
Abstract
l-Homoarginine (hArg) has recently emerged as a novel cardiovascular risk factor and to herald a poor prognosis in heart failure patients. Here, we report on the development and thorough validation of gas chromatography–mass spectrometry (GC–MS) and gas chromatography–tandem mass spectrometry (GC–MS/MS) methods for the quantitative determination of hArg in biological samples, including human plasma, urine and sputum. For plasma and serum samples, ultrafiltrate (10 µL; cutoff, 10 kDa) was used. For urine samples, native urine (10 µL) was used. For sputum, protein precipitation by acetone was performed. hArg is derivatized to its methyl ester tri(N-pentafluoropropionyl) derivative; de novo synthesized trideutero-methyl ester hArg is used as the internal standard (IS). Alternatively, [guanidino-15N2]-arginine can be used as an IS. Quantitative analyses were performed after electron-capture negative-ion chemical ionization by selected-ion monitoring in GC–MS and selected-reaction monitoring in GC–MS/MS. We obtained very similar hArg concentrations by GC–MS and GC–MS/MS, suggesting that GC–MS suffices for accurate and precise quantification of hArg in biological samples. In plasma and serum samples of the same subjects very close hArg concentrations were measured. The plasma-to-serum hArg concentration ratio was determined to be 1.12 ± 0.21 (RSD, 19 %), suggesting that blood anticoagulation is not a major preanalytical concern in hArg analysis. In healthy subjects, the creatinine-corrected urinary excretion of hArg varies considerably (0.18 ± 0.22 µmol/mmol, mean ± SD, n = 19) unlike asymmetric dimethylarginine (ADMA, 2.89 ± 0.89 µmol/mmol). In urine, hArg correlated with ADMA (r = 0.475, P = 0.040); in average, subjects excreted in the urine about 17.5 times more ADMA than hArg. In plasma of healthy humans, the concentration of hArg is of the order of 2 µM. hArg may be a low-abundance constituent of human plasma proteins. The GC–MS and GC-MS/MS methods we report in this article are useful to study the physiology and pathology of hArg in experimental and clinical settings.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
l-Homoarginine (hArg) is a non-essential, non-proteinogenic cationic amino acid, a homologous of l-arginine (Arg); the structural difference between hArg and Arg is a methylene (CH2, 14 Da) group (Fig. 1). hArg and Arg behave similarly in vivo and in vitro (Ryan and Wells 1964; Ryan et al. 1969; Drechsler et al. 2011). Arg plays an important role in metabolism and nutrition in growth, health and disease (Wu et al. 2009). Arg also serves as the substrate for nitric oxide synthases (NOS) which catalyze the conversion of Arg to nitric oxide (NO) and l-citrulline. hArg may also serve as a substrate for NOS; yet, hArg has also been suggested as an inhibitor of NOS (Moali et al. 1998, 2000; Bretscher et al. 2003). NO possesses various biological activities including inhibition of vascular inflammation and platelet aggregation, prevention of adhesion of immune cells, and vasodilation (Moncada and Higgs 1993; Leiper and Vallance 1999). Low plasma hArg concentrations were found to be associated with cardiovascular and all-cause mortality in patients (März et al. 2010; Pilz et al. 2011; Choe et al. 2013; Kayacelebi et al. 2014). Mean plasma and serum hArg concentrations are of the order of 2 µM. hArg homeostasis has been reported to be altered in pregnancy and it is assumed that hArg is involved in the pathology of preeclampsia (Valtonen et al. 2008; Khalil et al. 2013). Recently, lower hArg concentrations were measured in plasma of male smokers compared to male nonsmokers, while ADMA plasma concentrations were higher in smokers compared to nonsmokers (Sobczak et al. 2014).

Chemical structures, molecular formula and molecular weight (MW) of l-arginine and its homolog l-homoarginine
The biosynthesis of hArg and its biological activities in humans are incompletely understood (Ryan and Wells 1964; Ryan et al. 1969). l-Arginine:glycine amidinotransferase (AGAT; EC 2.1.4.1) has recently been demonstrated to play a pivotal role in hArg biosynthesis. AGAT catalyzes the transamination of l-lysine by glycine to form hArg (Davids et al. 2012a; Choe et al. 2013). Increased AGAT expression was found to be associated with increased hArg synthesis in a cell model (Choe et al. 2013).
Because of the particular scientific interest in Arg and its involvement in numerous biological processes, including protein synthesis and the Arg/NO pathway, a variety of chromatographic and mass spectrometric methods are currently available for Arg and its derivatives, including asymmetric dimethylarginine (ADMA) and symmetric dimethylarginine (SDMA) (Schwedhelm 2005; Horowitz and Heresztyn 2007; Schwedhelm et al. 2007; Teerlink 2007; Martens-Lobenhoffer and Bode-Böger 2014; Tsikas 2008; Wu et al. 2009). In principle, analytical methods currently available for Arg should also be useful for its homologue hArg. However, the concentration of hArg in biological samples, notably plasma, is severalfold lower than of Arg, and this may represent an analytical challenge. The emerging importance of hArg as a novel biomarker of cardiovascular diseases and its step motherly investigated biology prompted many scientists in recent years to develop specific and sensitive analytical methods for the quantitative determination of hArg in basic and clinical research.
The usefulness of liquid chromatography–tandem mass spectrometry (LC–MS/MS) for the analysis of hArg in biological samples (Atzler et al. 2011; Davids et al. 2012b), gas chromatography–mass spectrometry (GC–MS) and gas chromatography–tandem mass spectrometry (GC–MS/MS) methods for Arg and its relatives have been reviewed and discussed recently (Martens-Lobenhoffer et al. 2009; Martens-Lobenhoffer and Bode-Böger 2014).
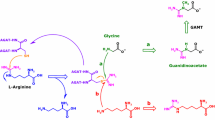
In previous work, we showed that GC–MS/MS allows for accurate and sensitive quantitative determination of Arg, ADMA and SDMA in human plasma and urine (Tsikas et al. 2003, 2011). Because no stable isotope-labeled analogs of ADMA were commercially available at that time, we synthesized [2H3]methyl ester ADMA (d3Me-d0ADMA) for use as internal standard (IS). In the present work, we report on the development and validation of GC–MS and GC–MS/MS methods for the quantitative determination of hArg in various biological samples. These approaches involve separate O-methylation of the carboxylic group of endogenous hArg using HCl in CH3OH to generate the methyl ester of endogenous hArg (i.e., d0Me-d0hArg), and of the carboxylic group of synthetic hArg using HCl in CD3OD to generate the [2H3]methyl ester of hArg (i.e., d3Me-d0hArg) for use as IS (Fig. 2). After methylation the samples containing d0Me-d0hArg and d3Me-d0hArg are combined to a single sample for subsequent derivatization (i.e., N-pentafluoropropionylation) with pentafluoropropionic anhydride (PFPA). The utility of these methods to measure hArg in human biological samples including plasma, serum, urine and sputum is demonstrated.

Schematic of the procedures used in the GC–MS analysis of homoarginine in biological fluids. PFP pentafluoropropionyl, PA peak area
Materials and methods
Materials and chemicals
l-Homoarginine hydrochloride and l-arginine hydrochloride were purchased from Sigma (Deisenhofen, Germany). PFPA was obtained from Pierce (Rockford, IL, USA). Tetradeuterated methanol (CD3OD, 99 % at 2H) was supplied by Aldrich (Steinheim, Germany). All other chemicals were obtained from Merck (Darmstadt, Germany). Vivaspin 2 Hydrosart cartridges (cutoff, 10 kDa) were supplied by Sartorius (Göttingen, Germany).
Biological samples: recovery of blood, plasma and ultrafiltrate
Blood (5–7 mL) was drawn from antecubital veins of healthy volunteers using syringes containing EDTA and put immediately on ice. Blood samples were centrifuged (1,500×g, 4 °C, 15 min) and the plasma generated was separated and analyzed immediately or stored at −78 °C until further analysis. Routinely, ultrafiltrates (approx. 200 µL) from 0.5-mL aliquots of plasma samples were obtained by centrifugation (15,000×g, 4 °C, 30 min) using the Vivaspin cartridges. Plasma ultrafiltrate (10 µL) was used for the quantification of plasma hArg. Urine from healthy volunteers was obtained from spontaneous micturition. Urine samples were analyzed immediately or stored at −20 °C until analysis.
Sputum was collected by a routine procedure, put immediately on ice and stored at −80 °C until analysis. After thawing on ice, portions of sputum were weighed (range 30–240 mg) and treated under vortexing with the fourfold volume of ice-cold acetone to inhibit enzymatic activity and precipitate proteins. After centrifugation (800×g, 2 °C, 5 min), 100-µL aliquots of the supernatants were evaporated to dryness under a stream of nitrogen, and the residues were subjected to analysis as described below.
Analytical methods
Procedures for the quantification of homoarginine in biological samples
Figure 2 shows schematically the procedures used for the quantitative determination of hArg in plasma and other biological samples using de novo synthesized d3Me-d0hArg. Aliquots (10 µL each) of plasma ultrafiltrate or native urine and of an aqueous solution of hArg (5 µM if not otherwise specified), serving as the IS, were transferred each into a 1.5-mL glass vial and the samples were evaporated to dryness by means of a nitrogen stream. The residues of biological samples were treated with 100-µL aliquots of a 2 M HCl solution in CH3OH. Analogous, the residue of the standard hArg solution was treated with a 100-µL aliquot of a 2 M HCl solution in CD3OD. Esterification was performed by heating the samples for 60 min at 80 °C. After cooling to room temperature, the methanolic solutions were combined. To ensure quantitative transfer, the residue of each sample was taken up with a 100-µL aliquot of CH3OH. Subsequently, solvents and reagents of the combined solutions were evaporated to dryness by a stream of nitrogen gas. The common residue was then treated with a 100-µL aliquot of a solution of PFPA in ethyl acetate (1:4, v/v), and the sample was heated for 30 min at 65 °C. After cooling to room temperature, the sample was evaporated to dryness, the residue was reconstituted with a 200-µL aliquot of 0.4 M borate buffer, pH 8.5, and the sample was mixed by vortexing immediately for 60 s with 200-µL aliquots of toluene. Aliquots (100 µL) of the upper organic layer were taken and transferred into glass vials, from which 1-µL aliquots were analyzed by GC–MS in the selected-ion monitoring (SIM) mode or by GC–MS/MS in the selected-reaction monitoring (SRM) mode as described below.
Method validation and comparison
Accuracy (recovery %) and precision (RSD %) of the method were determined using freshly obtained and pooled plasma and urine samples from spontaneous micturition from volunteers. Intra- and inter-assay precision was determined by working up in quadruplicate (plasma) or in duplicate (urine) of unspiked and spiked pooled plasma samples on four consecutive days. Recovery was calculated using the Formula (1):
whereas, [found] is the measured hArg concentration in the samples, [basal] is the measured concentration of hArg in the unspiked samples, and [added] is the concentration of hArg added to the sample.
For comparison, biological samples were analyzed for hArg by GC–MS and GC–MS/MS using the TSQ 7000 instrument, as well as by GC–MS using the DSQ instrument. These data were analyzed by linear regression and the Bland–Altman method.
Contribution of plasma proteins to homoarginine
To test the presence of hArg in plasma proteins, pooled plasma samples (100 µL) were incubated for 24 h with distilled water (900 µL) at room temperature, with 6 M HCl (900 µL) at room temperature, and with 6 M HCl (900 µL) at 110 °C in sealed glass vials. After sample ultrafiltration (15,000×g, 4 °C, 30 min), 10-µL aliquots of the ultrafiltrates were treated as described above except for the concentration of the IS hArg which was 0.5 µM. This procedure is the same recently applied to ADMA analysis in biological samples (Tsikas et al. 2010) and corresponds to generally valid protocols (Dai et al. 2014).
An alternative quantification mode for homoarginine
Due to the close physicochemical properties of hArg and Arg, we tested the utility of Arg as an IS for the quantification of hArg. Aliquots of aqueous solutions of hArg and Arg were mixed to obtain solutions containing hArg at the fixed concentration of 5 µM and Arg at the varying concentrations of 0, 25, 50 and 100 µM. Each two 10-µL aliquots were derivatized as described above and analyzed by GC–MS in the SIM mode.
GC–MS and GC–MS/MS conditions
Gas chromatography–mass spectrometry and GC–MS/MS analyses in the electron-capture negative-ion chemical ionization (ECNICI) mode were performed on a triple-stage quadrupole (TSQ) mass spectrometer model ThermoQuest TSQ 7000 (Finnigan MAT, San Jose, CA) directly interfaced with a Trace 2000 series gas chromatograph equipped with an autosampler AS 2000 (CE Instruments, Austin, TX). If not otherwise stated, the gas chromatograph was equipped with a 30-m-long fused-silica capillary column Optima 17 (0.25 mm I.D., 0.25-µm film thickness) from Macherey–Nagel (Düren, Germany). The following oven temperature program was used with helium (at a constant flow rate of 1 mL/min) as the carrier gas: 1 min at 90 °C, then increased to 225 and 320 °C at a rate of 15 and 30 °C/min, respectively. Interface (280 °C), injector (280 °C) and ion-source (180 °C) were kept at constant temperatures. Electron energy was set to 200 eV and electron current to 300 µA. Methane (530 Pa) and argon (0.13 Pa collision pressure) were used as reagent and collision gases, respectively. Routinely, collision energy and electron multiplier voltage were set to 18 eV and 1.6 kV, respectively.
Gas chromatography–mass spectrometry analyses in the ECNICI mode were also performed on a double-stage quadrupole (DSQ) mass spectrometer model ThermoQuest DSQ (ThermoFisher, Dreieich, Germany) directly interfaced with a Focus series gas chromatograph equipped with an autosampler AS 3000 (CE Instruments, Austin, TX). If not otherwise specified, the gas chromatograph was equipped with a 15-m-long fused-silica capillary column Optima 17 (0.25 mm I.D., 0.25 µm film thickness) from Macherey–Nagel (Düren, Germany). The following oven temperature program was used with helium (at a constant flow rate of 1 mL/min) as the carrier gas: 0.5 min at 90 °C, then increased to 210 and 320 °C at a rate of 15 and 35 °C/min, respectively. Interface, injector and ion-source were kept at 280, 280 and 250 °C, respectively. Electron energy was set to 70 eV and electron current to 100 µA. Methane (2.4 mL/min) was used as the reagent gas.
In both instruments, aliquots (1 µL from toluene extracts) were injected in the splitless mode by means of the respective autosampler. In quantitative analyses, the dwell time was 50 ms for each ion in GC–MS (SIM mode) and in GC–MS/MS (SRM mode).
Results
Method development and validation
Figures 3 and 4 show the GC–MS and GC–MS/MS spectra of unlabelled and deuterium-labeled hArg, respectively, after esterification and N-acylation by PFPA as described in “Materials and methods”. The GC–MS spectra indicate that under the chosen derivatization conditions hArg is converted to its methyl ester tri(N-pentafluoropropionyl) derivative (Fig. 2). The most intense ions in the GC–MS spectra were m/z 600 and 603 due to consecutive neutral loss of 2 HF (20 Da) molecules from unlabelled and labeled hArg, respectively. Subjection of the precursor ions [M–2xHF]− to collision-induced dissociation (CID) with argon (at a collision energy of 18 eV) resulted in similar product ion mass spectra (Figs. 3a, 4a). The most intense product ion was m/z 366 both for unlabelled and deuterium-labeled hArg (Figs. 3b, 4b). These findings suggest that the product ion m/z 366 does not carry the carboxylic group of hArg, but it contains the guanidine group. Expectedly, the derivative of deuterated hArg eluted a few seconds in front of the unlabelled hArg derivative. In quantitative analyses by GC–MS, the ions m/z 600 for unlabelled hArg and m/z 603 for deuterated hArg were detected in the SIM mode. In quantitative analyses by GC–MS/MS, the mass transitions m/z 600 to 366 for unlabelled hArg and of m/z 603 to 366 for deuterated hArg were detected in the SRM mode.

a GC–MS and b GC–MS/MS ECNICI spectra of the methyl ester tri(N-pentafluoropropionyl) derivative of unlabelled hArg. The parent ion at m/z 600 [M–2xHF]− was subjected to collision-induced dissociation (CID) with argon at a collision energy of 18 eV. The GC–MS/MS TSQ7000 instrument was used. The retention time of the GC peak from which the mass spectra were obtained was 6.80 min. Insets indicate the putative structures of the most intense ions

a GC–MS and b GC–MS/MS ECNICI spectra of the trideutero-methyl ester tri(N-pentafluoropropionyl) derivative of hArg. The parent ion at m/z 603 [M–2xHF]− was subjected to collision-induced dissociation (CID) with argon at a collision energy of 18 eV. The GC–MS/MS TSQ7000 instrument was used. The retention time of the GC peak from which the mass spectra were obtained was 6.78 min. Insets indicate the putative structures of the most intense ions
The results from the validation experiments are summarized in Tables 1 and 2 for plasma and Table 3 for urine. The imprecision (RSD, %) of the measurements ranged between 0.5 and 7.6 %. In the pooled plasma used in these experiments, the mean hArg plasma concentration was measured to be 1.41 ± 0.05 µM. The lowest hArg concentration added to plasma, i.e., 1 µM, (71 % of the hArg basal concentration) was quantitated with high accuracy and precision (Tables 1, 2). In the pooled urine sample used for method validation, endogenous hArg was measured at about 0.45 µM. The lowest concentration of hArg externally added to the urine sample was 0.2 µM (about 44 % of the basal level) (Table 3).
Linear regression analysis between found (y) and added (x) hArg concentrations revealed straight lines (r > 0.992) for plasma (Tables 1, 2) and urine (Table 3). These data indicate that hArg can be quantitated by these GC–MS and GC–MS/MS methods with high accuracy and precision using d3Me-d0hArg as IS in 10-µL aliquots of plasma ultrafiltrate or native urine (see also below).
Representative GC–MS and GC–MS/MS chromatograms from quantitative analyses of hArg in human plasma, urine and sputum samples are shown in Fig. 5. Unlike in human urine, in human plasma samples additional peaks from unknown, non-interfering substances appear in the chromatograms.

Partial chromatograms from analyses of hArg in human plasma by a GC–MS using instrument DSQ and by b GC–MS/MS using instrument TSQ. c Partial chromatograms from GC–MS analyses (DSQ instrument) of hArg in human urine. d Partial chromatograms from GC–MS analyses (DSQ instrument) of hArg in human sputum. d3Me-d0hArg was used as the internal standard at a final nominal concentration of 5 µM in plasma and urine. SIM was performed at m/z 600 for unlabelled hArg (upper traces) and m/z 603 for d3Me-d0hArg (lower traces). SRM was performed of the common product ion at m/z 366 which was produced by CID of the parent ions at m/z 600 for unlabelled hArg (upper traces) and m/z 603 for d3Me-d0hArg (lower traces). Endogenous hArg and internal standard were analyzed as their Me-(PFP)3 derivatives
Testing arginine as an internal standard for homoarginine
Selected-ion monitoring of the methyl ester pentafluoropropionyl derivatives of hArg (5 µM) and Arg (0, 25, 50, 100 µM) and plotting of the ratio of the measured peak areas of Arg and hArg (y) versus the Arg concentration (x) resulted in a straight line (r 2 = 0.9995) with the regression equation y = −0.11 + 0.158x (Fig. 6). The y-axis intercept is close to zero and the calculated slope of 0.158 deviates from the theoretical slope of 0.200 (corresponding to a response factor 1.0) by 21 % (i.e., 0.158/0.200 × 100). This means that at a molar basis hArg yields peak areas that are by 21 % smaller that those of Arg, presumably due to the longer retention time of the hArg derivative (relative retention time, 1.135). Thus, Arg is suitable as an IS for hArg. Calculation of hArg concentrations can be performed by multiplying the measured peak area ratio hArg-to-Arg by the concentration of Arg and by the correction factor of 1.266 (i.e., 0.2/0.158). For this quantification method, both de novo synthesized d3Me-d0Arg and commercially available stable isotope-labeled arginine such as [guanidino-15N2]arginine should be useful.

Arginine-to-homoarginine peak area ratio measured in aqueous solutions of the indicated varying concentrations of Arg and the fixed hArg concentration of 5 µM. Arg and hArg were measured simultaneously by GC–MS, i.e., SIM of m/z 586 for Arg and m/z 600 for hArg
Homoarginine concentrations in human plasma and serum
In EDTA plasma and serum samples obtained from blood of 10 healthy subjects of a previously reported study (Engeli et al. 2012) we measured hArg by GC–MS/MS. The plasma hArg concentrations (y) correlated (r 2 = 0.6997) with those measured in serum (x), however, there were also significant differences between plasma and serum concentrations in three subjects (Fig. 7). The hArg plasma-to-serum concentration ratio was determined to be (mean ± SD) 1.123 ± 0.211 (RSD, 19 %). Thus, in average plasma hArg concentrations were by about 12 % higher than serum hArg concentrations in these subjects. Considering both plasma and serum, the circulating hArg concentration in these subjects was determined to be 1.87 ± 0.67 µM (RSD, 36 %).

Plasma and serum concentrations of hArg in healthy subjects measured by GC–MS (DSQ instrument). d3Me-d0hArg was used as the IS at a final nominal concentration of 5 µM. SIM was performed of m/z 600 for unlabelled hArg (upper traces) and m/z 603 for d3Me-d0hArg (lower traces). Endogenous hArg and IS were analyzed as their Me-(PFP)3 derivatives
Homoarginine excretion in urine of healthy humans
In spot urine samples of 19 healthy subjects we measured hArg, ADMA, SDMA, the ADMA metabolite dimethyl amine (DMA) and creatinine by GC–MS. The creatinine-corrected excretion of these biochemical parameters are summarized in Table 4. Both in males and in females, hArg excretion varied greatly (range 37–816 nmol/mmol), unlike ADMA, SDMA and DMA. Males excreted less (by a factor of 1.3–2.3) hArg, ADMA, SDMA and DMA in the urine than females, presumably because males excreted in average 2 times more creatinine than females (12.7 ± 6.5 vs 6.0 ± 2.2 mM). In urine, hArg correlated with ADMA (r = 0.475, P = 0.040) and SDMA (r = 0.625, P = 0.004), but did not correlate with body mass index (BMI; data not shown). In average, the subjects excreted in the urine about 17.5 times more ADMA than hArg, although the plasma concentration of hArg is several times higher than the plasma concentration of ADMA, for instance about nine times higher in healthy pregnant women at 11–13 weeks of gestation (Khalil et al. 2013; Tsikas and Kayacelebi 2014). This finding suggests that hArg is effectively reabsorbed by the kidneys.
Contribution of plasma proteins to homoarginine
hArg plasma concentration in the samples incubated with 6 M HCl at room temperature was 1.34 ± 0.24 µM and tended to be higher than that measured in the pooled plasma samples incubated at 110 °C (1.76 ± 0.18 µM, P = 0.057) (Fig. 8). Analogous to ADMA (Tsikas and Beckmann 2009; Tsikas et al. 2010), hArg seems not be a considerable constituent of human plasma proteins including albumin. Yet, this issue remains to be investigated on plasma or serum samples from a large number of subjects.

Concentration of hArg in pooled human plasma upon incubation for 24 h at room temperature (RT) in the absence of HCl (distilled water was used instead of 6 M HCl, RT/H2O), with 6 M HCl (RT/HCl) and for 24 h at 110 °C with 6 M HCl
Comparison between GC–MS and GC–MS/MS for homoarginine in human plasma
Unspiked plasma samples from 369 humans and 92 plasma samples spiked with hArg at various concentrations (1, 2, 5, 7.5 and 10 µM) from a previous study (Khalil et al. 2013) were analyzed for hArg by GC–MS on the instrument DSQ and by GC–MS/MS on the instrument TSQ 7000. Linear regression analysis between the hArg concentrations measured by GC–MS (x) and those measured by GC–MS/MS (y) revealed high linearity (r = 0.997) and good agreement for the unspiked (82 %) and the spiked (89 %) plasma samples (Fig. 9). GC–MS analysis revealed lower hArg plasma concentrations than GC–MS/MS analysis (in average by 18 and 11 %). The Bland–Altman plotting revealed a systematic error (bias) of about 0.07 µM hArg between GC–MS and GC–MS/MS.

Comparison between GC–MS and GC–MS/MS for the analysis of hArg in unspiked plasma samples of 369 humans (a, b) and in 92 human plasma samples spiked with the hArg concentrations indicated by arrows (c) by linear regression analysis (a, c) and by the Bland–Altman method (b)
Discussion
For many decades, l-homoarginine (hArg) existed in the shadow of its homologous l-arginine (Arg). Yet, recent advances in analytical methodologies and the recognition of the immense importance of the l-Arg/NO pathway in the cardiovascular system brought hArg in the focus of current research. Measurement of hArg in blood (i.e., plasma or serum) revealed that hArg may play important role(s) in health and disease. For example, too low circulating hArg concentrations were found to be associated with cardiac morbidity and mortality (März et al. 2010; Drechsler et al. 2011; Pilz et al. 2011; Kayacelebi et al. 2014), like ADMA, another l-arginine analog and inhibitor of NOS activity. However, unlike ADMA (Horowitz and Heresztyn 2007; Teerlink 2007), the blood concentration of hArg seems not to be closely regulated. hArg blood concentrations in healthy and diseased subjects measured by reliable analytical methods were found to vary considerable in the range 0.5–5 µM. Given the emerging importance of hArg as a cardiovascular risk factor, our group aimed at developing sensitive and reliable analytical methods for the accurate quantification of hArg in biological samples including human plasma and urine. In this article, we report on the development and validation of GC–MS and GC–MS/MS methods for the precise and accurate quantitative determination of hArg in biological samples. This method was validated for human and urine samples. Its applicability is extendable to other biological samples such as saliva after appropriate treatment and in extracellular fluids that can be obtained by microdialysis techniques (data not shown).
The GC–MS-based methods involve a two-step derivatization procedure and the de novo synthesis of deuterium-labeled hArg for use as IS (Fig. 2). In the first derivatization step, the methyl esters of unlabelled hArg (d0Me-d0hArg) and deuterium-labeled hArg (d3Me-d0hArg) are prepared. In the subsequent second derivatization step, the N-pentafluoropropionyl (PFP) derivatives are produced. Our results suggest that the α-amino group and the two guanidine amino groups are N-pentafluoropropionylated. Thus, the final derivatives are d0Me-(PFP)3-d0hArg and d3Me-(PFP)3-d0hArg. Similar procedures have been shown to be useful for the GC–MS analysis of Arg (Tsikas et al. 2003; Albsmeier et al. 2007). However, because the concentrations of hArg in plasma and urine are several times lower than those of Arg, thorough examination of the utility of existing methods to hArg measurement was required. Our results show that, due to the 15 strong electron-capturing fluorine (F) atoms of d0Me-(PFP)3-d0hArg and d3Me-(PFP)3-d0hArg, ionization in the ECNICI mode produces anions in such high yield that sensitive and reliable quantitative measurement of hArg by GC–MS and GC–MS/MS is possible in very small volumes (routinely, 10 µL) of plasma ultrafiltrates and native urine samples.
The HCl-catalyzed methylation of the carboxylic group of hArg in deuterium-labeled methanol (CD3OD) was found to be excellently suitable for de novo synthesis of d3Me-d0hArg and its use as an IS for the quantitative determination of hArg in human plasma and urine samples by GC–MS and GC–MS/MS when analyzed as methyl ester pentafluoropropionyl derivatives. Alternatively, de novo synthesized d3Me-d0Arg or commercially available stable isotope-labeled analogs of arginine such as [guanidino-15N2]arginine should be useful as ISs for hArg. The results of our comparison studies on a large number of plasma samples, i.e., the measurement of hArg by GC–MS in the SIM mode and by GC–MS/MS in the SRM mode, demonstrate that hArg can be quantitated in small volumes of human plasma and urine by GC–MS as accurately and precisely as by GC–MS/MS.
It is well known that the concentration of many biomolecules may differ between plasma and serum, i.e., may depend on coagulation or anticoagulation. This is true for Arg of which the serum concentration is considerably higher than in plasma obtained from blood of human subjects, while ADMA concentrations are virtually identical in human plasma and serum (Teerlink 2007). In the present study, we investigated this potentially important preanalytical issue for hArg. Our results show that with respect to blood coagulation hArg ranks between ADMA and Arg. Another possibly important preanalytical issue in amino acid analysis is the ability of proteins to release their amino acids upon chemical or enzymatic proteolysis. In this regard, hArg resembles ADMA (Tsikas and Beckmann 2009; Tsikas et al. 2010). Quantitative hydrolysis of human plasma proteins and subsequent analysis by GC–MS were found to yield slightly higher hArg concentrations compared to nonhydrolyzed plasma proteins. From the analytical point of view, acid-catalyzed hydrolysis of proteins and peptides present in human plasma are unlikely to contribute to an appreciable extent to free hArg. From the biological standpoint, this issue remains to be investigated in detail.
The method, we reported in this article, provides hArg concentrations in human plasma and urine which are in accordance with hArg concentrations measured by others cited in this article. Our observations indicate that the procedures previously found by us to be optimum for ADMA are equally useful for hArg. This is most likely due to comparable concentrations of ADMA and hArg in human plasma and serum, which are several times smaller compared to Arg. In principle, Arg, hArg, ADMA and SDMA can be quantitated in a single run by GC–MS after a two-step derivatization procedure as described in this work. However, because of the higher Arg concentrations attenuation of the detector sensitivity is required for Arg analysis. Yet, this can be easily accomplished by modern GC–MS and GC–MS/MS instruments.
Conclusions
The GC–MS and GC–MS/MS methods reported in this article allow for accurate, precise and sensitive quantitative determination of homoarginine in various biological samples including human plasma and urine. Our current investigations indicate that these methods are a useful analytical tool in basic and in clinical research of hArg, an emerging cardiovascular risk factor. In healthy humans, the concentration of hArg in plasma and urine lies in the lowest µM-range. In healthy adult subjects, the hArg/ADMA ratio in plasma/serum is of the order of 5:1 (Tsikas and Kayacelebi 2014), while the hArg/ADMA ratio in the urine is of the order of 1:20. Obviously, the body seeks to save hArg and to eliminate ADMA, most likely because hArg is utilized in important pathways which are still undiscovered.
Abbreviations
- ADMA:
-
N G ,N G-Dimethyl-l-arginine
- AGAT:
-
Arginine:glycine amidinotransferase
- CID:
-
Collision-induced dissociation
- DMA:
-
Dimethyl amine
- DSQ:
-
Double-stage quadrupole
- ECNICI:
-
Electron-capture negative-ion chemical ionization
- GC–MS:
-
Gas chromatography–mass spectrometry
- GC–MS/MS:
-
Gas chromatography–tandem mass spectrometry
- hArg:
-
Homoarginine
- IS:
-
Internal standard
- LC–MS/MS:
-
Liquid chromatography–tandem mass spectrometry
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- PFP:
-
Pentafluoropropionyl
- PFPA:
-
Pentafluoropropionic anhydride
- SDMA:
-
Symmetric dimethylarginine
- SIM:
-
Selected-ion monitoring
- SRM:
-
Selected-reaction monitoring
- TSQ:
-
Triple-stage quadrupole
References
Albsmeier J, Schwedhelm E, Schulze F, Kastner M, Böger RH (2004) Determination of NG, NG-dimethyl-l-arginine, an endogenous NO synthase inhibitor, by gas chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 809(1):59–65
Atzler D, Mieth M, Maas R, Böger RH, Schwedhelm E (2011) Stable isotope dilution assay for liquid chromatography-tandem mass spectrometric determination of l-homoarginine in human plasma. J Chromatogr B 879:2294–2298
Bretscher LE, Li H, Poulos TL, Griffith OW (2003) Structural characterization and kinetics of nitric-oxide synthase inhibition by novel N5-(iminoalkyl)- and N5- (iminoalkenyl)-ornithines. J Biol Chem 278:46789–46797
Choe CU, Atzler D, Wild PS, Carter AM, Böger RH, Ojeda F, Simova O, Stockebrand M, Lackner K, Nabuurs C, Marescau B, Streichert T, Muller C, Luneburg N, De Deyn PP, Benndorf RA, Baldus S, Gerloff C, Blankenberg S, Heerschap A, Grant PJ, Magnus T, Zeller T, Isbrandt D, Schwedhelm E (2013) Homoarginine levels are regulated by l-arginine:glycine amidinotransferase and affect stroke outcome: results from human and murine studies. Circulation 128:1451–1461
Dai Z, Wu Z, Jia S, Wu G (2014) Analysis of amino acid composition in proteins of animal tissues and foods as pre-column o-phthaldialdehyde derivatives by HPLC with fluorescence detection. J Chromatogr B. doi:10.1016/j.jchromb.2014.03.025
Davids M, Ndika JD, Salomons GS, Blom HJ, Teerlink T (2012a) Promiscuous activity of arginine:glycine amidinotransferase is responsible for the synthesis of the novel cardiovascular risk factor homoarginine. FEBS Lett 586:3653–3657
Davids M, Swieringa E, Palm F, Smith DE, Smulders YM, Scheffer PG, Blom HJ, Teerlink T (2012b) Simultaneous determination of asymmetric and symmetric dimethylarginine, L-monomethylarginine, l-arginine, and L-homoarginine in biological samples using stable isotope dilution liquid chromatography tandem mass spectrometry. J Chromatogr B 900:38–47
Drechsler C, Meinitzer A, Pilz S, Krane V, Tomaschitz A, Ritz E, März W, Wanner C (2011) Homoarginine, heart failure, and sudden cardiac death in haemodialysis patients. Eur J Heart Fail 13:852–859
Engeli S, Tsikas D, Lehmann AC, Böhnke J, Haas V, Strauß A, Janke J, Gorzelniak K, Luft FC, Jordan J (2012) Influence of dietary fat ingestion on asymmetrical dimethylarginine in lean and obese human subjects. Nutr Metab Cardiovasc Dis 22:720–726
Horowitz JD, Heresztyn T (2007) An overview of plasma concentrations of asymmetric dimethylarginine (ADMA) in health and disease and in clinical studies: methodological considerations. J Chromatogr B 851:42–50
Kayacelebi AA, Nguyen TH, Neil C, Horowitz JD, Jordan J, Tsikas D (2014) Homoarginine and 3-nitrotyrosine in patients with takotsubo cardiomyopathy. Int J Cardiol. doi:10.1016/j.ijcard.2014.03.080
Khalil AA, Tsikas D, Akolekar R, Jordan J, Nicolaides KH (2013) Asymmetric dimethylarginine, arginine and homoarginine at 11–13 weeks gestation and preeclampsia: a case-control study. J Hum Hypertens 27:38–43
Leiper J, Vallance P (1999) Biological significance of endogenous methylarginines that inhibit nitric oxide synthases. Cardiovasc Res 43:542–548
Martens-Lobenhoffer J, Bode-Böger SM (2014) Mass spectrometric quantification of l-arginine and its pathway related substances in biofluids: the road to maturity. J Chromatogr B. doi:10.1016/j.jchromb.2013.10.030
Martens-Lobenhoffer J, Schwedhelm E, Tsikas D (2009) Quantification of arginine and its mono- and dimethylated analogs NMMA, ADMA and SDMA in biological fluids by LC-MS/MS: is LC superfluous? J Chromatogr B 877:3261–3266
März W, Meinitzer A, Drechsler C, Pilz S, Krane V, Kleber ME, Fischer J, Winkelmann BR, Böhm BO, Ritz E, Wanner C (2010) Homoarginine, cardiovascular risk, and mortality. Circulation 122:967–975
Moali C, Boucher JL, Sari MA, Stuehr DJ, Mansuy D (1998) Substrate specificity of NO synthases: detailed comparison of l-arginine, homo-l-arginine, their N omega-hydroxy derivatives, and N omega-hydroxynor-l-arginine. Biochemistry 37:10453–10460
Moali C, Brollo M, Custot J, Sari MA, Boucher JL, Stuehr DJ, Mansuy D (2000) Recognition of alpha-amino acids bearing various C = NOH functions by nitric oxide synthase and arginase involves very different structural determinants. Biochemistry 39:8208–8218
Moncada S, Higgs A (1993) The l-arginine-nitric oxide pathway. New Engl J Med 329:2002–2012
Pilz S, Tomaschitz A, Meinitzer A, Drechsler C, Ritz E, Krane V, Wanner C, Bohm BO, März W (2011) Low serum homoarginine is a novel risk factor for fatal strokes in patients undergoing coronary angiography. Stroke 42:1132–1134
Ryan WL, Wells IC (1964) Homocitrulline and homoarginine synthesis from lysine. Science 144:1122–1127
Ryan WL, Johnson RJ, Dimari S (1969) Homoarginine synthesis by rat kidney. Arch Biochem Biophys 131:521–526
Schwedhelm E (2005) Quantification of ADMA: analytical approaches. Vascular Med (London, England) 10(Suppl 1):S89–S95
Schwedhelm E, Maas R, Tan-Andresen J, Schulze F, Riederer U, Böger RH (2007) High-throughput liquid chromatographic-tandem mass spectrometric determination of arginine and dimethylated arginine derivatives in human and mouse plasma. J Chromatogr B 851:211–219
Sobczak A, Prokopowicz A, Radek M, Szula M, Zaciera M, Kurek J, Goniewicz ML (2014) Tobacco smoking decreases plasma concentration of the emerging cardiovascular risk marker, l-homoarginine. Circ J 78:1254–1258
Teerlink T (2007) HPLC analysis of ADMA and other methylated l-arginine analogs in biological fluids. J Chromatogr B 851:21–29
Tsikas D (2008) A critical review and discussion of analytical methods in the l-arginine/nitric oxide area of basic and clinical research. Anal Biochem 379:139–163
Tsikas D, Beckmann B (2009) Albumin from human serum does not contain asymmetric dimethylarginine (ADMA). Clin Biochem 42:1739–1740
Tsikas D, Kayacelebi AA (2014) Do homoarginine and asymmetric dimethylarginine act antagonistically in the cardiovascular system? Circ J 20
Tsikas D, Schubert B, Gutzki FM, Sandmann J, Frölich JC (2003) Quantitative determination of circulating and urinary asymmetric dimethylarginine (ADMA) in humans by gas chromatography-tandem mass spectrometry as methyl ester tri(N-pentafluoropropionyl) derivative. J Chromatogr B 798:87–99
Tsikas D, Engeli S, Beckmann B, Jordan J (2010) Asymmetric dimethylarginine (ADMA) is present in plasma proteins of healthy subjects at the low nmol-per-g-level. Nitric Oxide 22:316–317
Tsikas D, Beckmann B, Gutzki FM, Jordan J (2011) Simultaneous gas chromatography-tandem mass spectrometry quantification of symmetric and asymmetric dimethylarginine in human urine. Anal Biochem 413:60–62
Valtonen P, Laitinen T, Lyyra-Laitinen T, Raitakari OT, Juonala M, Viikari JS, Heiskanen N, Vanninen E, Punnonen K, Heinonen S (2008) Serum l-homoarginine concentration is elevated during normal pregnancy and is related to flow-mediated vasodilatation. Circ J 72:1879–1884
Wu G, Bazer FW, Davis TA, Kim SW, Li P, Marc Rhoads J, Carey Satterfield M, Smith SB, Spencer TE, Yin Y (2009) Arginine metabolism and nutrition in growth, health and disease. Amino Acids 37:153–168
Acknowledgments
We thank the staff of the Institute of Clinical Pharmacology, Hannover Medical School, for providing us with spot urine samples for the analysis of hArg and its relatives.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kayacelebi, A.A., Beckmann, B., Gutzki, FM. et al. GC–MS and GC–MS/MS measurement of the cardiovascular risk factor homoarginine in biological samples. Amino Acids 46, 2205–2217 (2014). https://doi.org/10.1007/s00726-014-1774-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-014-1774-3