the annual physical examination at the First Affiliated Hospital of Zhejiang University. All subjects were examined by a series of laboratory tests including assays for hepatitis B surface antigen (HBsAg), hepatitis B core antibody (HBcAb), hepatitis B e antigen (HBeAg), hepatitis B e antibody (HBeAb), hepatitis B surface antibody (HbsAb), hepatitis C surface antibody (HCVAb), serum alanine aminotransferase (ALT), and HBV DNA. Subjects who were co-infected with hepatitis A, hepatitis C, or hepatitis E virus were excluded from this study. In addition, subjects who were pregnant, had any type of cancer or autoimmune disease, or were positive for human immunodeficiency virus (HIV) infection were excluded. Finally, those subjects with an HBV-vaccination history were excluded. All subjects were unrelated to each other and had Chinese Han ethnicity. Informed written consent was obtained from all the participants, and the project was approved by the Ethical Committee of the First Affiliated Hospital of Zhejiang University School of Medicine.
The case subjects were selected on the basis of laboratory testing results as being “HBsAg positive”, “HBV DNA positive” or “both positive”. The subgroups of all subjects were further classified according to the diagnostic criteria published by the Chinese health authority (Chinese Society of and Chinese Society of Infectious Diseases 2011) as: (1) inactive HBV carrier: seropositive for HBsAg or HBV DNA, persistent normal concentration of ALT, or self-reported as not infected; (2) CHB: seropositive for HBsAg or HBV DNA, persistent or recurrently elevated concentration of ALT for >6 months; (3) hepatic cirrhosis: determined on the basis of medical records of each patient with typical symptoms and signs of ascites, hypersplenism, evaluated ALT, and typical result of imaging examination; or (4) HCC: determined on the basis of medical records of each patient with typical symptoms and signs of evaluated α-fetoprotein and typical result of imaging examination. The control group was defined as “both of HBsAg and HBV DNA negative” and had not received HBV vaccination. This group was separated into two subgroups: (1) healthy control: HBsAg, HBcAb, and HBsAb negative; and (2) spontaneous HBV clearance: HBsAg negative but HBcAb or HBsAb positive, as defined in a previously reported study on the same category (Guo et al. 2011). On the basis of these criteria, a total of 1657 cases and 1456 controls were included for the current study.
Genotyping and quality control
Genomic DNA was extracted from 3 ml of EDTA-treated peripheral venous blood using the Qiagen DNA purification kit according to the manufacturer’s instruction. All SNPs selected from the HLA region on chromosome 6 were genotyped using an Illumina iSelect custom genotyping array according to the Illumina Infinium HD Assay Ultra Manual. Following genotyping, quality control analyses were carried out on all genotyped SNPs, which excluded 68 SNPs with a call rate <97 % and 44 SNPs with P values for Hardy–Weinberg equilibrium (HWE) <10−5 in all samples. Finally, a total of 140 SNPs were included for the association analyses reported in this communication, which included 62 SNPs in HLA-DP, 76 SNPs in HLA-DQ, and 2 SNPs in HLA-DRB1. Additional 96 SNPs selected from different chromosomes by the manufacturer were also included in the iSelect array and used for population structure analysis of all samples examined in this study.
Quality control of each subject sample was further determined by the call rate of all genotyped SNPs. Under this QC step, we removed 9 case and 16 control samples with a call rate of <97 %. Next, we re-checked the clinical information on each subject and excluded 24 control subjects who were born after the year 1992, when the national vaccine project was initiated, and who had received vaccination for HBV.
Finally, a total of 1648 case and 1416 control samples remained with various HBV-related phenotypes for the 140 SNPs. A detailed description of those subjects and their corresponding demographic characteristics is shown in Table 1, which included 394 HBV carriers, 878 CHB patients, 301 HC patients, and 75 HCC patients for the case group and 667 persons with spontaneous HBV clearance and 749 healthy individuals for the control group.
Genetic association analysis
For individual SNP-based association analysis, we used the PLINK program (v. 1.07) (Purcell et al. 2007) to perform multiple logistic regression under the additive genetic model with sex, age, and the first three PCs (to control for population stratification) as covariates. Pair-wise LD values as well as haplotypes and LD blocks were calculated using Haploview (v. 4.2) with the default parameters (Barrett et al. 2005). By computing score statistics under the same genetic model and covariates, we calculated haplotype P values as well as the score for each haplotype and a global P value for each LD block using the R package “haplo.stats” (v. 1.6.8) function “haplo.score” (Schaid et al. 2002). We used “haplo.stats” function “haplo.cc” to calculate odds ratios and 95 % confidence intervals for each haplotype, with the most common haplotype within each block as the baseline. For the most frequent one, we took the second most frequent haplotype as the baseline, as reported (Mbarek et al. 2011a). Only statistically significant results of individual SNPs and major haplotypes with a frequency of ≥5 % after Bonferroni correction for multiple testing are reported in this communication.
To determine the relations among detected genetic associations between LD blocks, we performed conditional analysis by testing all block-included SNPs conditional on each other (Dixon et al. 2014; Mbarek et al. 2011a), which yielded a matrix of conditional P values. To visualize the interaction patterns for associations, we constructed an ideograph with color indicating the independent effect of the blocks and SNPs.
To determine whether these SNPs are associated with the progression of HBV-related diseases, we conducted further association analysis for the following subgroups: HBV carriers vs. CHB and CHB vs. HC. Because of the relative small sample (N = 75), the HCC subgroup was excluded from this part of the analysis. Further, we conducted association analysis for the following subgroups: (a) HBV clearance vs. HBV carriers; (b) HBV clearance vs. CHB; and (c) HBV clearance vs. HC.
Both the conditional analysis and subgroup-based association analyses were performed using PLINK under the additive genetic model with sex, age, and the first three PCs as covariates.
Results
Individual SNP-based association analysis
Of 140 SNPs analyzed in this study, 76 showed significant association with HBV infection after Bonferroni correction for multiple testing (i.e., the adjusted P value is 0.05/140 = 3.57 × 10−4) (Table 2). We not only confirmed significant associations of 12 SNPs in the HLA region (i.e., rs3179779, rs3180554, rs3180553, rs1042190, rs2301220, rs9277341, rs3135021, rs7770370, rs9277533, rs9277534, rs9277535, and rs10484569), which were reported to be significant in the discovery stage of the previous GWAS on HBV (Kim et al. 2013; Kamatani et al. 2009; Mbarek et al. 2011b), but also found 41 novel SNPs in HLA-DP and 23 novel SNPs in HLA-DQ to be significantly associated with HBV infection. Of the 76 detected significant SNPs, 55 reached genome-wide significance (P < 5.0 × 10−8) (Dudbridge and Gusnanto 2008; Gao et al. 2008; Pe’er et al. 2008).
Interestingly, we found that 7 of the 11 SNPs with the most significant associations are located in the 3′-UTR region of HLA-DPB1 (i.e., rs931, rs928, rs9277532, rs9277533, rs9277534, rs9277535, and rs9277536; see block 5 of Table 2 for details), with P values of 2.06 × 10−16 to 5.49 × 10−18. Moreover, these seven SNPs were also found to be significant in subsequent association analyses between the viral-clearance subgroup from the control group and subgroups of HBV carriers, CHB, or HC from the case samples. Detailed association results among these subgroups analysis are shown in Supplementary Table 1.
Also, in the HLA-DP gene cluster, we found four nonsynonymous SNPs to be significantly associated with HBV infection (Table 2). This includes SNPs rs1042190 (P = 7.77 × 10−11) and rs1042178 (P = 2.30 × 10−11) in HLA-DPA1, encoding the HLA-DP α1 chain of class II histocompatibility antigen, and rs1042169 (P = 3.87 × 10−15) and rs14362 (P = 3.98 × 10−5) in HLA-DPA1/B1 and HLA-DPB1, encoding the HLA-DP β1 chain of HLA class II histocompatibility antigen. Of the four nonsynonymous SNPs, three were predicted by the PolyPhen-2 program (Adzhubei et al. 2010) to be “probably damaging,” suggesting they are likely to be functional (Table 3).
Haplotype-based association analysis
By using Haploview (Gabriel et al. 2002), we found five LD blocks for 66 of the 76 SNPs significantly associated with HBV infection (Table 4; Fig. 2). The corresponding position of each block with its included SNPs and gene locations is shown in Table 2 and Fig. 1. As shown in Table 4, there are two LD blocks in the HLA-DQ cluster, with Block 1 consisting of 9 SNPs in HLA-DQA1/B1 and Block 2 consisting of 12 SNPs in HLA-DQA2/B2, whereas in the HLA-DP cluster, there are three blocks, with Block 3 consisting of 35 SNPs in HLA-DPA1, Block 4 consisting of 2 SNPs in the HLA-DPA1/B1 overlapping region, and Block 5 consisting of 8 SNPs in HLA-DPB1.

a Scatter plot of the 76 SNPs associated significantly with HBV infection; b relation of block and gene locus; c LD structures for 76 SNPs in the HLA-DP/DQ clusters, which were drawn by Haploview. The details of these SNPs and block information are shown in Supplementary Figure 1 with D′ in each box

Five LD block maps based on D′ value were constructed by Haploview with the genotypes of all case and control samples included. Blocks 1 and 2 are located in the HLA-DQ gene and Blocks 3, 4, and 5 in the HLA-DP gene
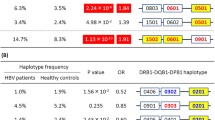
Following determination of LD blocks and major haplotypes (defined as a frequency ≥5 %) within each block, we performed association analysis using the Haplo.stats program to determine their associations with HBV infection (Table 4). All five LD blocks were significantly associated with HBV infection, with a global P value of 4.51 × 10−21, 4.0 × 10−9, 3.42 × 10−8, 8.86 × 10−16, and 1.75 × 10−16 for Blocks 1 to 5, respectively. On the basis of the positive haplotype scores, we identified one risky major haplotype for HBV infection from each block: GGGCAAAAA in Block 1 (haplotype score 5.968; P = 2.40 × 10−9), AAGTAGAAGCGG in Block 2 (haplotype score 6.454; P = 1.09 × 10−10), CACGAGAAAAGAAGATGGCCCGAGGGAGAGAGAGG in Block 3 (haplotype score 5.675; P = 1.39 × 10−8, AG in Block 4 (haplotype score 8.13; P = 4.31 × 10−16), and CACAAGGA in Block 5 (haplotype score 7.613; P = 2.67 × 10−14). In contrast, we detected at least one major haplotype from each block that showed a protective effect against HBV infection: GGAAAGGAG (haplotype score −7.176; P = 7.19 × 10−13) and AGGCAAAGG (haplotype score −5.683; P = 1.32 × 10−8) in Block 1, GGAAGAGGAGAA (haplotype score −6.371; P = 1.88 × 10−10) in Block 2, ATAAGAGCGGATGATAAAGGAACAAAGCGAGAGCA (haplotype score −5.591; P = 2.25 × 10−8) in Block 3, CA (haplotype score −7.881; P = 3.25 × 10−15) in Block 4, and CGGCGAAG (haplotype score −7.45; P = 9.34 × 10−14) in Block 5 (Table 4). Detailed haplotype-based association results and their corresponding frequency <0.05 are presented in Supplementary Table 2.
Conditional association analysis
To further investigate the relations among the five LD blocks and determine whether the significant associations with HBV infection detected in the HLA-DP/DQ gene clusters were attributable to certain SNP(s) or block(s), we conducted a conditional analysis on the 66 initial significant SNPs from the five LD blocks using these SNPs themselves as covariates. The results from such analyses are presented in Fig. 3, where we use the horizontal signal set as covariates and the vertical signal set as the adjusted results.

a Pattern of the interaction between gene clusters HLA-DQ and HLA-DP. b, c Pattern of the interaction within gene clusters HLA-DQ and HLA-DP. d Pattern of the interaction of the five LD blocks with the horizontal signal set as covariates and the vertical signal set as the result after adjustments. White indicates significant association with HBV infection; light grey indicates mixed region; dark grey indicates no significant association after adjustment by other block(s); black indicates no significant association after adjustment by SNPs within the same block
To determine whether variants in the HLA-DP and HLA-DQ clusters contribute to HBV infection jointly or independently, we analyzed the P value matrix at the gene cluster level (Fig. 3a). By using the SNPs in HLA-DQ as covariates (horizontal signal), we found it had no impact on the association of SNPs in HLA-DP with HBV infection (vertical signal; presented as white). Likewise, the association of SNPs in HLA-DQ with HBV infection remained significant even after adjustment for the SNPs in the HLA-DP cluster. This clearly indicates that our detected significant associations of SNPs in HLA-DQ and HLA-DP with HBV infection are independent. In other words, the SNPs in these two gene clusters independently contribute to HBV infection.
Next, within each gene cluster, we wanted to determine which LD block contributes most to the detected association signal (Fig. 3b, c). As stated earlier, in the HLA-DP locus, there are Block 3 (in HLA-DPA1), Block 4 (in HLA-DPA1/B1 overlapping region), and Block 5 (in HLA-DPB1) (Fig. 3b). When the SNPs in Block 3 were considered as covariates, the association of SNPs in Blocks 4 and 5 with HBV infection remained significant (shown in white in the figure). When we used SNPs in Block 4 as covariates, SNPs in Block 5 remained significantly associated with HBV infection (white), but not for the SNPs in Block 3 (dark gray). Further, we detected no significant association between SNPs in Block 4 or Block 3 with HBV infection when SNPs in Block 5 were used as covariates (dark grey). From these analyses, we thus concluded that the variants in Block 5 within HLA-DPB1 contribute to the most significant effect on the association of variants in this locus with HBV infection.
With the same approach, we analyzed the relation between Block 1 (i.e., HLA-DQA1/B1) and Block 2 (HLA-DQA2/B2) in the HLA-DQ cluster, which revealed a significant association of variants in Block 1 with HBV infection regardless of whether we adjusted for SNPs in Block 2 or vice versa (Fig. 3c). This indicates that the variants in Blocks 1 and 2 contribute independently to the detected association of HLA-DQ locus with HBV infection.
To validate these conditional analysis results, we used the same approach to analyze all individual SNPs within each LD block and found that none of the SNPs showed a significant association with HBV infection when conditioned on other SNPs for any given LD block (Fig. 3d). This confirms that the SNPs within the same LD block are in strong LD with each other and that these LD blocks represent the primary genetic unit of this region in determining the susceptibility of the HLA-DQ/DP locus to HBV infection.
Further association analysis among different subgroups
To determine whether SNPs in the HLA-DP and HLA-DQ clusters contribute to the progression of HBV-related diseases, we conducted association analysis of HBV carriers vs. CHB and CHB vs. HC, which revealed that none of the analyzed SNPs in the HLA-DP and HLA-DQ clusters was significant (data not shown). Thus, we conclude that the SNPs in the HLA-DQ/DP clusters are unlikely to influence the progression of HBV-related diseases, at least in our current investigated samples.
Further, we performed association analysis between the viral-clearance subgroup from the control sample and the subgroups of CHB, LC, or IHBV carriers from the case sample. These analyses revealed that a great number of SNPs between the HBV clearance and nine SNPs (located primarily in the 3′-UTR region of HLA-DPB1) between the HBV clearance and LC groups were significant, and no SNP was significant between the HBV clearance and IHBV carriers (see Supplementary Table 1).
Discussion
In this study, we not only confirmed but also greatly expanded the association of SNPs in the HLA-DP/HLA-DQ gene clusters on chromosome 6 with HBV infection by genotyping and analyzing 140 SNPs from the region in 1648 hepatitis cases and 1416 controls. With conditional analysis, we demonstrated that the variants in the HLA-DP and HLA-DQ clusters contribute independently to HBV infection and the variants in HLA-DPB1 play a leading role in HBV infection. Furthermore, we conducted association analysis between HBV carriers vs. CHB subjects and CHB vs. HC subjects and found that the variants in this genomic region were not significantly associated with the progression of HBV-related diseases except for HBV infection.
During the past several years, both GWAS and candidate gene-based association studies of the HLA region on chromosome 6 have been conducted with different ethnic populations (Al-Qahtani et al. 2014; Nishida et al. 2012; Li et al. 2011; Hu et al. 2012; Vermehren et al. 2012; Mbarek et al. 2011a; Kamatani et al. 2009). One of the most convincing findings was identification of SNPs in the HLA-DP and HLA-DQ clusters as susceptibility loci for HBV infection (Jiang et al. 2014). However, within this complex genomic region, almost all previous studies examined only a limited number of SNPs, and the detailed relation between the effects of those SNPs in each cluster with HBV infection has not been fully investigated. For example, to the best of our knowledge, whether the variants in the HLA-DP and HLA-DQ clusters contribute to HBV infection either jointly or independently has rarely been addressed in the literature. However, determination of the effects of the two gene clusters on HBV infection is highly significant for understanding the mechanisms underlying such infection.
Significant associations of SNPs in the HLA-DP cluster with HBV infection have been replicated by several studies with different samples, which include SNPs rs3077 in HLA-DPA1 and rs9277535 in HLA-DPB1 (Jiang et al. 2014). In the current study, we not only confirmed the association of rs9277535 with HBV infection, but also revealed additional novel SNPs that are significantly associated with HBV infection. Of the SNPs identified in this study, the seven top SNPs significantly associated with HBV infection are all located in the 3′-UTR region of HLA-DPB1 gene with P values of 10−17 to 10−18, which are much smaller than 5.0 × 10−8, a commonly accepted P value for genome-wide significance (Dudbridge and Gusnanto 2008; Gao et al. 2008; Pe’er et al. 2008). In addition, we predict with the PolyPhen-2 program (Adzhubei et al. 2010) that three missense SNPs (i.e., rs1042190, rs1042169, and rs14362) in the HLA-DP cluster are likely to be functional, which deserve further investigation in the near future.
Through haplotype-based association analysis and the following conditional analysis, we demonstrated that SNPs in Block 5 within the HLA-DPB1 locus, which contains Blocks 3–5, play an essential role in determining the observed association of this region with HBV infection. Moreover, significant association of SNPs in Block 5 with HBV infection was confirmed by independent association analysis between the viral-clearance and CHB groups. Thus, our current study not only revealed more SNPs within the HLA-DP gene cluster to be associated significantly with HBV infection, but also demonstrated the SNPs in Block 5 (HLA-DPB1) as the main susceptibility ones for HBV infection.
Independent of the HLA-DP cluster, SNPs rs2856718 and rs7453920 in the HLA-DQ cluster have been reported to be associated with HBV infection (Mbarek et al. 2011a; Hu et al. 2012). Another SNP, rs9275319, in the HLA-DQ cluster was also found to be associated with HBV infection in a Chinese population (Jiang et al. 2013). In addition to confirming the significant association of the SNPs rs2856718 and rs9275319 in the HLA-DQ cluster reported by others (Mbarek et al. 2011a; Al-Qahtani et al. 2014; Nishida et al. 2012; Li et al. 2011; Hu et al. 2012), our fine mapping analysis reported here revealed 21 more novel SNPs to be associated significantly with HBV infection. Moreover, by using haplotype-based association and conditional analysis, we found highly significant associations between SNPs or haplotypes in the HLA-DQ cluster with HBV infection. Together, our results not only validated, but also extended the significant association of SNPs and haplotypes in the HLA-DQ gene cluster with HBV infection.
We also investigated the potential association of SNPs in HLA-DP/DQ with the progression of HBV-related diseases. By comparing the HBV carrier and CHB groups, our results were in accordance with the Caucasian study or another study with Chinese populations, where no significant association of SNPs in HLA-DP/DQ with the progression of HBV-related diseases was detected (Vermehren et al. 2012; Li et al. 2011). There has been controversy regarding the association of the HLA region and the transformation of CHB to HC or HCC according to the reported studies on Chinese populations (Hu et al. 2012; Jiang et al. 2013; Chen et al. 2013) and eastern Asian or Saudi Arab populations (Al-Qahtani et al. 2014). Specifically, in this study, we observed no significant association of SNPs in the HLA-DP/DQ region with the progression of CHB to HC. Because of the small sample size, no association analysis was performed with our HCC samples. Thus, future study with a large HCC sample is greatly needed to determine whether there exists any significant association between the SNPs in these two gene clusters and the progression from simple HBV infection to HCC.
In sum, there are three main findings from this study. First, by genotyping significantly more SNPs from the HLA-DP/DQ gene clusters on chromosome 6 in a Chinese Han population with a large sample and employing various association analysis approaches, we not only validated a significant association of a limited number of SNPs in this region with HBV infection, but also revealed many more SNPs that are significantly associated with HBV infection. Interestingly, we showed that almost all top SNPs associated significantly with HBV infection are all located in the 3′-UTR region of HLA-DPB1, indicating this is an important region for future molecular study to define how the HLA-DP/DQ clusters are involved in HBV infection at the molecular level. Second, through a series of conditional analyses of associated P values of SNPs or haplotypes within this region with HBV infection, we discovered for the first time that the HLA-DP and HLA-DQ regions are independently involved in the process underlying HBV infection. Finally, our results indicate that the variants in this genomic region are less likely to be associated with the progression of HBV-related diseases. Because of the relative small sample for the HCC group, no association analysis could be performed of SNPs in this genomic region with HCC. Thus, a large sample of patients with HCC is greatly needed to determine whether this region is indeed related to the progression from CHB to HCC. Similarly, more molecular analysis of SNPs in the 3′-UTR of HLA-DPB1 is necessary to determine how these variants contribute to the process underlying HBV infection.
References
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249. doi:10.1038/nmeth0410-248
Ahn SH, Han KH, Park JY, Lee CK, Kang SW, Chon CY, Kim YS, Park K, Kim DK, Moon YM (2000) Association between hepatitis B virus infection and HLA-DR type in Korea. Hepatology 31(6):1371–1373. doi:10.1053/jhep.2000.7988
Al-Qahtani AA, Al-Anazi MR, Abdo AA, Sanai FM, Al-Hamoudi W, Alswat KA, Al-Ashgar HI, Khalaf NZ, Eldali AM, Viswan NA, Al-Ahdal MN (2014) Association between HLA Variations and Chronic Hepatitis B Virus Infection in Saudi Arabian Patients. PLoS One 9(1):e80445. doi:10.1371/journal.pone.0080445
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21(2):263–265
Chen K, Shi W, Xin Z, Wang H, Zhu X, Wu X, Li Z, Li H, Liu Y (2013) Replication of genome wide association studies on hepatocellular carcinoma susceptibility Loci in a chinese population. PLoS One 8(10):e77315. doi:10.1371/journal.pone.0077315
Chinese Society of H, Chinese Society of Infectious Diseases CMA (2011) The guideline of prevention and treatment for chronic hepatitis B (2010 version). Zhonghua Gan Zang Bing Za Zhi 19(1):13–24. doi:10.3760/cma.j.issn.1007-3418.2011.01.007
Cui Y, Jia J (2013) Update on epidemiology of hepatitis B and C in China. J Gastroenterol Hepatol 28(Suppl 1):7–10. doi:10.1111/jgh.12220
Desombere I, Willems A, Leroux-Roels G (1998) Response to hepatitis B vaccine: multiple HLA genes are involved. Tissue Antigens 51(6):593–604
Dixon PH, Wadsworth CA, Chambers J, Donnelly J, Cooley S, Buckley R, Mannino R, Jarvis S, Syngelaki A, Geenes V, Paul P, Sothinathan M, Kubitz R, Lammert F, Tribe RM, Ch’ng CL, Marschall HU, Glantz A, Khan SA, Nicolaides K, Whittaker J, Geary M, Williamson C (2014) A comprehensive analysis of common genetic variation around six candidate loci for intrahepatic cholestasis of pregnancy. Am J Gastroenterol 109(1):76–84. doi:10.1038/ajg.2013.406
Dudbridge F, Gusnanto A (2008) Estimation of significance thresholds for genomewide association scans. Genet Epidemiol 32(3):227–234. doi:10.1002/gepi.20297
Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D (2002) The structure of haplotype blocks in the human genome. Science 296(5576):2225–2229
Gao X, Starmer J, Martin ER (2008) A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet Epidemiol 32(4):361–369. doi:10.1002/gepi.20310
Guo X, Zhang Y, Li J, Ma J, Wei Z, Tan W, O’Brien SJ (2011) Strong influence of human leukocyte antigen (HLA)-DP gene variants on development of persistent chronic hepatitis B virus carriers in the Han Chinese population. Hepatology 53(2):422–428. doi:10.1002/hep.24048
Hu L, Zhai X, Liu J, Chu M, Pan S, Jiang J, Zhang Y, Wang H, Chen J, Shen H, Hu Z (2012) Genetic variants in human leukocyte antigen/DP-DQ influence both hepatitis B virus clearance and hepatocellular carcinoma development. Hepatology 55(5):1426–1431. doi:10.1002/hep.24799
Jiang YG, Wang YM, Liu TH, Liu J (2003) Association between HLA class II gene and susceptibility or resistance to chronic hepatitis B. World J Gastroenterol 9(10):2221–2225
Jiang DK, Sun J, Cao G, Liu Y, Lin D, Gao YZ, Ren WH, Long XD, Zhang H, Ma XP, Wang Z, Jiang W, Chen TY, Gao Y, Sun LD, Long JR, Huang HX, Wang D, Yu H, Zhang P, Tang LS, Peng B, Cai H, Liu TT, Zhou P, Liu F, Lin X, Tao S, Wan B, Sai-Yin HX, Qin LX, Yin J, Liu L, Wu C, Pei Y, Zhou YF, Zhai Y, Lu PX, Tan A, Zuo XB, Fan J, Chang J, Gu X, Wang NJ, Li Y, Liu YK, Zhai K, Hu Z, Liu J, Yi Q, Xiang Y, Shi R, Ding Q, Zheng W, Shu XO, Mo Z, Shugart YY, Zhang XJ, Zhou G, Shen H, Zheng SL, Xu J, Yu L (2013) Genetic variants in STAT4 and HLA-DQ genes confer risk of hepatitis B virus-related hepatocellular carcinoma. Nat Genet 45(1):72–75. doi:10.1038/ng.2483
Jiang X, Ma Y, Cui W, Li MD (2014) Association of variants in HLA-DP on chromosome 6 with chronic hepatitis B virus infection and related phenotypes. Amino Acids 46(8):1819–1826. doi:10.1007/s00726-014-1767-2
Kamatani Y, Wattanapokayakit S, Ochi H, Kawaguchi T, Takahashi A, Hosono N, Kubo M, Tsunoda T, Kamatani N, Kumada H, Puseenam A, Sura T, Daigo Y, Chayama K, Chantratita W, Nakamura Y, Matsuda K (2009) A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nat Genet 41(5):591–595. doi:10.1038/ng.348
Kim YJ, Kim HY, Lee JH, Yu SJ, Yoon JH, Lee HS, Kim CY, Cheong JY, Cho SW, Park NH, Park BL, Namgoong S, Kim LH, Cheong HS, Shin HD (2013) A genome-wide association study identified new variants associated with the risk of chronic hepatitis B. Hum Mol Genet 22(20):4233–4238. doi:10.1093/hmg/ddt266
Li J, Yang D, He Y, Wang M, Wen Z, Liu L, Yao J, Matsuda K, Nakamura Y, Yu J, Jiang X, Sun S, Liu Q, Song Q, Chen M, Yang H, Tang F, Hu X, Wang J, Chang Y, He X, Chen Y, Lin J (2011) Associations of HLA-DP variants with Hepatitis B virus infection in Southern and Northern Han Chinese Populations: a multicenter case-control study. PLoS One 6(8):e24221. doi:10.1371/journal.pone.0024221
Lin TM, Chen CJ, Wu MM, Yang CS, Chen JS, Lin CC, Kwang TY, Hsu ST, Lin SY, Hsu LC (1989) Hepatitis B virus markers in Chinese twins. Anticancer Res 9(3):737–741
Mbarek H, Ochi H, Urabe Y, Kumar V, Kubo M, Hosono N, Takahashi A, Kamatani Y, Miki D, Abe H, Tsunoda T, Kamatani N, Chayama K, Nakamura Y, Matsuda K (2011a) A genome-wide association study of chronic hepatitis B identified novel risk locus in a Japanese population. Hum Mol Genet. doi:10.1093/hmg/ddr301
Mbarek H, Ochi H, Urabe Y, Kumar V, Kubo M, Hosono N, Takahashi A, Kamatani Y, Miki D, Abe H, Tsunoda T, Kamatani N, Chayama K, Nakamura Y, Matsuda K (2011b) A genome-wide association study of chronic hepatitis B identified novel risk locus in a Japanese population. Hum Mol Genet 20(19):3884–3892. doi:10.1093/hmg/ddr301
Nishida N, Sawai H, Matsuura K, Sugiyama M, Ahn SH, Park JY, Hige S, Kang JH, Suzuki K, Kurosaki M, Asahina Y, Mochida S, Watanabe M, Tanaka E, Honda M, Kaneko S, Orito E, Itoh Y, Mita E, Tamori A, Murawaki Y, Hiasa Y, Sakaida I, Korenaga M, Hino K, Ide T, Kawashima M, Mawatari Y, Sageshima M, Ogasawara Y, Koike A, Izumi N, Han KH, Tanaka Y, Tokunaga K, Mizokami M (2012) Genome-Wide Association Study Confirming Association of HLA-DP with Protection against Chronic Hepatitis B and Viral Clearance in Japanese and Korean. PLoS One 7(6):e39175. doi:10.1371/journal.pone.0039175
Pe’er I, Yelensky R, Altshuler D, Daly MJ (2008) Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol 32(4):381–385. doi:10.1002/gepi.20303
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81(3):559–575. doi:10.1086/519795
Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA (2002) Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet 70(2):425–434. doi:10.1086/338688
Strachan T, Read A (2010) Human molecular genetics. 4 edn. Garland Science
Sun J, Hou JL (2010) Management of chronic hepatitis B: experience from China. J Viral Hepat 17(Suppl 1):10–17. doi:10.1111/j.1365-2893.2010.01274.x
Szmuness W, Harley EJ, Prince AM (1975) Intrafamilial spread of asymptomatic hepatitis B. Am J Med Sci 270(2):293–304
Vermehren J, Lotsch J, Susser S, Wicker S, Berger A, Zeuzem S, Sarrazin C, Doehring A (2012) A common HLA-DPA1 variant is associated with hepatitis B virus infection but fails to distinguish active from inactive Caucasian carriers. PLoS One 7(3):e32605. doi:10.1371/journal.pone.0032605
Xi-Lin Z, Te D, Jun-Hong L, Liang-Ping L, Xin-Hui G, Ji-Rong G, Chun-Yan G, Zhuo L, Ying L, Hui L (2006) Analysis of HLA-DQB1 gene polymorphisms in asymptomatic HBV carriers and chronic hepatitis B patients in the Chinese Han population. Int J Immunogenet 33(4):249–254. doi:10.1111/j.1744-313X.2006.00607.x
Zang GQ, Xi M, Feng ML, Ji Y, Yu YS, Tang ZH (2004) Curative effects of interferon-alpha and HLA-DRB1 -DQA1 and -DQB1 alleles in chronic viral hepatitis B. World J Gastroenterol 10(14):2116–2118
Acknowledgments
We thank all the volunteers who participated in this study. We also thank the many clinical research staff and clinicians in the First Affiliated Hospital of Zhejiang University School of Medicine for helping us to recruit these participants.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This study was in part supported by the National Science and Technology Major Project (No. 2012ZX10002004) and the Chinese High Technology Research and Development program (No. 2012AA020204).
Conflict of interest
The authors declare no conflict of interest related to this paper.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tao, J., Su, K., Yu, C. et al. Fine mapping analysis of HLA-DP/DQ gene clusters on chromosome 6 reveals multiple susceptibility loci for HBV infection. Amino Acids 47, 2623–2634 (2015). https://doi.org/10.1007/s00726-015-2054-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-015-2054-6