
Abstract
Maintenance of amino acid homeostasis is important for healthy cellular function, metabolism and growth. Intracellular amino acid concentrations are dynamic; the high demand for protein synthesis must be met with constant dietary intake, followed by cellular influx, utilization and recycling of nutrients. Autophagy is a catabolic process via which superfluous or damaged proteins and organelles are delivered to the lysosome and degraded to release free amino acids into the cytoplasm. Furthermore, autophagy is specifically activated in response to amino acid starvation via two key signaling cascades: the mammalian target of rapamycin (mTOR) complex 1 (mTORC1) and the general control nonderepressible 2 (GCN2) pathways. These pathways are key regulators of the integration between anabolic (amino acid depleting) and catabolic (such as autophagy which is amino acid replenishing) processes to ensure intracellular amino acid homeostasis. Here, we discuss the key roles that amino acids, along with energy (ATP, glucose) and oxygen, are playing in cellular growth and proliferation. We further explore how sophisticated methods are employed by cells to sense intracellular amino acid concentrations, how amino acids can act as a switch to dictate the temporal and spatial activation of anabolic and catabolic processes and how autophagy contributes to the replenishment of free amino acids, all to ensure cell survival. Relevance of these molecular processes to cellular and organismal physiology and pathology is also discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amino acids and autophagy are mutually dependent on each other. Amino acids are primarily acquired by means of dietary intake and are transported into cells through the plasma membrane-spanning transporters (Poncet and Taylor 2013). Free intracellular amino acids not only serve as a source of metabolites and energy, but also directly contribute to the tight regulation of two pathways, the mTORC1 and GCN2/eIF2 (eukaryotic initiation factor 2) cascades that integrate anabolic and catabolic signals (Fig. 1). These pathways regulate protein translation (albeit to a different degree, with differential sensitivities and of distinct subsets of genes), as well as control the cellular demand for amino acids by concomitantly regulating autophagy-dependent catabolism (Proud 2014; Jewell and Guan 2013; Laplante and Sabatini 2012; Meijer and Dubbelhuis 2004). Autophagy is the general term used to encompass three distinct mechanisms of intracellular lysosome-dependent degradation that are referred to as microautophagy, chaperone-mediated autophagy (CMA) and macroautophagy (Ravikumar et al. 2010b). The specific autophagic substrates and mechanisms of their transport for degradation differ between these different types of autophagy. They all, however, converge on the lysosomes where the autophagic substrates are degraded into their constituent parts by a battery of lysosomal hydrolytic enzymes (Ravikumar et al. 2010b). Transporters on the lysosomal membrane are then responsible for exporting free amino acids back into the cytoplasm. The inhibitory effects of amino acids on autophagy were demonstrated over three decades ago (Seglen et al. 1980; Seglen and Gordon 1984) and there has since been considerable progress in our understanding of the mechanistic details. In this review, we will discuss how amino acids regulate autophagy and discuss the physiological importance of these mechanisms to maintain cellular function.

Overview of physiological processes regulated by amino acids. Schematic representation of biological processes, including autophagy, controlled by amino acid sufficiency or deprivation conditions through the mTORC1 and GCN2 pathways
mTORC1 is the key hub coordinating the availability of amino acids and autophagy. It is selective in its ability to sense different amino acids, amongst which leucine, arginine and glutamine (Hara et al. 1998) are the most important regulators of mTORC1 activity and autophagy. At the molecular level, the proximal regulators of mTORC1 activity are the tuberous sclerosis complex (TSC2)/Rheb (Ras homolog enriched in brain) axis and the Rag small GTPase axis. While inputs such as growth factors, energy and oxygen availability signal to mTORC1 via the TSC2 complex, the primary input for amino acids is mediated by the Rag GTPases (Jewell and Guan 2013; Laplante and Sabatini 2012; Meijer and Codogno 2008). Rag GTPases reside on the cytoplasmic surface of the lysosome and promote the recruitment and retention of mTORC1 to this site during amino acid availability (Sancak et al. 2010). The lysosomal localization of mTORC1 is generally considered to be required for its activation by Rheb (Buerger et al. 2006; Sancak et al. 2008). There has been a rapid expansion in the understanding of the amino acid-dependent regulation of mTORC1 in recent years and a number of amino acid-sensing mechanisms have been postulated which will be described in more detail below.
mTORC1 directly and potently regulates macroautophagy by interacting with the autophagy-initiating protein complex, consisting of Unc-51-like autophagy activating kinase 1 (ULK1), which is phosphorylated and thereby repressed by mTORC1, leading to inhibition of macroautophagy. Inactivation of mTORC1 by stress conditions (including amino acid starvation) leads to the activation of this ULK1-containing pro-autophagic complex, which promotes a signaling cascade to positively regulate the formation, elongation, maturation and finally the degradation of autophagosomes (see below for more details and recent reviews, Lamb et al. 2013; Sarkar 2013b; Klionsky and Schulman 2014). Similar to macroautophagy, CMA and microautophagy can be activated by stress conditions. Moreover, the classical Autophagy-related (Atg) proteins, which were initially discovered in yeast as regulators of macroautophagy can also govern the proper functioning of CMA and microautophagy. The exact molecular mechanisms that initiate CMA and particularly microautophagy (Li et al. 2012), however, are poorly understood. Nonetheless, degradation of autophagy substrates in all these scenarios leads to the release of free amino acids and therefore, all forms of autophagy cooperate to maintain a critical level of intracellular amino acids. Notably, the localization of mTORC1 to the lysosomes allows extremely tight spatial coupling between autophagy-derived free amino acids with cellular growth and proliferation.
Intracellular amino acid concentrations also regulate another serine/threonine kinase, GCN2. During amino acid starvation, GCN2 is activated by increase in ‘uncharged’ transfer RNAs (tRNA), which are those not bound to their target amino acid. The GCN2-dependent phosphorylation of eukaryotic initiation factor 2α (eIF2α) leads to a decrease in the functional complex required for delivering methionine to the ribosome for initiating translation. Consequently, global protein translation is inhibited, however, concomitantly the translation of a subset of mRNAs is in fact increased (Kilberg et al. 2005). These targets are enriched in those encoding amino acid biosynthesis regulators, amino acid transporters and autophagy mediators (Kilberg et al. 2005; Talloczy et al. 2002; Kilberg et al. 2009; B’Chir et al. 2013). As such, GCN2 activation promotes a cellular program aimed at restoring intracellular levels of amino acids.
Despite their mutual role in regulating amino acid homeostasis, the extent to which the mTORC1 and GCN2/eIF2α pathways cooperate is not clear. Although they are generally considered to act independently, there is evidence of reciprocal regulatory processes and complex feedback loops that may impact on wider signaling processes. It is likely that the severity of amino acid starvation as well as differences in amino acid dependency in different cell types dictate the degree of co-operation between these pathways.
Amino acid homeostasis
Intracellular levels of amino acids are maintained by the balance between their influx, utilization (including incorporation into proteins or as metabolic intermediates) and recycling (Fig. 2). There are 20 proteinogenic amino acids, nine of which are classified in mammals as essential due to their inability to be synthesized by cells. A primary fate of free amino acids is their incorporation into newly translated proteins (Proud 2014). Free intracellular amino acids are loaded onto their corresponding tRNA molecule (which is then referred to as ‘charged’) and are then brought into contact with specific, complementary sequences of mRNA at the ribosomes. As such, amino acids are sequentially incorporated into growing polypeptides. Amino acids serving as building blocks for the synthesis of new proteins are fundamentally essential for cellular homeostasis (to replace old or damaged proteins or to maintain the levels of proteins with a short half-life), growth and proliferation. Amino acids are also important metabolic intermediates to support cellular function. For example, glutamate is an excitatory neurotransmitter with well-studied influence on cognitive function, arginine is a precursor for nitric oxide that is an important regulator for vasodilation, and tryptophan is a precursor for the neurotransmitter serotonin. Amino acids thus contribute to an extremely diverse range of cellular processes and cells employ a number of mechanisms to sense and maintain their homeostatic levels.

Amino acid homeostasis. Intracellular levels of amino acids are maintained by a constant influx via the transporters localized on the plasma membrane, utilization (protein translation and metabolism) and recycling (via lysosome-dependent autophagy and ubiquitin–proteasome system). Ala alanine, Arg arginine, αKG α-ketoglutarate, Gln glutamine, Gly glycine, Leu leucine, NOS nitric oxide synthase, PAT1 proton-assisted amino acid transporter 1, Pro proline, SLC solute-linked carrier, SNAT2 sodium-coupled neutral amino acid transporter 2, TCA cycle tricarboxylic acid cycle
Extracellular amino acid influx
Dietary intake accounts for the majority of amino acids in the body. Transport of serum amino acids into cells is an active process that is facilitated by plasma membrane-localized solute-linked carriers (SLC) (Fig. 2). Specifically, members of the SLC1, SLC6, SLC7 (in co-operation with SLC3), SLC16, SLC36, SLC38 and SLC43 families regulate amino acid transport (Poncet and Taylor 2013), although classical nomenclature groups them into systems, such as Systems A, N and L, system y+ and ASC, among others, based on their substrates and mode of transport (Collarini and Oxender 1987; Fotiadis et al. 2013). Solute carriers are typically large, multiple membrane-spanning proteins. Amino acids bind to the extracellular domain of these transporters, thereby causing a conformational change that allows transport of the amino acid into the cell. Transport is usually via symport (co-transport), anti-port or exchange mechanisms which, respectively, result in net increase or maintenance of the intracellular levels of amino acids. Importantly, transport is often reliant on electrochemical and proton gradients across the membrane (Collarini and Oxender 1987; Poncet and Taylor 2013). Many symporters, including the neutral amino acid (particularly glutamine) transporters, such as SLC1A5 and SLC38A2 (SNAT2) couple amino acid and Na+ transport, while SLC36A4 is a proton-assisted transporter (thus also commonly known as PAT4). Members of the SLC7 family can cooperate with SLC3 family to form heterodimers that are involved in amino acid exchange; for example, SLC7A5 (classically referred to as LAT1) dimerises with SLC3A2 (also referred to as CD98/4F2) to regulate the influx of leucine with a concomitant efflux of glutamine (Poncet and Taylor 2013) (Fig. 2).
Individual amino acids can typically enter the cell via more than one transporter and the expression of these amino acid transporters is tissue and cell-type specific (Collarini and Oxender 1987; Broer 2008). Furthermore, the abundance of amino acids can dictate the expression of specific transporters via a process referred to as adaptive regulation. For example, the cationic amino acid transporter SLC7A1 (CAT1) is a sodium-independent transporter of arginine and lysine. The negligible expression of CAT1 in quiescent liver cells is quickly and efficiently upregulated in response to anabolic stimuli (Liu and Hatzoglou 1998). The low basal levels of CAT1 are thought to be an adaptive response to conserve energy and resources since high levels of arginase (an enzyme that converts arginine to ornithine) in the liver would quickly use up any arginine imported into the cell (Liu and Hatzoglou 1998). Conversely, amino acid deprivation can equally drive an increase in the expression of transporters (Fig. 1), particularly those under the control of the GCN2/eIF2α pathway, including SLC38A2 (SNAT2/System A) and the aforementioned CAT1 (Malmberg and Adams 2008; Fernandez et al. 2002; Kamata et al. 2014). However, chronic starvation of amino acids can also decrease the expression of these transporters in a tissue-specific manner (Kamata et al. 2014), demonstrating complex regulation of these transporters.
In addition to their role in the transport of amino acids across the plasma membrane, there is evidence that amino acid transporters may have signaling capacity and as such are referred to as transceptors (Hundal and Taylor 2009; Pinilla et al. 2011). Competitive inhibition of SNAT2-dependent amino acid uptake, for example, leads to reduced intracellular concentration of amino acids but cell size and mTORC1 activity were increased (Pinilla et al. 2011). This suggests that mammalian amino acid transporters may indeed act as transceptors. Further to this, more comprehensive studies in yeast have demonstrated that ligand binding and the subsequent conformational changes in the amino acid sensors Gap1 and Ssy1 are required for transceptor capacity while transport functioning itself is not required; indeed Ssy1 has low to no transporter capacity (reviewed in Poulsen et al. 2005; Hundal and Taylor 2009; Rubio-Texeira et al. 2010). Expression and localization of the amino acid permease, Gap1, is regulated by amino acid availability; in the absence of amino acids, Gap1 is up-regulated. Upon the re-addition of amino acids, Gap1 acts as a transceptor to activate protein kinase A (PKA), which then promotes expression of genes required for growth in nitrogen-replete conditions and promotes the internalization and degradation of Gap1 to carefully control signaling (Donaton et al. 2003; Kriel et al. 2011). Binding of amino acid to Ssy1 induces conformational changes that subsequently promote transcription of a number of amino acid permeases (reviewed in Poulsen et al. 2005; Hundal and Taylor 2009).
In summary, the specific influx of amino acids depends on general cellular health (including maintenance of electrochemical gradients) and is likely to depend on cellular function which dictates the concentration and composition of amino acids required for proper function and growth. Mechanisms exist to ensure appropriate levels of amino acid transporters which, in turn, mediate efficient and carefully regulated influx of extracellular amino acids.
Intracellular metabolism of amino acids
Amino acids are metabolic intermediates for a myriad of processes involved in energy production and biosynthesis of molecules required for growth and proliferation. As mentioned above, cellular demands for amino acids are cell- and tissue-specific; the liver is the primary site for the urea cycle and so it tightly regulates arginine influx and arginase levels (Liu and Hatzoglou 1998), whereas glutamine is an important energy source feeding the tricarboxylic acid (TCA) cycle particularly in glycolysis-dependent tumor cells (DeBerardinis et al. 2008). Furthermore, signaling via mTORC1 regulates cellular metabolism by inducing expression of genes involved in glycolysis, pentose phosphate pathway, lipid biogenesis and pyrimidine biosynthesis (Duvel et al. 2010; Ben-Sahra et al. 2013). In addition, the conversion of arginine to nitric oxide (NO) by nitric oxide synthase is important for vasodilation and blood flow, but increased NO levels can inhibit autophagosome formation via both mTORC1-dependent and independent mechanisms (Sarkar et al. 2011). Therefore, amino acids can directly and indirectly impact on cellular metabolism with important implications for mTORC1 activity and autophagy.
Recycling of amino acids via protein degradation pathways
Degradation of proteins is the most important intracellular mechanism to release free amino acids both under steady-state conditions and during cellular stress (Fig. 2). There are two major protein degradation pathways in the cell; the ubiquitin–proteasome system (UPS) and autophagy. Both systems participate in the degradation of polypeptides but with varying degrees of selectivity. The UPS is responsible for the degradation of short-lived, soluble proteins. Proteins destined for degradation by the UPS are initially labeled with ubiquitin. Multiple ubiquitin molecules are typically conjugated to a protein (which is referred to as poly-ubiquitylation) via specialized enzymes that activate (E1), transfer (E2) and conjugate (E3) ubiquitin to lysines within the target protein. Multiple E1, E2 and E3 enzymes exist and contribute to the specificity of the UPS system (Korolchuk et al. 2010). Ubiquitylated proteins are delivered to the proteasome, an ATP-dependent protease complex which catalyzes the cleavage of proteins into oligopeptides that are further broken down into amino acids in the cytoplasm (Glickman and Ciechanover 2002; Marques et al. 2009; Korolchuk et al. 2010). These free amino acids directly contribute to the intracellular levels of amino acids. Indeed the proteasome is required to maintain translation during periods of amino acid starvation prior to the autophagy-dependent increase in amino acid availability (Vabulas and Hartl 2005). Interestingly, there is some selectivity in the starvation-induced proteasomal degradation and newly synthesized proteins are largely protected from degradation which would ensure the efficient translation of the adaptive response proteins (Vabulas and Hartl 2005). Although outside the main focus of this review, UPS is relevant to autophagy due to multiple cross-talk mechanisms that exist between these degradative systems (Korolchuk et al. 2010; Ravikumar et al. 2010b) (see below).
Autophagy
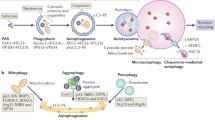
In contrast to the UPS, autophagy predominantly regulates the turnover of long-lived proteins, lipids and entire organelles. The three main types of autophagy (microautophagy, CMA and macroautophagy) differ in their substrate selectivity, kinetics and regulatory mechanisms, however, they all share a common end-point: delivery of targets to the lysosome for degradation (Fig. 3). Lysosomes contain acid hydrolases that are produced in the endoplasmic reticulum and trafficked through the Golgi to the lysosome. These enzymes are active only in the acidic environment of the lysosomal lumen. The low pH (pH 5) of the lysosomal lumen is maintained by Vacuolar-type H+-ATPase (V-ATPase) proton pumps on the lysosomal membrane. Cargo specificity and their delivery to the lysosome differentiate the three known types of autophagy.

Different types of autophagy. Schematic overview of three main types of autophagy: microautophagy, macroautophagy and chaperone-mediated autophagy (CMA), all of which deliver their respective cargo to the lysosome for degradation. Top panel shows two ubiquitin-like conjugation systems involving Atg proteins that regulate the initiation of macroautophagy
Microautophagy
Microautophagy is a process whereby cytoplasmic proteins are (selectively and non-selectively) engulfed via invagination of the lysosomal membrane and directly delivered to the degradative lumen (Fig. 3). Dynamic modification of the target protein as well as lipid composition and organization of the lysosomal membrane generate areas on the lysosomal surface that are prone to invagination, and it is at these sites that microautophagy occurs. The molecular mechanisms that regulate microautophagy, including substrate recognition and targeting, are poorly understood especially in mammalian cells. Studies carried out in yeast indicate microautophagy can broadly be characterized into five stages: invagination of the lysosomal membrane, vesicle formation, vesicle expansion, vesicle scission and finally vesicle degradation (Li et al. 2012). Selective microautophagy has been observed, for example, of mitochondria, nucleus and peroxisomes. Both mTORC1-dependent processes and Atg proteins are important for upregulation of microautophagy. More recently, microautophagy has been observed during mouse development. Specifically, in the visceral endoderm of mouse embryos, early endosomes are delivered to cathepsin B (lysosomal enzyme)-positive apical vacuoles where they are engulfed and degraded by these vacuoles (Kawamura et al. 2012). Due to the ultrastructural mechanism of microautophagy it is a difficult cellular process to study in vitro and to-date has relied largely on electron microscopic techniques, although future advances in super-resolution microscopy may provide powerful tools to gain further insights.
Chaperone-mediated autophagy
Chaperone-mediated autophagy (CMA) is unique amongst all the autophagy processes because it is exclusively a selective process and as such contributes to cellular homeostasis by the specific temporal turnover of regulatory proteins (comprehensively reviewed in Cuervo and Wong 2014). The cytosolic targets of CMA all contain a consensus pentapeptide motif, such as KFERQ or similar (ultimately the charge of the sequence is important for the interaction with chaperones) (Dice 1990). This motif facilitates binding of substrates to the chaperone, heat shock cognate protein 70 (hsc70). A number of co-chaperones and regulatory proteins cooperate to deliver CMA substrates to the lysosome where they promote an interaction with the cytosolic portion of the transmembrane lysosomal-associated membrane protein 2A (Lamp2A) (Fig. 3) (Cuervo and Wong 2014). Lamp2A usually exists as a monomer that then multimerizes and interacts with the intra-lysosomal-resident heat shock protein 90 (hsp90). This dynamic multi-protein complex facilitates protein unfolding and transport (which also involves lysosomal hsc70) into the lysosomal lumen where the protein substrate is exposed to the hydrolytic enzymes and degraded (Cuervo and Wong 2014). While the molecular mechanisms regulating CMA are not well understood, the levels of Lamp2A are important to ensure efficient CMA. Indeed, Lamp2A is transcriptionally upregulated during oxidative stress (Kiffin et al. 2004) and long-term amino acid starvation (Dice 2000). On the contrary, exposure to high-fat diet has inhibitory effects on CMA due a decrease in the levels of Lamp2A, owing to its reduced stability at the lysosomal membrane (Rodriguez-Navarro et al. 2012). Likewise, age-related diminution in Lamp2A levels without any obvious perturbations in its transcriptional rate has been associated with a decline in the activity of CMA in aged animals (Kiffin et al. 2007). However, modulating Lamp2A levels in the liver of aging mice improves hepatic function by preventing the reduction in receptor abundance with age (Zhang and Cuervo 2008). Pharmacological activation of CMA may be of therapeutic relevance in certain contexts, and has been recently shown to be achieved by retinoic acid derivatives, such as with atypical retinoid 7 (AR7), guanidine retinoid 1 (GR1) and guanidine retinoid 2 (GR2) (Anguiano et al. 2013).
Macroautophagy
Macroautophagy (which we will refer to as ‘autophagy’ in the following sections of this review) is the most extensively studied autophagic pathway and it was classically considered to be non-selective for bulk degradation of organelles and long-lived proteins. In recent years, however, macroautophagy has been shown to be cargo-specific; a function that has been implicated to have an important role in cellular and organismal physiology. For example, the selective degradation of damaged or excess mitochondria (through a process-termed mitophagy) directly impacts on cellular function and metabolism (Ashrafi and Schwarz 2013). While autophagy occurs in all cells at a basal level for maintaining energy and tissue homeostasis, cellular function and energy demands often dictate to which degree autophagic flux differs from the basal state. Autophagy can be further activated above basal levels in response to a range of cellular stressors including amino acid, nutrient, oxygen or energy deprivation, as well as with chemical inducers (Sarkar 2013b).
The molecular mechanisms and pathways controlling autophagy are well-studied compared to CMA and microautophagy. The autophagic machinery is evolutionarily conserved from yeast to mammals where several Atg proteins orchestrate distinct stages of the pathway, such as initiation, elongation, maturation and fusion. Autophagy is classically governed by mTORC1, which negatively regulates this process (Fig. 4). In amino acid-rich conditions, mTORC1 binds to, phosphorylates and thereby inactivates the autophagy initiators ULK1 and Atg13, which are present in a complex with focal adhesion kinase family interacting protein of 200 kDa (FIP200) and Atg101 (Ganley et al. 2009; Jung et al. 2009; Hosokawa et al. 2009). In the absence of activating stimuli, autophagy is induced through the dissociation of mTORC1 from the ULK1 complex, thus relieving the inhibition of ULK1. ULK1 is then responsible for phosphorylation of a range of regulatory proteins including itself, as well as of Atg13, FIP200 and the mTORC1 component Raptor (which further suppresses mTORC1 activity and promotes efficient autophagy) (Jung et al. 2009; Dunlop et al. 2011). It is clear that the occurrence of complex reciprocal phosphorylation events between mTORC1 and the downstream autophagy mediators ensure appropriate levels of anabolic versus catabolic processes.

Amino acid-dependent regulation of autophagy by mTORC1 and GCN2 signalling pathways. Schematic representation of the cellular signalling pathways governed by amino acids in the regulation of autophagy. Elevation in the levels of intracellular amino acid inhibits autophagy by suppressing the mTORC1 pathway and activating the GCN2 pathway. AMPK AMP-activated protein kinase, ATF4 activating transcription factor 4, Atg autophagy-related protein, CHOP C/EBP homologous protein, 4E-BP1 eukaryotic translation initiation factor 4E-binding protein 1, eIF2 eukaryotic initiation factor 2, ERK1/2 extracellular-signal-regulated kinase 1/2, FIP200 focal adhesion kinase family interacting protein of 200 kDa, GCN2 general control nonderepressible 2, GDP guanosine-5′-diphosphate, GPCR G-protein coupled receptor, GSK-3β glycogen synthase kinase-3β, GTP guanosine-5′-triphosphate, IGF insulin-like growth factor, IPMK inositol polyphosphate multikinase, IR insulin receptor, MAP4K3 mitogen-activated protein kinase kinase kinase kinase 3, mTORC1 mammalian target of rapamycin complex 1, PAT1 proton-assisted amino acid transporter 1, PI3K phosphatidylinositol-4,5-bisphosphate 3-kinase, p70S6K ribosomal S6 kinase of 70 kDa, Rheb ras homolog enriched in brain, SLC solute-linked carrier, TFEB transcription factor EB, tRNA transfer RNA, TSC1/2 tuberous sclerosis complex 1/2, ULK1 Unc-51-like autophagy activating kinase 1, Vps vacuolar protein sorting
Although the exact mechanisms are not fully understood, recent advances have been made into how ULK1 is then able to activate the class III phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) complex I [comprising of vacuolar protein sorting 34 (Vps34), Beclin1 and Vps15 in complex with either UVRAG (UV radiation resistance-associated gene protein) or Atg14L] and promote autophagosome synthesis (Russell et al. 2013; Fogel et al. 2013). Specifically, ULK1 was shown to phosphorylate Beclin1 at serine 14 (in complex with either Atg14L or UVRAG) and activate Vps34 in response to amino acid starvation (Russell et al. 2013), while Beclin1 phosphorylation at serine 90 and 93 was shown to be dependent on Atg14L (not UVRAG) and was required for autophagy (Fogel et al. 2013). Regardless of the mechanism, proper activation of the lipid kinase Vps34 is required for the formation of phosphatidylinositol 3-phosphate (PI3P) at the growing phagophore (immature autophagosome) membrane, possibly contributing to autophagosome synthesis. The formation of autophagosomes involves the sequestration of lipid membranes likely from multiple sources, although the primary contributory membrane in the cell is a matter of intense research. Indeed, a number of intracellular sources including the endoplasmic reticulum (Axe et al. 2008), Golgi (Ge et al. 2013), mitochondria (Hailey et al. 2010; Hamasaki et al. 2013) and plasma membrane (Ravikumar et al. 2010a; Moreau et al. 2012) have all been shown to contribute membrane to the growing autophagosome.
The expansion of phagophores is further regulated by two interconnected ubiquitin-like conjugation systems involving the generation of Atg5–Atg12–Atg16L complex and phosphatidylethalomine (PE)-conjugated microtubule-associated protein 1 light chain 3 (LC3) (Fig. 3) (Ohsumi 2001). The first system involves the conjugation of Atg5 with Atg12, in which Atg7 (E1 ubiquitin activating enzyme-like) activates Atg12 (ubiquitin-like protein), which is then transferred to Atg10 (E2 ubiquitin conjugating enzyme-like), and is finally linked covalently to Atg5. Along with Atg16L1, the Atg5-Atg12 conjugate then forms a large complex (Atg15–Atg12–Atg16L1), which localizes to the phagophore where it possibly dictates the site of the second conjugation event (Mizushima et al. 1998, 2003). In the second system, mammalian LC3 is initially synthesized as a cytosolic protein but its C-terminus is rapidly cleaved thereafter by the cysteine protease Atg4 to expose a glycine residue (and is then referred to as LC3-I). LC3-I is then lipidated by conjugation with PE by the E1-like and E2-like enzymes, Atg7 and Atg3, respectively. The lipidated LC3 protein (referred to as LC3-II) can then be recruited onto the expanding phagophore and remains on the autophagosome membrane throughout its lifespan (Kabeya et al. 2000; Tanida et al. 2004). During maturation, autophagosomes engulf portions of the cytoplasm that are to be degraded, including potentially toxic protein aggregates and surplus or damaged proteins, lipids and organelles. In addition, certain autophagic substrates are selectively targeted for degradation by adaptors such as sequestosome-1 (SQSTM1/p62) and Neighbor of BRCA1 gene protein 1 (NBR1) (Pankiv et al. 2007; Kirkin et al. 2009; Johansen and Lamark 2011). Autophagosomes are transported along the microtubule network towards the microtubule-organizing center (MTOC). En route to the MTOC and prior to their fusion with lysosomes, autophagosomes can also fuse with endosomes (late and recycling) to form amphisomes (Berg et al. 1998). During starvation, the number of amphisomes increases. The subsequent fusion of autophagosomes and amphisomes with lysosomes results in the formation of autolysosomes where degradation of autophagy substrate and liberation of free amino acids occur. The trafficking and fusion of autophagosomes with late endosomes and lysosomes are mediated by a number of protein families, including microtubule motors (Korolchuk et al. 2011; Monastyrska et al. 2009), Rab GTPases (Jager et al. 2004); (Gutierrez et al. 2004; Bento et al. 2013) and SNAP (Soluble NSF Attachment Protein) Receptor (SNARE) proteins (Nair et al. 2011; Fader et al. 2009; Renna et al. 2011; Furuta et al. 2010).
Cross-talk between degradation pathways
The ability to specifically regulate protein and organelle turnover is crucial to maintain amino acid and therefore general cellular homeostasis. As such, mechanisms of cross-talk and co-operation have evolved between different degradation pathways to ensure tight control. For example, autophagy and CMA are both activated in response to nutrient deprivation; however, this occurs in a temporally divergent manner. Autophagy activity reaches a peak in a matter of hours, whereas CMA activity persists and peaks only after 24 h. Autophagy therefore is the primary recycling mechanism to restore appropriate levels of amino acids following acute starvation while CMA participates in the highly selective turnover of specific proteins. CMA is, however, activated in cells that are defective in macroautophagy or UPS, potentially as a compensatory mechanism (Koga et al. 2011; Kaushik et al. 2008). Although cells with reduced expression of Lamp2A and diminished functional CMA response can up-regulate autophagy, they are still more prone to cellular stress indicating that both types of autophagy are required to maintain proper cellular integrity and function (Massey et al. 2006). Furthermore, acute inhibition of the UPS can also lead to an up-regulation of autophagy, possibly providing a compensatory mechanism to prevent or alleviate the potential accumulation of toxic or damaged substrates as demonstrated in fruit flies (Pandey et al. 2007). Indeed, accumulation of protein aggregates can lead to endoplasmic reticulum stress, activation of the unfolded protein response and subsequent up-regulation of autophagy through transcription of autophagy-mediating genes by ATF4 (activating transcription factor 4) (Zhu et al. 2010; Milani et al. 2009). ATF4-dependent regulation of autophagy genes is also activated in response to amino acid starvation which will be discussed further in the next sections. Conversely, defective autophagy or CMA can perturb the efficiency of UPS as a result of the accumulation of aggregation-prone proteins such as p62 (Korolchuk et al. 2009). It is important to consider that chronic perturbation of any of these systems is likely to impact on the availability/stability of regulatory proteins for the others with a direct impact on intracellular amino acid availability. Protein turnover is vitally important to maintain intracellular levels of amino acids and the regulation of these recycling mechanisms is directly dictated by the availability of free amino acids, which we will discuss in the following sections.
Sensing of amino acid sufficiency by mTORC1 and GCN2
As discussed above, intracellular levels of amino acids are regulated by both influx from the extracellular environment and recycling of intracellular resources (Fig. 2). The primary aim of these mechanisms is to ensure that intracellular levels of free, available amino acids are at an appropriate concentration for promoting anabolic processes in the cell, including protein translation, energy production and ultimately drive cellular growth and proliferation. Amino acid sufficiency is ‘sensed’ via two major signaling pathways that are controlled by the serine/threonine protein kinases: mTORC1 and GCN2. A number of molecular players contribute to these signaling pathways to govern the regulation of autophagy by amino acids (Table 1). Both direct and indirect mechanisms have been demonstrated through which these pathways integrate amino acid availability with anabolic processes and, importantly for this review, control autophagy.
mTORC1 signaling pathway
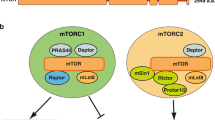
mTOR is an evolutionarily conserved serine/threonine kinase that exists in two multi-protein complexes, mTORC1 and mTORC2. The two complexes share some core proteins including mTOR and GβL/Lst8 but also have unique binding partners; mTORC1 further contains Raptor while mTORC2 complexes with mSIN1 and Rictor (Laplante and Sabatini 2012). A primary role for mTORC2 is in the regulation of the actin cytoskeleton via members of the Rho small GTPase family (Oh and Jacinto 2011). While amino acids may possibly impact on mTORC2 signaling, its role in autophagy is not established (Oh and Jacinto 2011; Tato et al. 2011) and thus this pathway will not be discussed in depth in this review. Instead, mTORC1 plays a critical role in the control of autophagy by amino acids, as well as by other upstream signals including growth factors (Fig. 4).
Growth factors are the most potent regulators of mTORC1 and signal via the PI3K/Akt signaling cascade to phosphorylate a subunit of the tuberous sclerosis complex (TSC), which consists of TSC2 in complex with TSC1 and TBC1D7 (Dibble et al. 2012). The TSC2 subunit has a GTPase activating protein (GAP) domain, which is active against (i.e., it enhances the hydrolysis of GTP to GDP) and thereby inhibits the small GTPase Rheb (Tee et al. 2003; Garami et al. 2003; Inoki et al. 2003; Zhang et al. 2003). Rheb is considered to be a master activator of mTORC1. By means of its farnesylation, Rheb resides on intracellular membranes, most importantly on lysosomes (Sancak et al. 2008; Buerger et al. 2006) where, when bound to GTP, it activates mTORC1 (Long et al. 2005a). The exact mechanism via which Rheb promotes the activity of mTORC1 as well as the mechanisms and regulators of GTP loading onto Rheb remain elusive (reviewed in Avruch et al. 2006, 2009). The TSC complex has emerged as a central integrator of diverse cellular inputs to regulate mTORC1 activity. In addition to growth factors, other regulators include the hypoxia-induced REDD1 (regulated in development and DNA damage responses 1), AMPK (AMP-activated protein kinase; which is activated in response to energy deprivation), Erk (extracellular-signal-regulated kinase) and GSK-3β (glycogen synthase kinase-3β), all of which have been shown to phosphorylate TSC2 as the primary mechanism in regulating its activity (Huang and Manning 2008) (Fig. 4).
Amino acids are both necessary and sufficient to activate mTORC1 and subsequently repress autophagy. Activation of mTORC1 by growth factors requires the presence of amino acids, which allows this signaling pathway to coordinate growth-promoting signals with the availability of nutrients. A direct role for amino acids in mTOR signaling is supported by the well-documented observation that treatment of cells with protein translation inhibitors (such as cycloheximide), which contribute to increased intracellular concentration of amino acids, can activate mTORC1 and inhibit autophagy even in complete starvation conditions (Beugnet et al. 2003; Watanabe-Asano et al. 2014). Leucine and glutamine are particularly important mediators of mTORC1 in addition to arginine which has a potent, but less studied role in the regulation of mTORC1. The TSC/Rheb signaling input to mTORC1 is classically considered to be insensitive to amino acids (Smith et al. 2005; Long et al. 2005b; Roccio et al. 2006). However, recent studies suggest that intracellular localization of TSC complex and consequently its ability to suppress the activity of mTORC1 is affected by amino acids, implicating one mechanism via which amino acids can control the activation of mTORC1 by growth factors (Demetriades et al. 2014). The main pathway, however, by which amino acids, in particular leucine and glutamine regulate mTORC1 is via the Rag small GTPases (Sancak et al. 2008; Kim et al. 2008) and a plethora of regulatory proteins (Fig. 4; Table 1). The Rag GTPases exist as an obligate heterodimer (RagA or RagB with RagC or RagD) and are resident on the membrane of lysosomes through the interaction with a multi-protein complex termed Ragulator, consisting of p18, p14, MP1, HBXIP and C7orf59 (Sancak et al. 2010). Rag GTPase-dependent recruitment of mTORC1 to lysosomal membrane is associated with specific nucleotide loading of the Rag heterodimer; RagA/B loaded with GTP and RagC/D loaded with GDP. The Ragulator complex has guanine exchange factor (GEF) activity towards RagA/B, that is, it enhances GTP loading. Thus, the Ragulator complex not only supports the localization of Rag but also its function (Bar-Peled et al. 2012). In addition, leucyl tRNA synthetase has GAP activity towards RagD (Han et al. 2012) and the tumor suppressor folliculin in complex with FNIP1/2 (folliculin-interacting protein 1/2) shows GAP activity towards RagC/D (Tsun et al. 2013). Together, these proteins cooperate to ensure Rag GTPases are in the right conformation to promote mTORC1 localisation and activity. Inactivation of the Rag GTPases and therefore mTORC1 is mediated in mammalian cells by GATOR-1 and -2 complexes [and yeast homologues lml1, Npr2 and Npr3 (Panchaud et al. 2013)] that have GAP activity for RagA and RagB (Bar-Peled et al. 2013). Another study, however, was unable to detect any amino acid-dependent changes in nucleotide loading of Rag GTPases although they did observe that nucleotide binding could modulate the Rag–mTOR interaction (Oshiro et al. 2014). There has been rapid progress in the understanding of the amino acid-dependent Rag-mediated regulation of mTORC1 and a number of amino acid-sensing mechanisms have been postulated (described in more detail below). The molecular mechanisms underlying amino acid sensing remain poorly understood, but represent an exciting future area of research.
A number of other pathways have been shown to positively regulate mTORC1 in response to amino acids (Fig. 4), although the exact mechanisms remain largely elusive. For example, inositol polyphosphate multikinase (IPMK) seemingly helps to regulate the Raptor–mTOR interaction to suppress mTORC1 activity under amino acid deficiency, whereas the presence of amino acids converts this complex into a low-affinity state and thereby allowing lysosomal relocation of mTORC1 for activation (Kim et al. 2011). In addition, mitogen-activated protein kinase kinase kinase kinase 3 (MAP4K3) acts via an unknown mechanism to activate mTORC1 (Findlay et al. 2007). The G-protein coupled taste sensing receptor T1R1/T1R3 modulates mTORC1 and autophagy downstream of amino acids through the activation of Erk1/2 (Wauson et al. 2012). Although the contribution of Erk1/2 phosphorylation to mTORC1 activity via a direct mechanism is debatable (Gulati et al. 2008), studies have demonstrated a role for amino acid-dependent activation of phospholipase C and a subsequent increase in intracellular Ca2+ to maximally activate mTORC1 (Wauson et al. 2012; Gulati et al. 2008).
The contribution of these diverse amino acid-dependent mTORC1-activating mechanisms may reflect cell type, metabolic state, extracellular environment, cell cycle or differentiation state. Tight spatial and temporal regulation between amino acid availability, mTORC1 and autophagy is, however, fundamentally important, particularly for postnatal survival. Autophagy-deficient newborn mice die quickly after birth due to their inability to survive the starvation condition (Kuma et al. 2004). Similarly, newly born mice expressing constitutively active RagA die due to persistent mTORC1-dependent inhibition of autophagy and the resulting glucose deprivation (Efeyan et al. 2013).
Regulation of mTORC1 and autophagy by solute-linked carriers
As we have already mentioned, some amino acids (leucine, glutamine and arginine) show a greater contribution to mTORC1 activity; however, no individual amino acid is sufficient for mTORC1 activation and therefore autophagy inhibition. These observations suggest a more general reliance on other amino acids and their transporters. Recent work has identified a number of solute carriers that participate in modulating mTORC1 activity and autophagy (Fig. 4; Table 1), although it should be noted that information garnered regarding the participation of amino acid transporters to the regulation of autophagy is largely indirect and rather extrapolated from their involvement in mTORC1 (and GCN2) activity.
Many studies have concentrated on the important anabolic effects of leucine (which we will discuss in the next sections), and indeed this amino acid has a dominate role in the regulation of mTORC1 and autophagy (Dodd and Tee 2012). In addition, glutamine is one of the most abundant intracellular amino acids and, in cooperation with leucine, represents a key regulator of mTORC1 activity and autophagy. Glutamine is an important cellular energy source, particularly in cancer cells and their in vitro culture models including HeLa cells, where the high intracellular levels are maintained by SLC1A5. This glutamine gradient was found to be the rate-limiting step in mTORC1 activation by leucine; the SLC7A5 (LAT1)/SLC3A2 heterodimer couples the import of leucine with a concomitant efflux of glutamine (Nicklin et al. 2009). This study indicates that individual amino acids may impact on mTORC1 differentially, i.e., while leucine directly acts on the mTORC1 pathway, the role of glutamine is rather as a facilitator to maximally activate mTORC1.
The proton-assisted transporters SLC36A1 (PAT1) and SLC36A4 (PAT4) positively regulate mTORC1 activity despite their substrates (including glycine, alanine and proline) not necessarily being well-documented mTORC1 activators. Specifically, PAT1 and PAT4 were identified to regulate growth in a genetic screen in Drosophila (Heublein et al. 2010). Subsequent studies have implicated that the turnover of PAT4 by the small GTPase Rab12 can impact mTORC1 activity and autophagy possibly via an indirect effect because its internalization consequently reduces the influx and therefore the intracellular levels of amino acids (Matsui and Fukuda 2013). Lysosomally localized SLC36A1/PAT1 can form a complex with Rag GTPases on the lysosomal surface and was shown to participate in regulating amino acid-dependent mTORC1 localization (Ogmundsdottir et al. 2012). Interestingly, this report suggests that amino acids and growth factors may cooperate much more closely in regulating mTORC1 than previously thought: activation of the PI3K/Akt/Rheb pathway was found to promote endocytosis and increase lysosomal localization of SLC36A1/PAT1, which is expected to facilitate amino acid-dependent mTORC1 activation (Ogmundsdottir et al. 2012). This observation also highlights the currently unexplored area of the trafficking and turnover of amino acid transporters as a mechanism of mTORC1 regulation. It is possible that a dynamic relationship exists between plasma membrane and intracellular pools of amino acid transporters, and deciphering the mechanisms that regulate this cross-talk will undoubtedly contribute to our understanding.
The exact mechanisms via which the lysosomal amino acid transporters contribute to the activation of mTORC1, and conversely the suppression of autophagy are not clear. The above study provides an arguably logical explanation that amino acids exported from the lysosome would be ‘sensed’ by mTORC1, which is localized on the cytoplasmic surface of the lysosomal compartments due the increased local concentration of amino acids in the proximity of lysosome-resident mTORC1. Another report, however, demonstrates that amino acid sensing occurs via what has been termed as an ‘inside-out’ mechanism and that there is direct coupling between intra-lysosomal amino acids and mTORC1 activity (Zoncu et al. 2011). Specifically, intra-lysosomal amino acids appear to promote mTORC1 activation through a mechanism involving ATP hydrolysis and the consequential conformational change of the lysosomal V-ATPase. The V-ATPase was found to directly interact with the Rag GEF complex, the Ragulator, and co-immunoprecipitated with Ragulator and Rag GTPases. Amino acids weaken the interaction between Ragulator and the V-ATPase V1 domain, but have no effect on the interaction with the V0 domain. These amino acid-sensitive interactions are required for proper nucleotide loading of the Rag GTPases, recruitment of mTORC1 to the lysosome and the consequent activation of mTORC1 (Zoncu et al. 2011). Furthermore, this study observed that overexpression of PAT1 actually prevented mTORC1 activation in response to amino acids due to leaching of amino acids from the lysosome. Future investigations will uncover more detailed understanding of this fascinating inside-out mechanism of amino acid sensing.
One particular aspect that will be of interest for future studies is the relative contribution of extracellular amino acids versus autophagy-derived lysosomal amino acids in activating mTORC1. For example, can these pools of amino acids be differentiated? In periods of high autophagic flux (such as during amino acid starvation), one would expect an increase in the concentration of intra-lysosomal amino acids. Does this mean that under these conditions the requirement for the extracellular amino acids eventually becomes minimal? Indeed, while acute amino acid starvation (for 1 h), or deprivation of individual amino acids particularly with leucine or arginine, leads to mTORC1 inhibition, long-term starvation leads to re-activation of mTORC1 (Yu et al. 2010). This scenario raises further questions of fundamental interest. Are amino acids sensed in a concentration-dependent manner under these conditions? Would targeting leucine-rich proteins to the lysosome for degradation re-activate mTORC1 more rapidly? It is possible that transport of amino acids across lysosomal membranes is significantly more dynamic than previously considered and that bidirectional movement of amino acids, as well as proper maintenance of electrochemical gradients participates in the signaling cascades regulating mTORC1 activity? Further investigation into lysosomal transporters is warranted in light of these questions.
Amino acid metabolism, mTORC1 and autophagy
As mentioned above, glutamine is particularly important in rapidly growing cells where it not only serves as a key energy store but also cooperates with leucine to activate mTORC1. Glutamine can be converted to α-ketoglutarate, which is an intermediate in the tricarboxylic acid (TCA) cycle and a regulator of mTORC1 activity and autophagy (Fig. 4; Table 1). In this biochemical reaction, glutamine is converted first to glutamate (catalyzed by glutaminase; GLS) and then to α-ketoglutarate (via glutamate dehydrogenase; GDH) by transamination processes. Interestingly, GDH has been shown to contribute to leucine sensing, which inhibits autophagy through mTORC1 activation (Lorin et al. 2013). In fact, leucine can bind to GDH and promote the production of α-ketoglutarate and the activation of mTORC1 (Duran et al. 2012). The specific mechanism of mTORC1 activation by α-ketoglutarate is not clear lest to say that RagA/B is loaded with GTP and thereby positively regulates the lysosomal recruitment of mTORC1 and concomitantly the inhibition of autophagy (Duran et al. 2012). The downstream participation of prolyl hydroxylases has been recently demonstrated although how they regulate Rag GTPase activity is not clear (Duran et al. 2013). Interestingly, mTORC1 has recently been shown to actively regulate the metabolism of intracellular glutamine by increasing the activity of GDH, demonstrating that sophisticated mechanisms are employed to regulate cellular homeostasis (Csibi et al. 2013). Specifically, mTORC1 promotes the ubiquitination and proteasomal degradation of cAMP-responsive element binding 2 (CREB2), which transcriptionally regulates the expression of SIRT4 (a negative regulator of GDH activity) (Csibi et al. 2013). These studies provide interesting future perspectives in the light of cell- and tissue-specific cellular metabolism and the fact that metabolism is altered in many diseases, most notably in cancer (where glutamine addiction is a common occurrence). Understanding the impact of altered metabolism on mTORC1 and autophagy could have important future implications.
Co-regulators of autophagy and mTOR in response to amino acids
Amino acids also control autophagy through the regulation of the transcription factor EB (TFEB) by lysosome-resident mTORC1 (Fig. 4). The activity of nuclear TFEB positively impacts on the expression of genes regulating lysosome biogenesis and autophagy by binding to the CLEAR (coordinated lysosomal expression and regulation) motif in their promoter region (Settembre et al. 2011, 2012). Recent studies have demonstrated that localization (and therefore inactivation) of TFEB to the lysosome is regulated via a mechanism that involves the activation of Rag GTPases and lysosome-resident mTORC1 which phosphorylates TFEB under nutrient-rich conditions (Settembre et al. 2012; Martina and Puertollano 2013). These phosphorylation events facilitate the interaction between TFEB and the cytosolic chaperone protein 14-3-3 which retains and therefore inactivates TFEB in the cytoplasm (Martina et al. 2012). During amino acid limitation conditions, inactivation of mTORC1 leads to re-localization of TFEB (non-phosphorylated form) to the nucleus where it causes the expression of multiple lysosomal and autophagy-related genes, including lysosomal hydrolases and membrane transporters, as well as p62 and LC3B, respectively (Settembre et al. 2011). This allows cellular adaptation to diminished amino acid levels by increasing the lysosomal and autophagic compartments in order to maintain a critical level of energy and metabolites for surviving the starvation condition.
The class III PI3K, Vps34 is a lipid kinase required for the induction of autophagy as we have discussed above. The Vps34 complex, containing Atg14, Vps15 and Beclin-1, functions in the initiation of autophagy. During amino acid/nutrient sufficiency in mammalian cells, mTORC1-mediated phosphorylation-dependent inactivation of the Atg14-containing Vps34 complex suppresses autophagosome biogenesis (Fig. 4). On the contrary, depletion of amino acids causes inhibition of mTORC1, thereby relieving the brake on this pro-autophagic complex to stimulate autophagy (Yuan et al. 2013). Therefore, mTORC1 inhibits autophagy by phosphorylating and inactivating both the autophagy-initiating ULK1 (as mentioned in previous section) and Vps34 complexes. In addition, Vps34 has also been shown to participate in amino acid-dependent activation of mTORC1 (Nobukuni et al. 2005; Byfield et al. 2005), possibly downstream of amino acid-induced Ca2+ influx (Gulati et al. 2008). Vps34 may exist [as in yeast (Kihara et al. 2001)] in two populations, with and without Beclin-1 (yeast Vps30) association.
The autophagy adaptor protein p62 also contributes to both amino acid sensing and the regulation of autophagy. Recent reports have indicated that p62 is required for maximal mTORC1 activity in response to amino acids. p62 can interact with the mTORC1 subunit, raptor, TRAF6 (TNF receptor associated factor 6) and Rag GTPases to promote mTORC1 recruitment to lysosomes where it is activated. Specifically, p62 promotes the formation of the active Rag heterodimers and together with TRAF6, p62 is postulated to promote recruitment of mTORC1 to lysosomal membranes via its interaction with raptor (Duran et al. 2011). Via an incompletely understood mechanism, TRAF6-dependent ubiquitylation of mTOR is then required for amino acid-dependent activation of mTORC1 (Linares et al. 2013). In terms of autophagy regulation, p62 itself is a well-known autophagy substrate although it is an important mediator for selective targeting of substrates (usually ubiquitylated) for autophagic (and proteasomal) degradation (Johansen and Lamark 2011).
Another intriguing mechanism via which amino acid sufficiency has been implicated in regulating multiple pathways is via leucyl-transfer RNA (tRNA) synthetase, the enzyme required to load leucine onto its tRNA (Han et al. 2012; Bonfils et al. 2012). Leucyl tRNA synthetase was found to translocate to the lysosome and bind Rag GTPases in a leucine-dependent manner where it acts as a GAP to promote the GTPase activity of RagC/D, which is required for mTORC1 activity (Han et al. 2012) (Fig. 4). Leucyl tRNA synthetase mutants, incapable of binding to leucine, could not activate mTORC1. Likewise, its knockdown inhibited mTORC1 and stimulated autophagy (Han et al. 2012). In the absence of amino acids, there is an increase in the cytosolic concentration of uncharged tRNAs, which activates the GCN2/eIF2 signaling pathway, another pivotal central node the regulation of anabolism and catabolism. The growing roles of GCN2/eIF2 in mediating autophagic responses to fluctuations in the levels of amino acids are described below.
Amino acid-dependent regulation of autophagy via GCN2
mTORC1 and GCN2/eIF2 are both regulated by amino acid sufficiency and, in turn carefully control autophagy (Figs. 1, 4). In addition, they promote translation of different subsets of mRNA transcripts (Proud 2014), notably those with 5′ terminal oligopyrimidine (TOP) motif-containing mRNA (Thoreen et al. 2012) and those with sequences such as upstream open reading frames (uORF) and internal ribosomal entry sequences (IRES) in their 5′ untranslated region, respectively. GCN2 is a serine–threonine kinase that binds to uncharged tRNAs through a domain homologous to histidyl tRNA synthetase. During amino acid starvation, the concentration of tRNAs not bound to amino acids increases. GCN2-tRNA binding causes a protein conformation change, resulting in homodimerization and autophosphorylation of GCN2. Phosphorylated, active GCN2 subsequently phosphorylates eIF2α subunit on serine 51. Phosphorylated eIF2α has higher binding affinity for eIF2β; however, eIF2β is unable to load eIF2α with GTP (in its capacity as a GEF). This GTP-loading step is required for binding to and targeting of methionine-tRNA to the ribosome and as a result, translation initiation is globally reduced. Furthermore, amino acid removal can directly influence phosphorylation and activity of eIF2β; phosphorylation of serine 525 in the absence of amino acids inhibited its activity (Wang and Proud 2008). Fascinatingly, however, a subset of proteins are not repressed, rather translation of these proteins involved in what is referred to as the adaptive response, are induced. This subset of genes includes amino acid biosynthesis enzymes, amino acid transporters and regulators of autophagy and transcription factors including ATF4 and CHOP (C/EBP homologous protein) (Talloczy et al. 2002). An increase in ATF4 and CHOP enhances the transcription of autophagy proteins involved in the synthesis (Atg5, Atg12), maturation (LC3) and turnover of autophagosomes in response to amino acid starvation (Milani et al. 2009; Zhu et al. 2010; B’Chir et al. 2013), as well as that of the amino acid transporters such as CAT1 and SNAT2 and the translational regulator 4E-BP [reviewed in Kilberg et al. (2009)]. The GCN2/eIF2α pathway promotes a feedback mechanism by promoting expression of the phosphatase regulator Gadd34 (Growth arrest and DNA-damage-inducible protein) which inhibits eIF2α phosphorylation and activity to restore protein translation (Novoa et al. 2001). Adaptive regulation of eIF2 phosphorylation and translation is essential for mammalian and Drosophila development; deletion of the gadd34 homologue is embryonic lethal (Malzer et al. 2013; Harding et al. 2009). Together, the absence of amino acids sets in motion a signaling cascade aimed at reducing the demand for amino acids while concomitantly increasing intracellular synthesis, recycling and influx of amino acids.
Cross-talk between mTORC1 and GCN2/eIF2 in the regulation of amino acid homeostasis
Despite both mTORC1 and GCN2/eIF2 being regulated by amino acid availability, these pathways are largely considered to be independent; however, there are occurrences of documented cross-talk. For example, the expression of the transcription factor ATF4 can be enhanced via both the GCN2/eIF2 and insulin/mTORC1-regulated pathways (Adams 2007; Malmberg and Adams 2008). Furthermore, inhibition of tRNA charging by amino acid alcohols (which out-compete amino acids for binding to tRNA synthetases) was shown to perturb amino acid-dependent phosphorylation of p70S6K (Iiboshi et al. 1999). In the presence of amino acids, phosphorylation of GCN2 at serine 577 has been postulated to repress GCN2 activity by reducing its binding affinity for tRNAs and indeed mutation of S577 to alanine partially activates GCN2 (Cherkasova and Hinnebusch 2003). Moreover, rapamycin (inhibitor of mTOR) has been shown to induce the phosphorylation of eIF2α at S51 via a reduction in GCN2 phosphorylation at S577. Specifically, this study indicates that mTORC1 regulates the activity of a 2A-related protein phosphatase (Cherkasova and Hinnebusch 2003), although other studies have failed to see any effect of mTORC1 inhibition on eIF2 phosphorylation (Thoreen et al. 2012). In the liver of GCN −/− mice, leucine starvation (either acute for 1 h or chronic for 6 days) did not induce eIF2 phosphorylation (as expected) and furthermore phosphorylation of mTORC1 substrates, 4E-BP1 (eukaryotic translation initiation factor 4E-binding protein 1) and S6K (ribosomal S6 kinase) persisted, unlike control animals (Anthony et al. 2004). Protein synthesis in the liver therefore persevered in these mice, and this was observed to be at the expense of muscle growth/mass. This study indicates that GCN2 activity may act upstream of the inhibition of mTORC1 in the absence of amino acids (Anthony et al. 2004). It is important therefore to consider amino acid sufficiency as a systemic regulatory program in the body. It appears that the degree to which the mTOR and GCN2-regulated pathways cooperate and cross-talk is likely to depend on the type as well as the severity and the longevity of exposure to cellular stress.
Amino acids and autophagy: pathophysiological relevance
Metabolism
Autophagy is an important regulator of metabolism at both cellular and organismal levels. It provides a critical source of internal nutrients under conditions of starvation in order to maintain cellular integrity and survival. The metabolites derived from the breakdown of autophagy substrates in autolysosomes provide the resources for biosynthetic and energy-generating pathways. For example, autophagic degradation of proteins, lipids and carbohydrates results in the production of amino acids, fatty acids and sugars, respectively. When these metabolites are liberated in the cytoplasm, they not only contribute as anabolic substrates for biosynthesis, but are also catabolized for the generation of energy (Rabinowitz and White 2010).
Furthermore, autophagy has a vital role in cellular quality control by regulating the turnover of intracellular organelles, such as selective clearance of mitochondria (mitophagy), peroxisomes (pexophagy), ribosomes (ribophagy) and lipids (lipophagy) (Green and Levine 2014). Through all of these processes, autophagy also indirectly influences the cellular metabolic capabilities. The role of autophagy in metabolic homeostasis helps to fight against degenerative diseases and cancer in adults. In cancer, the impact of autophagy is paradoxical. Although autophagy limits the initiation of certain cancers, it can also support the growth of tumors (possibly by providing metabolic substrates) and aids in tumor resiliency once cancer occurs (White 2012). Modulation of autophagy may be of therapeutic relevance in different stages of cancer: inhibition of autophagy at an early stage for suppressing tumor growth, or activation of autophagy at a late stage for inflicting cytotoxicity in established tumors (Janku et al. 2011).
Defective autophagy is implicated in contributing to the development of various metabolic disorders, including obesity, insulin resistance and diabetes mellitus (Kim and Lee 2014). Likewise, lipids including cholesterol are degraded by autophagy (Singh et al. 2009), and impairment in this process has been recently attributed to the etiology of Niemann–Pick type C1 disease, a lipid storage disorder exhibiting neurodegeneration and liver dysfunction (Sarkar et al. 2013). Thus, autophagy has a multitude of physiological effects in maintaining metabolic homeostasis that possibly helps preventing the disease onset.
Neurodegeneration
Autophagy has a house-keeping role in the clearance of naturally occurring misfolded proteins in all tissues of an organism, thereby acting as a critical mediator in maintaining tissue homeostasis. Deregulation of intracellular protein degradation pathways, in particular, autophagy, has been implicated in neurodegeneration associated with the accumulation of ubiquitinated protein aggregates in the degenerating neurons (Nixon 2013). The strongest evidence linking autophagy to neurodegeneration comes from the studies where brain-specific deletion of essential autophagy genes, such as Atg5 or Atg7, causes neurodegenerative phenotype in the central nervous system (Hara et al. 2006; Komatsu et al. 2006). A number of mutant aggregation-prone proteins causing neurodegenerative diseases are predominantly removed by autophagy, and thus malfunction of this process leads to an increase in the load of these toxic species, resulting in neurodegeneration. Consequently, impaired autophagy reported in several neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease, is likely to act as a major contributing factor to the disease pathology (Carroll et al. 2013; Sarkar 2013b). In addition, perturbation in autophagy also impacts on the clearance of damaged mitochondria (mitophagy), and specific defects in the mechanism of mitophagy have been reported in Parkinson’s disease associated with Parkin and PINK1 mutations (Youle and Narendra 2011). In a nutshell, failure in autophagy (and mitophagy) causes a multitude of deleterious effects, including increase in protein aggregation, mitochondrial dysfunction and susceptibility to pro-apoptotic insults. Therefore, decline or impairment of autophagic activity due to aging or mutant proteins, respectively, can influence both longevity and neurodegeneration. Indeed, stimulation of autophagy by inhibiting mTORC1 or through modulation of mTOR-independent pathways has been demonstrated to have beneficial effects in various transgenic animal models of neurodegenerative diseases, as well as acts as a common denominator in promoting longevity through manipulations extending lifespans (Ravikumar et al. 2010b; Sarkar et al. 2009; Madeo et al. 2010). Small molecule modulators of autophagy are thus of considerable biomedical interest (reviewed in Sarkar 2013a, b; Rubinsztein et al. 2012).
Although a concrete link between alterations in amino acid homeostasis with the onset of neurodegeneration has not been established, exploratory links between non-protein amino acids and the sporadic forms of neurodegenerative diseases have been postulated (Rodgers 2014). Furthermore, dysfunction of the excitatory amino acid transporters (EAATs) through acute stimulation with their substrates has been shown to cause degeneration in rat striatal neurons, leading to Parkinsonism-like phenotype in vivo (Assous et al. 2014).
Senescence
Cellular senescence refers to a process whereby cells have undergone irreversible cell cycle arrest, but remain metabolically active. Generally, cellular senescence is considered to be a cell survival mechanism in the face of irreparable cellular damage that could otherwise lead to cellular transformation. Senescence can occur via replicative (telomere shortening) and stress-induced (oncogene activation, DNA damage response) mechanisms. The current working hypothesis is that induction of senescence occurs through a hyperactive growth mechanism involving overactive or ectopic mTORC1 activity (Blagosklonny 2003, 2008). The ability of cells to respond appropriately to extracellular and intracellular stimuli is generally stunted in aged cells. Indeed, we have recently observed that mTORC1 activity persists during acute amino acid starvation in replicative senescent fibroblasts but is completely inhibited in starved, quiescent and proliferating fibroblasts (Carroll, Korolchuk et al., unpublished observation) (Fig. 5). As a result, ectopic mTORC1 activity in the absence of appropriate levels of amino acids may include persistent protein translation (which may compromise quality) as well as increased intracellular stress (for example, increased mitochondrial biogenesis may directly contribute to oxidative damage via reactive oxygen species; ROS). Turning off mTORC1 is therefore just as important as turning it on (Efeyan et al. 2013). Indeed, the role of autophagy in senescence is currently unclear; inhibition and activation of autophagy have equally been demonstrated to participate in the induction of senescence. It is likely that the accumulation of cellular damage and stress will have complex downstream effects on mTORC1 and autophagy. For example, a range of stressors can influence levels of ROS, and increased cellular ROS production can directly damage autophagy proteins.

mTORC1 is refractory to amino acid starvation in senescent cells. Young primary human lung fibroblasts were maintained as sub-confluent cultures in full nutrient medium (proliferating) while quiescent cells (of the same age) were cultured until confluent, at which point proliferation ceases due to contact inhibition. Primary human lung fibroblasts were cultured until they reached senescence by serial passaging of sub-confluent cells. Replicative senescence was achieved by serial passage of cells until the population doubling (PD) was <0.1 per month, after which the cells were stained positive for senescence-associated β-galactosidase (Sen-β-Gal) and negative for proliferating marker Ki-67. The proliferating, quiescent and senescent cells were incubated for 1 h in medium with individual amino acids omitted as indicated. Cells were lysed and analyzed by immunoblot for mTORC1 substrates, phosphorylated S6K (threonine 389) and phosphorylated S6 (serine 235/236). Quantification of phosphorylated S6 is shown relative to total S6 protein levels
Aging and longevity
The functioning of autophagy is perturbed during aging with important implications for age-related disorders, such as neurodegeneration (as we have discussed above and reviewed). The repercussions of aging on autophagy (and vice versa) are complex; some studies have shown an increase in autophagy-related proteins with age while others have demonstrated a decrease in their expression or function (Kiffin et al. 2007; Simonsen et al. 2008; Lipinski et al. 2010; Wohlgemuth et al. 2010; Yang et al. 2014). Expression of the autophagy co-chaperone BAG3 (Bcl2-associated athanogene 3) is elevated in aged tissues (Gamerdinger et al. 2009) that has important implications for age-related neurodegeneration, such as Huntington’s disease. BAG3 in complex with HspB8 has been demonstrated to increase eIF2α phosphorylation via GCN2 and as such inhibits the translation of mutant huntingtin while simultaneously enhancing its autophagic clearance (Carra et al. 2009).
A direct link between amino acid (nutrient) availability and autophagy during aging has not been formally demonstrated. Mechanisms that influence longevity, however, have consistently identified a contributory role for autophagy pathways. Dietary restriction (DR; which is restriction of nutrients without causing malnutrition) for example is one of the most well-known interventions to improve lifespan and this paradigm is evolutionary conserved from yeast, C. elegans, Drosophila to mammals. A number of studies have demonstrated a central importance for amino acid availability to DR-dependent longevity (Grandison et al. 2009; Miller et al. 2005; De Marte and Enesco 1986). General dietary restriction of essential amino acids maximizes life span, albeit at the cost of reduced fecundity. A study in Drosophila, however, has demonstrated that the addition of methionine (and to a lesser extent tryptophan) to a standard DR diet maximizes both life span and increases fecundity (Grandison et al. 2009). Furthermore, deletion of autophagy genes abrogates DR-induced increase in lifespan, demonstrating that induction of autophagy contributes, at least partially, to this beneficial effect. Indeed, stimulation of autophagy increased longevity in mice (Eisenberg et al. 2009; Zheng et al. 2010). The TOR and GCN2/eIF2 pathways have both been implicated in longevity; inhibition of TORC1 activity by pharmacological agents (such as with rapamycin) (Harrison et al. 2009); (Bjedov et al. 2010) or genetic ablation (such as of S6K and insulin receptor) (Selman et al. 2009; Kimura et al. 1997) increased lifespan. It is thus clear that the mechanisms regulating intracellular amino acid levels are fundamentally important for healthy aging and longevity.
Conclusions and future perspectives
The ability to appropriately control intracellular levels of amino acids is fundamental for healthy cellular growth and survival. Organisms have developed sophisticated mechanisms to control intake, utilization and recycling of amino acids to ensure that sufficient concentrations are maintained to promote protein translation and support cellular function. The spatial coupling of amino acid sensing (by mTORC1) and recycling (through autophagy) at the lysosome ensures that cells can respond quickly and with a high degree of sensitivity to fluctuations in amino acid levels. The amino acid-sensing kinases, mTORC1 and GCN2, employ different mechanisms to sense intracellular levels of amino acids, and subsequently dictate differential downstream signaling cascades to control protein synthesis and autophagy, amongst many other processes. Although the degree (if any) to which these pathways cooperate has yet to be fully realized, together they provide an exquisite level of control over anabolic and catabolic processes (Figs. 1, 4). The ability to sense and appropriately respond to cellular stress such as amino acid starvation is commonly perturbed during the aging process with direct consequences for the disease progression. Interventions that affect mTORC1 and GCN2/eIF2 activity, as well as autophagy, have been shown to consistently alleviate symptoms of aging and cellular stress. Undoubtedly, our knowledge of amino acid homeostasis will continue to expand in the future with fundamentally important implications for health and disease.
In particular, we have discussed here that some amino acids, including leucine, glutamine and arginine contribute disproportionately to cellular growth. Although mechanisms have been demonstrated via which leucine and glutamine can contribute to mTORC1 activity and autophagy, the specific mechanisms via which arginine signals is not clear. Future work is likely to provide more information to delineate the exact roles and pathways that dictate the sensitivity and dependence of different cell lines to different amino acids. This is likely to include specific cellular function. For example, leucine is an essential amino acid and may act as a proxy for sufficient amino acid/nutrient availability, thus communicating to the cell that protein translation and growth can occur and minimizing the degradative process of autophagy. Arginine, however, is only conditionally essential in adult, fully differentiated cells. Nonetheless, it is essential during mammalian embryogenesis and to other organisms such as yeast and Drosophila; therefore, evolutionary conserved mechanisms of arginine sensing may exist.
Despite a particular importance for leucine, glutamine and arginine to cellular growth, a full complement of amino acids provides more potent activation of mTORC1. The strength of mTORC1 signal in the presence of amino acids is weak, whereas growth factors provide a much more robust signal to this growth-promoting kinase. Amino acids thus appear to be a permissive signal to the cell to carry out global protein translation. Further work will help elucidate the specific molecular mechanisms via which amino acids are sensed. In particular, does a common sensing mechanism exist for all amino acids or whether there are unique molecular players for individual amino acids? Furthermore, what are the underlying mechanisms of amino acid sensing, whether amino acids can modulate protein or molecular conformations or interactions, and are they indeed even sensed directly or rather through their metabolites? Despite the explosion of knowledge in this field, there are still exciting questions yet to be explored and understood regarding the fundamental ways in which cells balance growth and degradation. As we enter an era where ever advancing technology is becoming increasingly accessible, new investigative avenues are likely to expose this field even further.
References
Adams CM (2007) Role of the transcription factor ATF4 in the anabolic actions of insulin and the anti-anabolic actions of glucocorticoids. J Biol Chem 282:16744–16753
Anguiano J, Garner TP, Mahalingam M, Das BC, Gavathiotis E, Cuervo AM (2013) Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol 9:374–382
Anthony TG, Mcdaniel BJ, Byerley RL, Mcgrath BC, Cavener DR, Mcnurlan MA, Wek RC (2004) Preservation of liver protein synthesis during dietary leucine deprivation occurs at the expense of skeletal muscle mass in mice deleted for eIF2 kinase GCN2. J Biol Chem 279:36553–36561
Ashrafi G, Schwarz TL (2013) The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20:31–42
Assous M, Had-Aissouni L, Gubellini P, Melon C, Nafia I, Salin P, Kerkerian-Le-Goff L, Kachidian P (2014) Progressive Parkinsonism by acute dysfunction of excitatory amino acid transporters in the rat substantia nigra. Neurobiol Dis 65:69–81
Avruch J, Hara K, Lin Y, Liu M, Long X, Ortiz-Vega S, Yonezawa K (2006) Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene 25:6361–6372
Avruch J, Long X, Lin Y, Ortiz-Vega S, Rapley J, Papageorgiou A, Oshiro N, Kikkawa U (2009) Activation of mTORC1 in two steps: Rheb-GTP activation of catalytic function and increased binding of substrates to raptor. Biochem Soc Trans 37:223–226
Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT (2008) Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182:685–701
Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM (2012) Ragulator is a GEF for the Rag GTPases that signal amino acid levels to mTORC1. Cell 150:1196–1208
Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM (2013) A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340:1100–1106
B’chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A (2013) The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 41:7683–7699
Ben-Sahra I, Howell JJ, Asara JM, Manning BD (2013) Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339:1323–1328
Bento CF, Puri C, Moreau K, Rubinsztein DC (2013) The role of membrane-trafficking small GTPases in the regulation of autophagy. J Cell Sci 126:1059–1069
Berg TO, Fengsrud M, Stromhaug PE, Berg T, Seglen PO (1998) Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem 273:21883–21892
Beugnet A, Tee AR, Taylor PM, Proud CG (2003) Regulation of targets of mTOR (mammalian target of rapamycin) signalling by intracellular amino acid availability. Biochem J 372:555–566
Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, Partridge L (2010) Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab 11:35–46
Blagosklonny MV (2003) Cell senescence and hypermitogenic arrest. EMBO Rep 4:358–362
Blagosklonny MV (2008) Aging: ROS or TOR. Cell Cycle 7:3344–3354
Bonfils G, Jaquenoud M, Bontron S, Ostrowicz C, Ungermann C, De Virgilio C (2012) Leucyl-tRNA synthetase controls TORC1 via the EGO complex. Mol Cell 46:105–110
Broer S (2008) Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev 88:249–286
Buerger C, Devries B, Stambolic V (2006) Localization of Rheb to the endomembrane is critical for its signaling function. Biochem Biophys Res Commun 344:869–880
Byfield MP, Murray JT, Backer JM (2005) hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J Biol Chem 280:33076–33082
Carra S, Brunsting JF, Lambert H, Landry J, Kampinga HH (2009) HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2{alpha} phosphorylation. J Biol Chem 284:5523–5532
Carroll B, Hewitt G, Korolchuk VI (2013) Autophagy and ageing: implications for age-related neurodegenerative diseases. Essays Biochem 55:119–131
Cherkasova VA, Hinnebusch AG (2003) Translational control by TOR and TAP42 through dephosphorylation of eIF2alpha kinase GCN2. Genes Dev 17:859–872
Collarini EJ, Oxender DL (1987) Mechanisms of transport of amino acids across membranes. Annu Rev Nutr 7:75–90
Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, Jeong SM, Dempsey JM, Parkhitko A, Morrison T, Henske EP, Haigis MC, Cantley LC, Stephanopoulos G, Yu J, Blenis J (2013) The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 153:840–854
Cuervo AM, Wong E (2014) Chaperone-mediated autophagy: roles in disease and aging. Cell Res 24:92–104
De Marte ML, Enesco HE (1986) Influence of low tryptophan diet on survival and organ growth in mice. Mech Ageing Dev 36:161–171
Deberardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7:11–20
Demetriades C, Doumpas N, Teleman AA (2014) Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 156:786–799
Dibble CC, Elis W, Menon S, Qin W, Klekota J, Asara JM, Finan PM, Kwiatkowski DJ, Murphy LO, Manning BD (2012) TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell 47:535–546
Dice JF (1990) Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci 15:305–309
Dice JF (2000) Lysosomal pathways of protein degradation. Landes Bioscience, Texas
Dodd KM, Tee AR (2012) Leucine and mTORC1: a complex relationship. Am J Physiol Endocrinol Metab 302:E1329–E1342
Donaton MC, Holsbeeks I, Lagatie O, Van Zeebroeck G, Crauwels M, Winderickx J, Thevelein JM (2003) The Gap1 general amino acid permease acts as an amino acid sensor for activation of protein kinase A targets in the yeast Saccharomyces cerevisiae. Mol Microbiol 50:911–929
Dunlop EA, Hunt DK, Acosta-Jaquez HA, Fingar DC, Tee AR (2011) ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy 7:737–747
Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT (2011) p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell 44:134–146
Duran RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, Hall MN (2012) Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell 47:349–358
Duran RV, Mackenzie ED, Boulahbel H, Frezza C, Heiserich L, Tardito S, Bussolati O, Rocha S, Hall MN, Gottlieb E (2013) HIF-independent role of prolyl hydroxylases in the cellular response to amino acids. Oncogene 32:4549–4556
Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, Mackeigan JP, Finan PM, Clish CB, Murphy LO, Manning BD (2010) Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39:171–183
Efeyan A, Zoncu R, Chang S, Gumper I, Snitkin H, Wolfson RL, Kirak O, Sabatini DD, Sabatini DM (2013) Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 493:679–683
Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, Fussi H, Deszcz L, Hartl R, Schraml E, Criollo A, Megalou E, Weiskopf D, Laun P, Heeren G, Breitenbach M, Grubeck-Loebenstein B, Herker E, Fahrenkrog B, Frohlich KU, Sinner F, Tavernarakis N, Minois N, Kroemer G, Madeo F (2009) Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 11:1305–1314
Fader CM, Sanchez DG, Mestre MB, Colombo MI (2009) TI-VAMP/VAMP7 and VAMP3/cellubrevin: two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim Biophys Acta 1793:1901–1916
Fernandez J, Yaman I, Merrick WC, Koromilas A, Wek RC, Sood R, Hensold J, Hatzoglou M (2002) Regulation of internal ribosome entry site-mediated translation by eukaryotic initiation factor-2alpha phosphorylation and translation of a small upstream open reading frame. J Biol Chem 277:2050–2058
Findlay GM, Yan L, Procter J, Mieulet V, Lamb RF (2007) A MAP4 kinase related to Ste20 is a nutrient-sensitive regulator of mTOR signalling. Biochem J 403:13–20
Fogel AI, Dlouhy BJ, Wang C, Ryu SW, Neutzner A, Hasson SA, Sideris DP, Abeliovich H, Youle RJ (2013) Role of membrane association and Atg14-dependent phosphorylation in beclin-1-mediated autophagy. Mol Cell Biol 33:3675–3688
Fotiadis D, Kanai Y, Palacin M (2013) The SLC3 and SLC7 families of amino acid transporters. Mol Aspects Med 34:139–158
Furuta N, Fujita N, Noda T, Yoshimori T, Amano A (2010) Combinational soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Mol Biol Cell 21:1001–1010
Gamerdinger M, Hajieva P, Kaya AM, Wolfrum U, Hartl FU, Behl C (2009) Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J 28:889–901
Ganley IG, Du Lam H, Wang J, Ding X, Chen S, Jiang X (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 284:12297–12305
Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G (2003) Insulin activation of Rheb, a mediator of mTOR/S6 K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell 11:1457–1466
Ge L, Melville D, Zhang M, Schekman R (2013) The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife 2:e00947
Glickman MH, Ciechanover A (2002) The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82:373–428
Grandison RC, Piper MD, Partridge L (2009) Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature 462:1061–1064
Green DR, Levine B (2014) To be or not to be? How selective autophagy and cell death govern cell fate. Cell 157:65–75
Gulati P, Gaspers LD, Dann SG, Joaquin M, Nobukuni T, Natt F, Kozma SC, Thomas AP, Thomas G (2008) Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab 7:456–465
Gutierrez MG, Munafo DB, Beron W, Colombo MI (2004) Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci 117:2687–2697
Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141:656–667
Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, Yoshimori T (2013) Autophagosomes form at ER-mitochondria contact sites. Nature 495:389–393
Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK, Ha SH, Ryu SH, Kim S (2012) Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 149:410–424
Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J (1998) Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem 273:14484–14494
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889
Harding HP, Zhang Y, Scheuner D, Chen JJ, Kaufman RJ, Ron D (2009) Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc Natl Acad Sci USA 106:1832–1837
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460:392–395
Heublein S, Kazi S, Ogmundsdottir MH, Attwood EV, Kala S, Boyd CA, Wilson C, Goberdhan DC (2010) Proton-assisted amino-acid transporters are conserved regulators of proliferation and amino-acid-dependent mTORC1 activation. Oncogene 29:4068–4079
Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, Guan JL, Oshiro N, Mizushima N (2009) Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 20:1981–1991
Huang J, Manning BD (2008) The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J 412:179–190
Hundal HS, Taylor PM (2009) Amino acid transceptors: gate keepers of nutrient exchange and regulators of nutrient signaling. Am J Physiol Endocrinol Metab 296:E603–E613
Iiboshi Y, Papst PJ, Kawasome H, Hosoi H, Abraham RT, Houghton PJ, Terada N (1999) Amino acid-dependent control of p70(s6k). Involvement of tRNA aminoacylation in the regulation. J Biol Chem 274:1092–1099
Inoki K, Li Y, Xu T, Guan KL (2003) Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 17:1829–1834
Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL (2004) Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 117:4837–4848
Janku F, Mcconkey DJ, Hong DS, Kurzrock R (2011) Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 8:528–539
Jewell JL, Guan KL (2013) Nutrient signaling to mTOR and cell growth. Trends Biochem Sci 38:233–242
Johansen T, Lamark T (2011) Selective autophagy mediated by autophagic adapter proteins. Autophagy 7:279–296
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH (2009) ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20:1992–2003
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19:5720–5728
Kamata S, Yamamoto J, Kamijo K, Ochiai T, Morita T, Yoshitomi Y, Hagiya Y, Kubota M, Ohkubo R, Kawaguchi M, Himi T, Kasahara T, Ishii I (2014) Dietary deprivation of each essential amino acid induces differential systemic adaptive responses in mice. Mol Nutr Food Res. doi:10.1002/mnfr.201300758
Kaushik S, Massey AC, Mizushima N, Cuervo AM (2008) Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell 19:2179–2192
Kawamura N, Sun-Wada GH, Aoyama M, Harada A, Takasuga S, Sasaki T, Wada Y (2012) Delivery of endosomes to lysosomes via microautophagy in the visceral endoderm of mouse embryos. Nat Commun 3:1071
Kiffin R, Christian C, Knecht E, Cuervo AM (2004) Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell 15:4829–4840
Kiffin R, Kaushik S, Zeng M, Bandyopadhyay U, Zhang C, Massey AC, Martinez-Vicente M, Cuervo AM (2007) Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J Cell Sci 120:782–791
Kihara A, Noda T, Ishihara N, Ohsumi Y (2001) Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol 152:519–530
Kilberg MS, Pan YX, Chen H, Leung-Pineda V (2005) Nutritional control of gene expression: how mammalian cells respond to amino acid limitation. Annu Rev Nutr 25:59–85
Kilberg MS, Shan J, Su N (2009) ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol Metab 20:436–443
Kim KH, Lee MS (2014) Autophagy-a key player in cellular and body metabolism. Nat Rev Endocrinol 10:322–337
Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL (2008) Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 10:935–945
Kim S, Kim SF, Maag D, Maxwell MJ, Resnick AC, Juluri KR, Chakraborty A, Koldobskiy MA, Cha SH, Barrow R, Snowman AM, Snyder SH (2011) Amino acid signaling to mTOR mediated by inositol polyphosphate multikinase. Cell Metab 13:215–221
Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G (1997) daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277:942–946
Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, Mcewan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Overvatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T (2009) A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 33:505–516
Klionsky DJ, Schulman BA (2014) Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat Struct Mol Biol 21:336–345
Koga H, Martinez-Vicente M, Macian F, Verkhusha VV, Cuervo AM (2011) A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat Commun 2:386
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884
Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC (2009) Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 33:517–527
Korolchuk VI, Menzies FM, Rubinsztein DC (2010) Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 584:1393–1398
Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, Jahreiss L, Sarkar S, Futter M, Menzies FM, O’kane CJ, Deretic V, Rubinsztein DC (2011) Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol 13:453–460
Kriel J, Haesendonckx S, Rubio-Texeira M, Van Zeebroeck G, Thevelein JM (2011) From transporter to transceptor: signaling from transporters provokes re-evaluation of complex trafficking and regulatory controls: endocytic internalization and intracellular trafficking of nutrient transceptors may, at least in part, be governed by their signaling function. Bioessays 33:870–879
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N (2004) The role of autophagy during the early neonatal starvation period. Nature 432:1032–1036
Lamb CA, Yoshimori T, Tooze SA (2013) The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 14:759–774
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293
Li WW, Li J, Bao JK (2012) Microautophagy: lesser-known self-eating. Cell Mol Life Sci 69:1125–1136
Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT (2013) K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell 51:283–296
Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C, Yankner BA, Scherzer CR, Yuan J (2010) Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc Natl Acad Sci USA 107:14164–14169
Liu J, Hatzoglou M (1998) Control of expression of the gene for the arginine transporter Cat-1 in rat liver cells by glucocorticoids and insulin. Amino Acids 15:321–337
Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J (2005a) Rheb binds and regulates the mTOR kinase. Curr Biol 15:702–713
Long X, Ortiz-Vega S, Lin Y, Avruch J (2005b) Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J Biol Chem 280:23433–23436
Lorin S, Tol MJ, Bauvy C, Strijland A, Pous C, Verhoeven AJ, Codogno P, Meijer AJ (2013) Glutamate dehydrogenase contributes to leucine sensing in the regulation of autophagy. Autophagy 9:850–860
Madeo F, Tavernarakis N, Kroemer G (2010) Can autophagy promote longevity? Nat Cell Biol 12:842–846
Malmberg SE, Adams CM (2008) Insulin signaling and the general amino acid control response. Two distinct pathways to amino acid synthesis and uptake. J Biol Chem 283:19229–19234
Malzer E, Szajewska-Skuta M, Dalton LE, Thomas SE, Hu N, Skaer H, Lomas DA, Crowther DC, Marciniak SJ (2013) Coordinate regulation of eIF2alpha phosphorylation by PPP1R15 and GCN2 is required during Drosophila development. J Cell Sci 126:1406–1415
Marques AJ, Palanimurugan R, Matias AC, Ramos PC, Dohmen RJ (2009) Catalytic mechanism and assembly of the proteasome. Chem Rev 109:1509–1536
Martina JA, Puertollano R (2013) Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J Cell Biol 200:475–491
Martina JA, Chen Y, Gucek M, Puertollano R (2012) MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8:903–914
Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM (2006) Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci USA 103:5805–5810
Matsui T, Fukuda M (2013) Rab12 regulates mTORC1 activity and autophagy through controlling the degradation of amino-acid transporter PAT4. EMBO Rep 14:450–457
Meijer AJ, Codogno P (2008) Nutrient sensing: TOR’s Ragtime. Nat Cell Biol 10:881–883
Meijer AJ, Dubbelhuis PF (2004) Amino acid signalling and the integration of metabolism. Biochem Biophys Res Commun 313:397–403
Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, Harris AL (2009) The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res 69:4415–4423
Miller RA, Buehner G, Chang Y, Harper JM, Sigler R, Smith-Wheelock M (2005) Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell 4:119–125
Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y (1998) A protein conjugation system essential for autophagy. Nature 395:395–398
Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T (2003) Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci 116:1679–1688
Monastyrska I, Rieter E, Klionsky DJ, Reggiori F (2009) Multiple roles of the cytoskeleton in autophagy. Biol Rev Camb Philos Soc 84:431–448
Moreau K, Ravikumar B, Puri C, Rubinsztein DC (2012) Arf6 promotes autophagosome formation via effects on phosphatidylinositol 4,5-bisphosphate and phospholipase D. J Cell Biol 196:483–496
Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen WL, Griffith J, Nag S, Wang K, Moss T, Baba M, Mcnew JA, Jiang X, Reggiori F, Melia TJ, Klionsky DJ (2011) SNARE proteins are required for macroautophagy. Cell 146:290–302
Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, Mackeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO (2009) Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136:521–534
Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19:983–997
Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, Byfield MP, Backer JM, Natt F, Bos JL, Zwartkruis FJ, Thomas G (2005) Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci USA 102:14238–14243
Novoa I, Zeng H, Harding HP, Ron D (2001) Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol 153:1011–1022
Ogmundsdottir MH, Heublein S, Kazi S, Reynolds B, Visvalingam SM, Shaw MK, Goberdhan DC (2012) Proton-assisted amino acid transporter PAT1 complexes with Rag GTPases and activates TORC1 on late endosomal and lysosomal membranes. PLoS One 7:e36616
Oh WJ, Jacinto E (2011) mTOR complex 2 signaling and functions. Cell Cycle 10:2305–2316
Ohsumi Y (2001) Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2:211–216
Oshiro N, Rapley J, Avruch J (2014) Amino acids activate mammalian target of rapamycin (mTOR) complex 1 without changing Rag GTPase guanyl nucleotide charging. J Biol Chem 289:2658–2674
Panchaud N, Peli-Gulli MP, De Virgilio C (2013) Amino acid deprivation inhibits TORC1 through a GTPase-activating protein complex for the Rag family GTPase Gtr1. Sci Signal 6:ra42
Pandey UB, Nie Z, Batlevi Y, Mccray BA, Ritson GP, Nedelsky NB, Schwartz SL, Diprospero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447:859–863
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282:24131–24145
Pinilla J, Aledo JC, Cwiklinski E, Hyde R, Taylor PM, Hundal HS (2011) SNAT2 transceptor signalling via mTOR: a role in cell growth and proliferation? Front Biosci (Elite Ed) 3:1289–1299
Poncet N, Taylor PM (2013) The role of amino acid transporters in nutrition. Curr Opin Clin Nutr Metab Care 16:57–65
Poulsen P, Wu B, Gaber RF, Ottow K, Andersen HA, Kielland-Brandt MC (2005) Amino acid sensing by Ssy1. Biochem Soc Trans 33:261–264
Proud CG (2014) Control of the translational machinery by amino acids. Am J Clin Nutr 99:231S–236S
Rabinowitz JD, White E (2010) Autophagy and metabolism. Science 330:1344–1348
Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC (2010a) Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 12:747–757
Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC (2010b) Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90:1383–1435
Renna M, Schaffner C, Winslow AR, Menzies FM, Peden AA, Floto RA, Rubinsztein DC (2011) Autophagic substrate clearance requires activity of the syntaxin-5 SNARE complex. J Cell Sci 124:469–482
Roccio M, Bos JL, Zwartkruis FJ (2006) Regulation of the small GTPase Rheb by amino acids. Oncogene 25:657–664
Rodgers KJ (2014) Non-protein amino acids and neurodegeneration: the enemy within. Exp Neurol 253:192–196
Rodriguez-Navarro JA, Kaushik S, Koga H, Dall’armi C, Shui G, Wenk MR, Di Paolo G, Cuervo AM (2012) Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc Natl Acad Sci USA 109:E705–E714
Rubinsztein DC, Codogno P, Levine B (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11:709–730
Rubio-Texeira M, Van Zeebroeck G, Voordeckers K, Thevelein JM (2010) Saccharomyces cerevisiae plasma membrane nutrient sensors and their role in PKA signaling. FEMS Yeast Res 10:134–149
Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL (2013) ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 15:741–750
Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320:1496–1501
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141:290–303
Sarkar S (2013a) Chemical screening platforms for autophagy drug discovery to identify therapeutic candidates for Huntington’s disease and other neurodegenerative disorders. Drug Discov Today Technol 10:e137–e144
Sarkar S (2013b) Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem Soc Trans 41:1103–1130
Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC (2009) Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 16:46–56
Sarkar S, Korolchuk VI, Renna M, Imarisio S, Fleming A, Williams A, Garcia-Arencibia M, Rose C, Luo S, Underwood BR, Kroemer G, O’kane CJ, Rubinsztein DC (2011) Complex inhibitory effects of nitric oxide on autophagy. Mol Cell 43:19–32
Sarkar S, Carroll B, Buganim Y, Maetzel D, Ng AH, Cassady JP, Cohen MA, Chakraborty S, Wang H, Spooner E, Ploegh H, Gsponer J, Korolchuk VI, Jaenisch R (2013) Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep 5:1302–1315
Seglen PO, Gordon PB (1984) Amino acid control of autophagic sequestration and protein degradation in isolated rat hepatocytes. J Cell Biol 99:435–444
Seglen PO, Gordon PB, Poli A (1980) Amino acid inhibition of the autophagic/lysosomal pathway of protein degradation in isolated rat hepatocytes. Biochim Biophys Acta 630:103–118
Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, Gems D, Withers DJ (2009) Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326:140–144
Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A (2011) TFEB links autophagy to lysosomal biogenesis. Science 332:1429–1433
Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A (2012) A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31:1095–1108
Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD (2008) Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4:176–184
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ (2009) Autophagy regulates lipid metabolism. Nature 458:1131–1135
Smith EM, Finn SG, Tee AR, Browne GJ, Proud CG (2005) The tuberous sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin by amino acids and certain cellular stresses. J Biol Chem 280:18717–18727
Talloczy Z, Jiang W, Virgin HWT, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B (2002) Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA 99:190–195
Tanida I, Ueno T, Kominami E (2004) LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 36:2503–2518
Tato I, Bartrons R, Ventura F, Rosa JL (2011) Amino acids activate mammalian target of rapamycin complex 2 (mTORC2) via PI3K/Akt signaling. J Biol Chem 286:6128–6142
Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J (2003) Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol 13:1259–1268
Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM (2012) A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485:109–113
Tsun ZY, Bar-Peled L, Chantranupong L, Zoncu R, Wang T, Kim C, Spooner E, Sabatini DM (2013) The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell 52:495–505
Vabulas RM, Hartl FU (2005) Protein synthesis upon acute nutrient restriction relies on proteasome function. Science 310:1960–1963
Wang X, Proud CG (2008) A novel mechanism for the control of translation initiation by amino acids, mediated by phosphorylation of eukaryotic initiation factor 2B. Mol Cell Biol 28:1429–1442
Watanabe-Asano T, Kuma A, Mizushima N (2014) Cycloheximide inhibits starvation-induced autophagy through mTORC1 activation. Biochem Biophys Res Commun 445:334–339
Wauson EM, Zaganjor E, Lee AY, Guerra ML, Ghosh AB, Bookout AL, Chambers CP, Jivan A, Mcglynn K, Hutchison MR, Deberardinis RJ, Cobb MH (2012) The G protein-coupled taste receptor T1R1/T1R3 regulates mTORC1 and autophagy. Mol Cell 47:851–862
White E (2012) Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 12:401–410
Wohlgemuth SE, Seo AY, Marzetti E, Lees HA, Leeuwenburgh C (2010) Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life-long exercise. Exp Gerontol 45:138–148
Yang F, Chu X, Yin M, Liu X, Yuan H, Niu Y, Fu L (2014) mTOR and autophagy in normal brain aging and caloric restriction ameliorating age-related cognition deficits. Behav Brain Res 264C:82–90
Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12:9–14
Yu L, Mcphee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, Hailey DW, Oorschot V, Klumperman J, Baehrecke EH, Lenardo MJ (2010) Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465:942–946
Yuan HX, Russell RC, Guan KL (2013) Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy 9:1983–1995
Zhang C, Cuervo AM (2008) Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med 14:959–965
Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D (2003) Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol 5:578–581
Zheng S, Clabough EB, Sarkar S, Futter M, Rubinsztein DC, Zeitlin SO (2010) Deletion of the huntingtin polyglutamine stretch enhances neuronal autophagy and longevity in mice. PLoS Genet 6:e1000838
Zhu K, Dunner K Jr, Mcconkey DJ (2010) Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 29:451–462
Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM (2011) mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334:678–683
Acknowledgments
We thank Francisco Marques (Newcastle University) for maintaining and providing primary human fibroblasts and Tom DiCesare (BARC, Whitehead Institute for Biomedical Research) for assistance with illustrations. B.C and V.I.K are supported by Biotechnology and Biological Sciences Research Council (BBSRC), UK. S.S. is thankful to the laboratory of Rudolf Jaenisch and Whitehead Institute for Biomedical Research for funding. V.I.K and S.S are Former Fellows at Hughes Hall, University of Cambridge, UK.
Conflict of interest
The authors declare no conflict of interests.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Carroll, B., Korolchuk, V.I. & Sarkar, S. Amino acids and autophagy: cross-talk and co-operation to control cellular homeostasis. Amino Acids 47, 2065–2088 (2015). https://doi.org/10.1007/s00726-014-1775-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-014-1775-2