
Abstract
Protein–nucleic acid interaction is an important process in many biological phenomena. In this study, a fluorescence resonance energy transfer (FRET)-based protein–DNA binding assay has been developed, in which a fluorescent amino acid is genetically incorporated into a DNA-binding protein. A coumarin-containing amino acid was incorporated into a DNA-binding protein, and the mutant protein specifically produced a FRET signal upon binding to its cognate DNA labeled with a fluorophore. The protein–DNA binding affinity was then measured under equilibrium conditions. This method is advantageous for studying protein-nucleic acid interactions, because it is performed under equilibrium conditions, technically easy, and applicable to any nucleic acid-binding protein.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Recognition of nucleic acids by proteins is a crucial interaction in many biological phenomena, such as gene expression and regulation, nucleic acid transport, formation of the chromatin structure, DNA replication and repair, and translation of messenger RNAs. To characterize the interactions between proteins and nucleic acids, their affinities and specificities need to be determined. There are several methods available for determining protein–nucleic acid interactions, and each has advantages and disadvantages depending on the purpose of the studies (Cai and Huang 2012; Anderson et al. 2008; Hellman and Fried 2007). Traditional methods include electrophoretic mobility shift assay (EMSA), filter-binding assays, and footprinting assay. They are extremely sensitive and able to analyze very small amounts of nucleic acids because they use radioisotope-labeled nucleic acids. However, EMSA and filter-binding assay measure samples in a non-equilibrium state, and therefore, the binding affinity measured by these methods can be different from the real affinity (Fried and Liu 1994; Fried and Bromberg 1997; Vossen and Fried 1997; Woodbury and von Hippel 1983; Oehler et al. 1999). The footprinting assay has an important advantage in that it is carried out under conditions of binding equilibrium for proteins and nucleic acids, and its variants can provide quantitative information about their binding (Brenowitz et al. 1986). On the other hand, footprinting assays are difficult to perform, and incomplete binding results in indistinct footprint patterns. Another popular method is isothermal titration calorimetry (ITC), which is advantageous because it provides binding information under equilibrium conditions, and does not require protein and nucleic acid labeling (Oda and Nakamura 2000). A critical limitation of ITC is that it requires a considerably large amount of material, which causes difficulty in measuring the exact binding affinity especially for high-affinity binding complexes. In this report, we present a general method for measuring protein–nucleic acid interactions based on fluorescent resonance energy transfer (FRET) in which a fluorescent amino acid is genetically incorporated into a nucleic acid-binding protein.
Materials and methods
Protein expression and purification
Protein expression and purification were carried out as previously described. The plasmid, pBAD-CAP-K26TAG, was co-transformed with pEvol-CouRS into E. coli DH10B. Transformed cells grown on an agar plate were amplified in LB media supplemented with ampicillin (100 μg/mL) and chloramphenicol (35 μg/mL). The starter culture (2.5 mL) was used to inoculate 100 mL LB supplemented with ampicillin (100 μg/mL), chloramphenicol (35 μg/mL), and 1 mM CouA at 37 °C. Expression was induced at optical density 0.8 (550 nm) by adding 0.2 % l-arabinose. Cells were grown at 37 °C for 10–12 h and harvested by centrifugation. The target protein was purified by Strep-Tactin affinity chromatography under native conditions according to the manufacturer’s protocol (Novagen). The cells were resuspended in 10 mL lysis buffer (100 mM Tris–HCl, pH 8.0, 150 mM NaCl, 10 mM EDTA) and sonicated on ice. The cell debris was removed by centrifugation at 4 °C, and Strep-Tactin resin (400 μL) was added to the supernatant. The mixture was incubated for 30 min at 4 °C, and the protein-bound resin was washed with wash buffer (1 mL × 3, 100 mM Tris–HCl, pH 8.0, 150 mM NaCl, 10 mM EDTA). The protein was collected by elution with elution buffer (100 mM Tris–HCl, pH 8.0, 150 mM NaCl, 10 mM EDTA, 25 mM desthiobiotin).
DNA-binding assay
5′-FAM-labeled double-stranded DNA (dsDNA) was prepared from 5′-FAM-labeled single-stranded DNA (ssDNA) and its complementary ssDNA (unlabeled). The sequences of DNAs used in this experiment are shown in Fig. S1. The 5′-FAM-labeled DNA (10 pmol) was mixed with the unlabeled complementary strand (11 pmol) in 10 μL of 100 mM potassium acetate and 30 mM HEPES (pH 7.5). The mixture was heated at 90 °C for 2 min and cooled gradually (approximately 2 h) to room temperature to form a duplex. The 5′-FAM-labeled dsDNA (125 nM) was incubated with CAP-K26CouA (700 nM) in 10 mM 3-(N-morpholino)propanesulfonic acid (MOPS, pH 7.3) containing 0.2 mM cAMP, and fluorescence was scanned from 400 to 600 nm with excitation at 360 nm. To measure the dissociation constant, the 5′-FAM-labeled dsDNA (125 nM) containing the CAP-binding sequence was titrated with CAP-K26CouA (0, 50, 100, 150, 200, 250, 300, 400, 600, and 800 nM), and fluorescence was measured at 523 nm with excitation at 360 nm.
EMSA
The 5′-FAM-labeled dsDNA (125 nM) containing the CAP-binding sequence was incubated with CAP-WT and CAP-K26CouA at various concentrations (0, 50, 100, 150, 200, 250, 300, 400, 600, and 800 nM, total volume 10 μL) in 10 mM MOPS (pH 7.3) containing 0.2 mM cAMP for 10 min. The solutions were loaded on 10 % Tris/Borate/EDTA (TBE) gel (Invitrogen), and analyzed according to the manufacturer’s protocol (Invitrogen, TBE running buffer, 200 V, 45 min running time, room temperature). Fluorescence images were taken using a Typhoon 9210 variable mode imager.
Results
Recently, fluorescent amino acids have been genetically incorporated into proteins in bacteria (Wang et al. 2006; Speight et al. 2013), yeast (Summerer et al. 2006; Lee et al. 2009a), and mammalian cells (Chatterjee et al. 2013) using orthogonal aminoacyl-tRNA (aa-tRNA)/aminoacyl-tRNA synthetase (aa-RS) pairs. The amino acids were used to detect protein folding (Wang et al. 2006; Speight et al. 2013; Summerer et al. 2006), conformational change of a protein upon ligand binding (Lee et al. 2009a), and cellular localization of various proteins (Chatterjee et al. 2013). Each fluorescent amino acid has advantages and disadvantages in terms of brightness, compatible cells for their incorporation into a protein, environmental sensitivity, and synthetic easiness. Because these amino acids can be easily incorporated into proteins and fluorophore-labeled nucleic acids can be obtained from commercial sources. It was expected that the genetic incorporation of a fluorescent amino acid into a nucleic acid binding protein would allow us to monitor the interaction between the protein containing a fluorescent amino acid and a fluorophore-labeled nucleic acid by measuring FRET between the two biomolecules (Fig. 1).

Protein–DNA interaction assay design. The DNA-binding protein containing a fluorescent amino acid produces a FRET signal upon binding to its cognate DNA labeled with a fluorophore
To analyze protein–nucleic acid interaction by FRET, l-(7-hydroxycoumarin-4-yl)ethylglycine (CouA) was used as a fluorescent amino acid because bacterial cells could be used for its incorporation, and it could be readily prepared in two synthetic steps (Wang et al. 2006). For a nucleic acid-binding protein, catabolite activator protein (CAP) was chosen because the protein is functionally and structurally well characterized, and has a high affinity to its cognate DNA (De Crombrugghe et al. 1984; Berg and von Hippel 1988; Schultz et al. 1991; Parkinson et al. 1996). Based on the X-ray crystal structure of CAP complexed with its cognate DNA (Schultz et al. 1991; Parkinson et al. 1996), Lys-26 was selected for the incorporation of CouA. Lys-26 is positioned at the protein–DNA interface of the DNA recognition site and has been chosen for chemical derivatization with an electrophilic phenanthroline derivative to create a selective affinity-cleaving agent (Pendergrast et al. 1994) and for the incorporation of other unnatural amino acids (Lee and Schultz 2008; Lee et al. 2009b).
To incorporate CouA, an amber (TAG) mutation was introduced into the position for Lys-26 in the CAP gene containing a C-terminal Strep-tag. CouA was synthesized starting from N-Cbz-l-glutamic acid benzyl ester as previously described (Wang et al. 2006). CAP containing CouA was expressed in the presence of the evolved aa-tRNA/aa-RS (CouRS) pair and CouA in E. coli. Mutant and wildtype (WT) CAP were purified by Strep-Tactin affinity chromatography, and the yield of the mutant CAP was 3–6 mg/L (CAP-WT yield was 10–15 mg/L). Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis showed that the full-length CAP was expressed in the presence of CouA, while little CAP was expressed in the absence of CouA, and the fluorescence image confirmed the incorporation of the fluorescent amino acid (Fig. 2a). In addition, electrospray ionization mass spectroscopic (ESI–MS) analysis showed the selective incorporation of CouA with no incorporation of any natural amino acid (Fig. 2b).

SDS-PAGE and ESI–MS analyses of purified CAP-K26CouA. a Left coumassie-stained gel image; right fluorescence image. b ESI–MS results. The inset shows the deconvoluted spectrum: calculated 25,277, observed 25,276
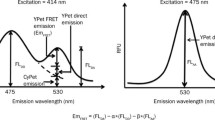
Next, we examined whether the CAP mutant (CAP-K26CouA) containing CouA generates a FRET signal upon binding to the cognate DNA, a 50-bp dsDNA labeled with fluorescein amidite (FAM) at the 5′-end. FAM was chosen as a fluorophore for DNA labeling because its absorption spectrum showed a significant overlap with the emission spectrum of CouA (Wang et al. 2006). The labeled DNA was treated with CAP-K26CouA and fluorescence was measured with excitation at 360 nm. Upon the addition of the CAP mutant, a significant fluorescence increase at 520 nm and decrease at 450 nm were observed (Fig. 3a). In contrast, in the same experiment performed using the DNA containing no CAP binding site, a slight fluorescence increase at 520 nm and no decrease at 450 nm were observed. The small fluorescence increase at 520 nm for the noncognate DNA was likely due to nonspecific binding of the CAP mutant to the DNA. These results showed that the CAP mutant containing CouA specifically produced a FRET signal upon binding to its cognate DNA labeled with FAM and could be used to measure protein–DNA interactions. We also used dsDNA labeled with FAM on both strands for the same experiment. However, we did not observe a significant FRET signal from this experiment for an unknown reason.

FRET experiments using CAP-K26CouA and 5′-FAM-labeled dsDNA. a Emission spectra from the complex of CAP-K26CouA (700 nM) and 5′-FAM-labeled dsDNA (125 nM) containing the CAP binding site with excitation at 360 nm. Noncognate DNA was also used for comparison. b 5′-FAM-labeled dsDNA (125 nM) containing the CAP binding site was titrated with CAP-K26CouA and fluorescence intensity at 523 nm was measured with excitation at 360 nm. Each data point represents an average based on assays conducted in triplicate
The FAM-labeled DNA with the CAP binding site was then titrated with CAP-K26CouA to measure the binding affinity. Fluorescence was measured at 523 nm with excitation at 360 nm. The fluorescence increased with increasing concentration of CAP-K26CouA and was saturated at 500 nM protein (Fig. 3a). We observed a small fluorescence increase from CAP-K26CouA itself because of the weak fluorescence of CouA at 523 nm, which was subtracted from the measurement. The dissociation constant (K d) calculated from this result was 191 nM. For comparison, the dissociation constant was also measured by EMSA (Fig. 4). This assay was performed for both CAP-WT and CAP-K26CouA, and the dissociation constants were 219 and 233 nM, respectively. The K d value for CAP-K26CouA measured by the FRET experiment was comparable to the value measured by EMSA. In these experiments, the difference was not significant even though the EMSA was in a non-equilibrium state (Fried and Liu 1994; Fried and Bromberg 1997; Vossen and Fried 1997; Woodbury and von Hippel 1983). More importantly, the dissociation constants for CAP-WT and CAP-K26CouA measured by EMSA were comparable, showing that the incorporation of CouA did not affect the binding affinity of CAP (Lee and Schultz 2008; Lee et al. 2009b).

EMSA of CAP-WT and CAP-K26CouA with 5′-FAM-labeled dsDNA
Discussion
Analysis of protein–nucleic acid interactions is an important topic in the study of many biological processes involving these interactions. Many methods have been developed and used to identify and measure these interactions, and each method has its unique advantages and disadvantages. The important factors for the evaluation of these methods include sensitivity, assay conditions (equilibrium or non-equilibrium), and requirement for protein and nucleic acid labeling. As a minor factor, the ease of use of the assay should also be considered. The method described in this report uses FRET to analyze protein–nucleic acid interactions. The key challenge in using this method is the incorporation of fluorophores for FRET into the biomolecules. Currently, fluorophore-labeled proteins cannot be obtained from commercial sources, whereas fluorophore-labeled nucleic acids are readily available. In our method, a fluorescent amino acid is genetically introduced into a nucleic acid-binding protein; the fluorescent amino acid is encoded by using an amber codon and an engineered aa-tRNA/aa-RS pair (Wang and Schultz 2005; Liu and Schultz 2010). This genetic incorporation method is technically easy, quantitative, and high yielding. In addition, this method can be applied to any position in any protein. Because the method uses fluorescence spectroscopy, it allows measurements under equilibrium conditions and provides excellent sensitivity.
In terms of sensitivity, EMSA and filter-binding assays are superior to this method, because they use radioisotope-labeled nucleic acids. However, these assays measure the interactions under non-equilibrium conditions and show discrepancies between measured and real binding affinities (Fried and Liu 1994; Fried and Bromberg 1997; Vossen and Fried 1997; Woodbury and von Hippel 1983; Oehler et al. 1999). This is because protein–nucleic acid complexes are often more stable in polyacrylamide gels (Fried and Liu 1994; Vossen and Fried 1997) and they can be dissociated during filtering process in the filter-binding assay (Woodbury and von Hippel 1983; Oehler et al. 1999). In the FRET-based assay reported here, the binding assay is carried out under equilibrium conditions, and the results reflect the real binding affinity. The incorporation of CouA into the DNA-binding protein did not affect its binding affinity to the cognate DNA as shown by the comparison of the binding affinity to that of the WT protein. In addition, the incorporation of unnatural amino acids into proteins in other reports showed negligible effects on protein function (Lee and Schultz 2008, 2009a, b; Park et al. 2012).
Because the method described here is based on FRET, it can be applied to high-throughput screening (HTS). For example, small molecules that perturb protein–DNA interactions can regulate the expression of genes involved in important biochemical processes and pathogenesis of human diseases, and therefore have great potential for drug discovery and applications in biochemical research (Boger et al. 2003). This FRET-based assay can be used to screen a large number of small molecules to identify compounds that inhibit a specific protein–DNA interaction, which is not possible for other protein-nucleic acid assays mentioned above.
A potential issue pertaining to this assay is that an oligomeric form of a nucleic acid-binding protein can produce a complex FRET signal. Although the FRET signal from multiple FRET donors might increase the overall FRET signal, experimental results should be carefully evaluated to better understand the protein-nucleic acid interactions identified by this method.
In summary, a FRET-based protein–DNA-binding assay has been developed in which a fluorescent amino acid is genetically incorporated into a DNA-binding protein. Since a fluorescent amino acid is genetically incorporated into a DNA-binding protein, the incorporation process is quantitative, technically easy, and high yielding. In addition, this method can measure protein–DNA interactions directly in solution while EMSA and filter-binding assays measure the interaction in non-equilibrium state. Furthermore, because the assay can be applied to any nucleic acid-binding protein, it could be a useful and general method for studying protein–nucleic acid interactions.
References
Anderson BJ, Larkin C, Guja K, Schildbach JF (2008) Using fluorophore-labeled oligonucleotides to measure affinities of protein–DNA interactions. Methods Enzymol 450:253–272
Berg O, von Hippel P (1988) Selection of DNA binding sites by regulatory proteins. II. The binding specificity of cyclic AMP receptor protein to recognition sites. J Mol Biol 200:709–723
Boger DL, Desharnais J, Capps K (2003) Solution-phase combinatorial libraries: modulating cellular signaling by targeting protein–protein or protein–DNA interactions. Angew Chem Int Ed Engl 42:4138–4176
Brenowitz M, Senear DF, Shea MA, Ackers GK (1986) Quantitative DNase footprint titration: a method for studying protein–DNA interactions. Methods Enzymol 130:132–181
Cai Y-H, Huang H (2012) Advances in the study of protein–DNA interaction. Amino Acids 43:1141–1146
Chatterjee A, Guo J, Lee HS, Schultz PG (2013) A genetically encoded fluorescent probe in mammalian cells. J Am Chem Soc 135:12540–12543
De Crombrugghe B, Busby S, Buc H (1984) Cyclic AMP receptor protein: role in transcription activation. Science 224:831–838
Fried MG, Bromberg JL (1997) Factors that affect the stability of protein–DNA complexes during gel electrophoresis. Electrophoresis 18:6–11
Fried MG, Liu G (1994) Molecular sequestration stabilizes CAP-DNA complexes during polyacrylamide gel electrophoresis. Nucleic Acids Res 22:5054–5059
Hellman LM, Fried MG (2007) Electrophoretic mobility shift assay (EMSA) for detecting protein–nucleic acid interactions. Nat Protoc 2:1849–1861
Lee HS, Schultz PG (2008) Biosynthesis of a site-specific DNA cleaving protein. J Am Chem Soc 130:13194–13195
Lee HS, Guo J, Lemke EA, Dimla RD, Schultz PG (2009a) Genetic incorporation of a small, environmentally sensitive, fluorescent probe into proteins in Saccharomyces cerevisia. J Am Chem Soc 131:12921–12923
Lee HS, Dimla RD, Schultz PG (2009b) Protein–DNA photo-crosslinking with a genetically encoded benzophenone-containing amino acid. Bioorg Med Chem Lett 19:5222–5224
Liu CC, Schultz PG (2010) Adding new chemistries to the genetic code. Annu Rev Biochem 79:413–444
Oda M, Nakamura H (2000) Thermodynamic and kinetic analyses for understanding sequence-specific DNA recognition. Genes Cells 5:319–326
Oehler S, Alex R, Barker A (1999) Is nitrocellulose filter binding really a universal assay for protein–DNA interactions? Anal Biochem 268:330–336
Park N, Ryu J, Jang S, Lee HS (2012) Metal ion affinity purification of proteins by genetically incorporating metal-chelating amino acids. Tetrahedron 68:4649–4654
Parkinson G, Wilson C, Gunaseker A, Ebright YW, Ebright RH (1996) Structure of the CAP-DNA complex at 2.5 Å resolution: a complete picture of the protein–DNA interface. J Mol Biol 260:395–408
Pendergrast PS, Ebright YW, Ebright RH (1994) High-specificity DNA cleavage agent: design and application to kilobase and megabase DNA substrates. Science 265:959–962
Schultz SC, Shields GC, Steitz TA (1991) Crystal structure of a CAP-DNA complex: the DNA is bent by 90 degrees. Science 253:1001–1007
Speight LC, Muthusamy AK et al (2013) Efficient synthesis and in vivo incorporation of acridon-2-ylalanine, a fluorescent amino acid for lifetime and forster resonance energy transfer/luminescence resonance energy transfer studies. J Am Chem Soc 135:18806–18814
Summerer D, Chen S, Wu N, Deiters A, Chin JW, Schultz PG (2006) A genetically encoded fluorescent amino acid. Proc Natl Acad Sci USA 103:9785–9789
Vossen KM, Fried MG (1997) Sequestration stabilizes lac repressor-DNA complexes during gel electrophoresis. Anal Biochem 245:85–92
Wang L, Schultz PG (2005) Expanding the genetic code. Angew Chem Int Ed Engl 44:34–66
Wang J, Xie J, Schultz PG (2006) A genetically encoded fluorescent amino acid. J Am Chem Soc 128:8738–8739
Woodbury CP, von Hippel PH (1983) On the determination of deoxyribonucleic acid-protein interactions parameters using the nitrocellulose filter-binding assay. Biochemistry 22:4730–4737
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (2014003870), and the Sogang University Research Grant (201010048.01). We would like to thank P. G. Schultz for providing us the plasmids.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Handling Editor: D. Tsikas.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Park, H., Kang, H., Ko, W. et al. FRET-based analysis of protein-nucleic acid interactions by genetically incorporating a fluorescent amino acid. Amino Acids 47, 729–734 (2015). https://doi.org/10.1007/s00726-014-1900-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-014-1900-2