
Abstract
Bombesin (BBN) is a peptide exhibiting high affinity for the gastrin-releasing peptide receptor (GRPR), which is overexpressed on several types of cancers. Various GRPR antagonists and agonists have been labeled with radiometals for positron emission tomography (PET) imaging of GRPR-positive tumors. However, unfavorable hepatobiliary excretion such as high intestinal activity may prohibit their clinical utility for imaging abdominal cancer. In this study, the modified BBN peptide with a new hydrophilic linker was labeled with 68Ga for PET imaging of GRPR-expressing PC-3 prostate cancer xenograft model. GRPR antagonists, MATBBN (Gly-Gly-Gly-Arg-Asp-Asn-d-Phe-Gln-Trp-Ala-Val-Gly-His-Leu-NHCH2CH3) and ATBBN (d-Phe-Gln-Trp-Ala-Val-Gly-His-Leu-NHCH2CH3), were conjugated with 1,4,7-triazacyclononanetriacetic acid (NOTA) and labeled with 68Ga. Partition coefficient and in vitro stability were also determined. GRPR binding affinity of both tracers was investigated by competitive radioligand binding assay. The in vivo receptor targeting potential and pharmacokinetic of 68Ga-NOTA-MATBBN were also evaluated in PC-3 prostate tumor model and compared with those of 68Ga-NOTA-ATBBN. NOTA-conjugated BBN analogs were labeled with 68Ga within 20 min with a decay-corrected yield ranging from 90 to 95 % and a radiochemical purity of more than 98 %. The specific activity of 68Ga-NOTA-MATBBN and 68Ga-NOTA-ATBBN was at least 16.5 and 11.9 GBq/μmol, respectively. The radiotracers were stable in phosphate-buffered saline and human serum. 68Ga-NOTA-MATBBN was more hydrophilic than 68Ga-NOTA-ATBBN, as indicated by their log P values (−2.73 ± 0.02 vs. −1.20 ± 0.03). The IC50 values of NOTA-ATBBN and NOTA-MATBBN were similar (102.7 ± 1.18 and 124.6 ± 1.21 nM). The accumulation of 68Ga-labeled GRPR antagonists in the subcutaneous PC-3 tumors could be visualized via small animal PET. The tumors were clearly visible, and the tumor uptakes of 68Ga-NOTA-MATBBN and 68Ga-NOTA-ATBBN were determined to be 4.19 ± 0.32, 4.00 ± 0.41, 2.93 ± 0.35 and 4.70 ± 0.40, 4.10 ± 0.30, 3.14 ± 0.30 %ID/g at 30, 60, and 120 min, respectively. There was considerable accumulation and retention of 68Ga-NOTA-ATBBN in the liver and intestines. In contrast, the abdominal area does not have much retention of 68Ga-NOTA-MATBBN. Biodistribution data were in accordance with the PET results, showing that 68Ga-NOTA-MATBBN had more favorable pharmacokinetics and higher tumor to background ratios than those of 68Ga-NOTA-ATBBN. At 1 h postinjection, the tumor to liver and intestine of 68Ga-NOTA-MATBBN were 8.05 ± 0.56 and 21.72 ± 3.47 and the corresponding values of unmodified counterpart were 0.85 ± 0.23 and 3.45 ± 0.43, respectively. GRPR binding specificity was demonstrated by reduced tumor uptake of radiolabeled tracers after coinjection of an excess of unlabeled BBN peptides. 68Ga-NOTA-MATBBN exhibited GRPR-targeting properties both in vitro and in vivo. The favorable characterizations of 68Ga-NOTA-MATBBN such as convenient synthesis, specific GRPR targeting, high tumor uptake, and satisfactory pharmacokinetics warrant its further investigation for clinical cancer imaging.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Radiolabeled peptides targeting receptors overexpressed on tumor for imaging have been extensively investigated due to easy good manufacturing practices, quick diffusion, and low immunogenicity (Ambrosini et al. 2011; Graham and Menda 2011). Gastrin-releasing peptide receptor (GRPR) is overexpressed in a variety of human tumors, which provides a potential molecular target for diagnosis and therapy of cancers (Carroll et al. 2000; Fleischmann et al. 2005; Reubi et al. 2004).
Bombesin (BBN), a natural GRPR-targeting peptide, is involved in regulating exocrine secretion, smooth muscle contraction, and gastrointestinal hormone release (Reubi et al. 2005). Although various BBN analogs have already been labeled and used for positron emission tomography (PET) imaging of GRPR-positive tumors, few compounds have been tested in clinical research (Chen et al. 2004; Hoffman et al. 2003; Zhang et al. 2006; Dumont et al. 2013; Ananias et al. 2011). The main drawback of most BBN-based radiotracers is the relatively high intestinal activity, which may limits its potential clinical applications (Dimitrakopoulou-Strauss et al. 2007; Dijkgraaf et al. 2012; Liu et al. 2012; Lears et al. 2011; Schuhmacher et al. 2005).
Gly-Gly-Gly-Gly-Arg-Asp-Asn is a new hydrophilic linker, which significantly improves the PET image quality of GRPR tracers. Our previous study showed that radiolabeled modified GRPR antagonist (18F-FP-Gly-Gly-Gly-Arg-Asp-Asn-d-Phe-Gln-Trp-Ala-Val-Gly-His-Leu-NHCH2CH3) had more favorable pharmacokinetics than 18F-FP-ATBBN (18F-FP-d-Phe-Gln-Trp-Ala-Val-Gly-His-Leu-NHCH2CH3) (Yang et al. 2011). However, the synthesis of such 18F-labeled compound through prosthetic groups is complicated and time-consuming.
Recently, the application of 68Ga-labeled peptides has attracted considerable interest for cancer imaging because of its excellent physical characteristics (Fani et al. 2008; de Sá et al. 2010; Breeman et al. 2011). 68Ga decays by 89 % through positron emission of 1.92 MeV (max. energy) and is available from a 68Ge/68Ga generator (68Ge, t 1/2 = 270.8 day), without the need of an onsite cyclotron. With a half-life of 68 min, it is also appropriate for studying the pharmacokinetics of many peptides. In addition, biomolecules are labeled with 68Ga via macrocyclic chelators, which allows possible kit formulation and wide availability of the corresponding imaging probes.
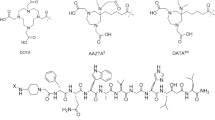
Herein, we labeled the 1,4,7-triazacyclononanetriacetic acid (NOTA) conjugates of MATBBN and ATBBN with 68Ga (Fig. 1), the in vitro receptor binding and in vivo tumor imaging properties of 68Ga-NOTA-MATBBN and 68Ga-NOTA-ATBBN were also investigated in a PC-3 human prostate tumor model.

Chemical structure of 68Ga-labeled NOTA conjugates of BBN analogs
Materials and methods
p-SCN-Bn-NOTA was purchased from Macrocyclics (Dallas, TX). MATBBN (Gly-Gly-Gly-Arg-Asp-Asn-d-Phe-Gln-Trp-Ala-Val-Gly-His-Leu-NHCH2CH3) and ATBBN (d-Phe-Gln-Trp-Ala-Val-Gly-His-Leu-NHCH2CH3) peptides were kindly gifted from Professor Shawn Chen (The National Institute of Biomedical Imaging and Bioengineering, USA). 125I-[Tyr4]BBN (74 TBq/mmol, 2,000 Ci/mmol) was purchased from Perkin-Elmer. 68Ga was obtained from a 68Ge/68Ga generator (ITG isotope technologies Garching GmbH, Germany) eluted with 0.1 N HCl. All other commercially obtained chemicals were of analytical grade and used without further purification.
A Waters high-performance liquid chromatography (HPLC) system with a Waters 2998 photodiode array detector (PDA) using a preparative C18 HPLC column (Xbridge C18 5 μm, 250 × 19 mm, Waters) was used for peptide conjugate purification. The flow rate was 20 mL/min, and the mobile phase was changed from 95 % solvent A (0.1 % trifluoroacetic acid in water) and 5 % solvent B (0.1 % trifluoroacetic acid in acetonitrile) (0–2 min) to 35 % solvent A and 65 % solvent B at 21 min. The UV absorbance was monitored at 218 nm, and the identification of the peptides was confirmed based on the UV spectrum of peptides obtained with the PDA detector.
The radiolabeled compounds were analyzed by RP-HPLC on Waters Breeze system. A C18 column (5 μm, 250 × 4.6 mm, Phenomenex) was used at a flow rate of 1 mL/min with the following buffer system: buffer A, 0.1 % v/v trifluoroacetic acid in H2O; buffer B, 0.1 % v/v trifluoroacetic acid in acetonitrile; and a gradient of 95 % buffer A at 0–2 min to 35 % buffer A at 35 min. The radioactivity of the eluate was monitored using a Radiomatic 610TR flow scintillation analyzer (Perkin-Elmer).
Mass spectra were obtained with a Waters LC–MS system (Waters, Milford, MA) that included an Acquity UPLC system coupled to a Waters Q-Tof Premier high-resolution mass spectrometer.
NOTA conjugation of peptides
MATBBN and ATBBN peptides were conjugated with NOTA under standard SCN-amine reaction conditions as previously described (Lang et al. 2011). Briefly, a solution of 6 μmol of peptide (ATBBN or MATBBN) was mixed with 8 μmol of p-SCN-Bn-NOTA in sodium bicarbonate buffer (pH 9.0). After stirring at room temperature overnight, the NOTA-conjugated peptides were isolated by preparative HPLC. The desired fractions were combined and lyophilized to afford the final product as a white powder. NOTA-MATBBN was obtained in 50 % yield with 18.1 min retention time on analytical HPLC. Matrix-assisted laser desorption/ionization (MALDI) time-of-flight (TOF) mass spectrometry (MS) measured m/z 1,991.58 for [MH]+ (C89H127N27O24S, calculated molecular weight, 1,991.23). NOTA-ATBBN was obtained in 55 % yield with 21.4 min retention time on analytical HPLC. MALDI-TOF MS measured m/z 1,435.54 for [MH]+ (C69H95N17O15S, calculated molecular weight, 1,434.69).
68Ga radiolabeling
Fresh 68Ga activity was eluted from the 68Ge/68Ga generator with 0.1 M HCl at 0.5 mL per fraction into the 1.5-mL polypropylene tubes. The fraction containing the most radioactivity (185 MBq) was added to 0.2 mL 1 M HEPES buffer and 20 μg NOTA-MATBBN or NOTA-ATBBN in 20 μL 0.2 M pH 4 sodium acetate buffer. The mixture was heated at 100 °C for 10 min. After cooling, the reaction mixture was diluted with 10 mL deionized water and then loaded into a Varian BOND ELUT C18 column. The cartridge was washed again with 10 mL water, and the desired labeled peptide was eluted with 0.3 mL 10 mM HCl in ethanol. For in vitro and in vivo studies, the eluate was diluted to <5 % ethanol in PBS and passed through a 0.22-μm Millipore filter into a sterile multidose vial.
For quality control purposes, a portion of the product was diluted and injected into an analytical C18 HPLC column to assay for radiochemical purity. The retention times for 68Ga-NOTA-MATBBN and 68Ga-NOTA-ATBBN were 18.6 and 20.3 min, respectively (Fig. 2).

Radio-HPLC chromatograms of 68Ga-NOTA-MATBBN (top) and 68Ga-NOTA-ATBBN (bottom)
Octanol/water partition coefficient
The radiolabeled peptide was dissolved in a mixture of phosphate buffer (0.5 mL, pH 7.4) and n-octanol (0.5 mL) at 25 °C. The mixture was vigorously stirred by a vortex mixer for 2 min followed by centrifugation. Samples of 100 μL were taken from each layer, radioactivity was measured in a well-type γ-counter (Perkin-Elmer), and log P values were calculated.
In vitro stability studies
The stability of the radiolabeled BBN analogs was investigated in human plasma at various incubation times (0–120 min) at 37 °C. PBS was used as a control. After incubation, the serum was passed through a Sep-Pak C18 cartridge, washed with 0.5 mL PBS buffer, and then eluted with 0.5 mL ACN containing 0.1 % TFA. The PBS control was diluted with 0.5 mL acetonitrile. The elution fractions and the PBS control were analyzed by analytical HPLC.
Competitive cell binding assay
The PC-3 human prostate cancer cell line was cultured in RPMI 1,640 medium supplemented with 10 % (v/v) fetal calf serum. Cells were grown in tissue culture flasks at 37 °C in a humidified atmosphere containing 5 % CO2 and routinely passed using 0.25 % trypsin/ethylenediaminetetraacetic acid.
The in vitro GRPR binding affinity of NOTA-MATBBN and NOTA-ATBBN was measured via displacement cell binding assays using 125I-[Tyr4]BBN as the radioligand. Experiments were performed on GRPR-expressing PC-3 human prostate carcinoma cells following a previously described method (Yang et al. 2011). IC50 (the best-fit 50 % inhibitory concentration) values were determined using GraphPad Prism 4 (GraphPad Software, Inc.) by fitting the data with nonlinear regression. Experiments were performed with triplicate samples.
MicroPET imaging
Male BALB/c nude mice were injected subcutaneously in the right flank with 0.2 mL of a PC-3 cell suspension (2 × 107 cells/mL). Two or three weeks after inoculation of the tumor cells, mice were subjected to PET imaging studies. All animal experiments were approved by the local animal welfare committee and performed according to Chinese national regulations.
PET scans and image analysis were performed using an Inveon microPET (Siemens Medical Solutions). Each PC-3 tumor-bearing mouse was injected intravenously with about 3.7 MBq (100 μCi) 68Ga-NOTA-MATBBN or 68Ga-NOTA-ATBBN under isoflurane anesthesia (n = 4/group). For static PET, 5-min scans were acquired at 30 min, 1, and 2 h after injection. Scans were reconstructed using Inveon Acquisition Workplace software (version 1.4; Siemens Preclinical Solutions), using a three-dimensional ordered-subset expectation maximization/maximum a posteriori algorithm with the following parameters: matrix, 128 × 128 × 159; pixel size, 0.86 × 0.86 × 0.8 mm; and β-value, 1.5, with uniform resolution.
For the blocking experiment, PC-3 tumor-bearing mice were coinjected with blocking dose (10 mg/kg body weight) of unlabeled peptides (MATBBN or ATBBN) and 3.7 MBq 68Ga-NOTA-MATBBN or 68Ga-NOTA-ATBBN, respectively. Five-minute static PET scans were acquired at 60 min postinjection (n = 4/group).
For each microPET scan, regions of interest (ROIs) were drawn over the tumor, and major organs using vendor software ASI Pro 6.7.1.1 on decay-corrected whole-body coronal images. The radioactivity concentration (accumulation) within a tumor or an organ was obtained from mean pixel values within the multiple ROI volume. These values were then divided by the administered activity to obtain (assuming a tissue density of 1 g/mL) an image ROI-derived percent injected dose per gram (%ID/g).
Biodistribution studies
Male athymic nude mice bearing PC-3 xenografts were injected with 0.74 MBq (20 μCi) of 68Ga-NOTA-MATBBN or 68Ga-NOTA-ATBBN to evaluate the distribution of the tracer in the tumor tissues and major organs. At 0.5, 1, and 2 h after injection of the tracer, the mice were killed and dissected. Blood, tumor, and major organs were collected and wet-weighed. The radioactivity in the tissue was measured by a γ-counter (Perkin-Elmer). The amount of radioactivity was determined with the γ-counter to calculate uptake as the percentage injected dose per gram of tissue (%ID/g). In addition, mice that received a coinjection of unlabeled peptide (10 mg/kg body weight) and the corresponding radiolabeled compound (n = 4/group) were killed at 1 h after injection to determine nonspecific tissue uptake.
Statistical analysis
Quantitative data are expressed as mean ± SD. Means were compared using one-way analysis of variance (ANOVA) and Student’s t test. P values <0.05 were considered statistically significant.
Results
Chemistry and radiochemistry
The labeling was done within 20 min, with a decay-corrected yield ranging from 90 to 95 % and a radiochemical purity of more than 98 %. The specific activity of 68Ga-NOTA-MATBBN and 68Ga-NOTA-ATBBN were at least 16.5 and 11.9 GBq/μmol, respectively. The logP octanol/water values for 68Ga-NOTA-MATBBN and 68Ga-NOTA-ATBBN were −2.73 ± 0.02 and −1.20 ± 0.03, respectively. 68Ga-NOTA-MATBBN and 68Ga-NOTA-ATBBN were stable in PBS and human serum at 37 °C. After 2 h, the radiochemical purities were >95 % with HPLC analysis.
Cell binding assay
The affinity of NOTA-MATBBN and NOTA-ATBBN for GRPR was determined by performing competitive binding assay with 125I-[Tyr4]BBN as the radioligand. The results of these assays are summarized in Fig. 3. Binding of 125I-[Tyr4]BBN peptide to GRPR was displaced by BBN analogs in a concentration-dependent manner. The IC50 values of the NOTA conjugates of ATBBN and MATBBN were 102.7 ± 1.18 and 124.6 ± 1.21 nM, respectively.

Competition bindings of 125I-[Tyr4]BBN with NOTA-ATBBN (left) and NOTA-MATBBN (right), respectively
MicroPET imaging
Representative coronal microPET images of PC-3 tumor-bearing mice (n = 4/group) at different times after intravenous injection of 3.7 MBq (100 μCi) 68Ga-NOTA-MATBBN and 68Ga-NOTA-ATBBN are shown in Figs. 4 and 5. The tumors were clearly visible with high contrast at all the time points examined for 68Ga-NOTA-MATBBN. However, there was considerable accumulation and retention of 68Ga-NOTA-ATBBN in normal organs such as liver and intestines. In contrast, 68Ga-NOTA-MATBBN showed little retention in the abdominal area.

a Decay-corrected whole-body coronal microPET images of PC-3 tumor-bearing mice at 30, 60, and 120 min after injection of 3.7 MBq 68Ga-NOTA-MATBBN, 60 min after injection of 3.7 MBq 68Ga-NOTA-MATBBN with MATBBN as blocking agent (10 mg/kg body weight). Tumors are indicated by arrows. b Quantification of 68Ga-NOTA-MATBBN in PC-3 tumor, liver, kidneys, and muscle at 30, 60, and 120 min after injection of 3.7 MBq 68Ga-NOTA-MATBBN, 60 min after injection of 3.7 MBq 68Ga-NOTA-MATBBN with MATBBN as blocking agent (10 mg/kg body weight). ROIs are shown as mean %ID/g ± SD

a Decay-corrected whole-body coronal microPET images of PC-3 tumor-bearing mice at 30, 60, and 120 min after injection of 3.7 MBq 68Ga-NOTA-ATBBN, 60 min after injection of 3.7 MBq 68Ga-NOTA-ATBBN with ATBBN as blocking agent (10 mg/kg body weight). Tumors are indicated by arrows. b Quantification of 68Ga-NOTA-ATBBN in PC-3 tumor, liver, kidneys, and muscle at 30, 60, and 120 min after injection of 3.7 MBq 68Ga-NOTA-ATBBN, 60 min after injection of 3.7 MBq 68Ga-NOTA-ATBBN with ATBBN as blocking agent (10 mg/kg body weight). ROIs are shown as mean %ID/g ± SD
Activity accumulation in the tumor and major organs in the microPET scans were quantified by measuring the ROIs that encompassed the entire organ on the coronal images. The tumor uptake was determined to be 4.19 ± 0.32, 4.00 ± 0.41, and 2.93 ± 0.35 %ID/g for 68Ga-NOTA-MATBBN and 4.70 ± 0.40, 4.10 ± 0.30, and 3.14 ± 0.30 %ID/g for 68Ga-NOTA-ATBBN at 30, 60, and 120 min, respectively. The liver uptake of 68Ga-NOTA-MATBBN was very low, with the highest being about 1 %ID/g at 30 min after injection. In contrast, the liver uptake of 68Ga-NOTA-ATBBN was 3.23 ± 0.57 %ID/g at the same time point. Prominent uptake of 68Ga-NOTA-MATBBN was also observed in kidneys at early time points, suggesting that this tracer is mainly excreted through the renal–urinary route.
Representative coronal images of PC-3 tumor mice at 1 h after injection of 68Ga-NOTA-MATBBN and 68Ga-NOTA-ATBBN in the presence of MATBBN (10 mg/kg) or ATBBN (10 mg/kg) are also shown in Figs. 4 and 5. The tumor uptakes of 68Ga-NOTA-MATBBN and 68Ga-NOTA-ATBBN were significantly inhibited by MATBBN (from 4.00 ± 0.41 to 0.55 ± 0.08 %ID/g) and by ATBBN (from 4.10 ± 0.30 to 0.78 ± 0.15 %ID/g) at 60 min postinjection, respectively.
Biodistribution studies
The biodistribution studies of 68Ga-labeled BBN analogs were performed in nude mice bearing PC-3 tumors. The results are summarized in Tables 1 and 2. Both tracers showed decreased uptakes from 0.5- to 2-h time point in the PC-3 tumors and all the examined organs. For example, the tumor uptake of 68Ga-NOTA-MATBBN was 4.12 ± 0.28 %ID/g at 0.5 h and 2.85 ± 0.38 %ID/g at 2 h. The kidney uptake decreased from 4.46 ± 0.24 %ID/g at 0.5 h after injection to 2.40 ± 0.32 %ID/g at 2 h after injection. The other normal organs showed less than 1 %ID/g uptake of 68Ga-NOTA-MATBBN at 1 h postinjection. In contrast, 68Ga-NOTA-ATBBN had relatively high uptakes in the liver (6.01 ± 1.03 %ID/g), pancreas (7.84 ± 0.65 %ID/g), and small intestine (1.68 ± 0.66 %ID/g) at 1-h time point.
Due to the rapid clearance, the T/NT ratios increased with time for both tracers. Tumor-to-tissue ratios in PC-3 tumor-bearing mice from 30 to 120 min after injection were markedly higher for 68Ga-NOTA-MATBBN than for 68Ga-NOTA-ATBBN in the same tumor model.
The tumor uptakes of 68Ga-NOTA-MATBBN and 68Ga-NOTA-ATBBN significantly decreased from 3.93 ± 0.25 and 4.24 ± 0.28 to 0.47 ± 0.16 and 0.66 ± 0.10 %ID/g in the presence of only MATBBN or ATBBN at 1 h postinjection, respectively.
Discussion
Molecular imaging probes based on GRPR-targeting peptides, such as BBN derivatives, have attracted intensive research attention in the past decade. A few radiolabeled BBN peptides and its derivatives have proved significant insight into the biological nature of tumor over the last decade (Sancho et al. 2011; Laverman et al. 2012; Ambrosini et al. 2011). Although most of the BBN-peptide-derived molecular probes were developed as receptor agonists due to a higher receptor-mediated cell uptake and internalization in vitro, radiolabeled GRPR antagonists may be superior targeting agents to agonist ligands because of better in vivo behaviors (Yang et al. 2011; Nanda et al. 2012).
Previous study showed that a modified GRPR antagonist tracer, 18F-FP-MATBBN, had better imaging qualities and pharmacokinetics than the corresponding unmodified tracer 18F-FP-ATBBN (Yang et al. 2011). Since the use of radiometal chelator complexes for labeling is easy and convenient, we labeled the NOTA conjugate of MATBBN with 68Ga in this study. The in vitro and in vivo characteristics of 68Ga-NOTA-MATBBN were directly compared with those of the 68Ga-labeled unmodified GRPR antagonist (68Ga-NOTA-ATBBN). 68Ga-labeled BBN analogs conjugates could be easily prepared with high labeling yield through solid-phase extraction. The radiochemical purity and the specific activity were satisfactory. Compared with our previously reported 18F-labeled BBN analogs, the production of 68Ga-NOTA-MATBBN is simpler.
Roivainen et al. (2013) reported that a 68Ga-labeled GRPR antagonist, 68Ga-RM2 (68Ga-DOTA-4-amino-1-carboxymethyl-piperidine-d-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2) is a promising novel PET tracer for the imaging of GRPR. Preclinical studies revealed that the tumor uptake of 68Ga-RM2 in male nude mice bearing PC-3 tumors was 14.11 %ID/g at 1 h postinjection (Mansi et al. 2011). We have already performed the microPET study in the nude mice bearing PC-3 tumors with 68Ga-RM2; results showed that the tumor uptake was nearly 3 %ID/g (data not shown). The difference may originated from the species of animal and measuring device.
In the study, we also observed significant differences in abdomen imaging. Since GRPR expression in the liver is very low, it is likely that diminution of liver accumulation of 68Ga-NOTA-ATBBN may reflect saturation of the hepatobiliary elimination pathway. In contrast, 68Ga-NOTA-MATBBN is mainly excreted through the renal–urinary route and resulting in higher tumor to background contrast due to high hydrophilicity of the linker.
The results of the biodistribution study further validated the PET quantification. The tumor uptakes of both tracers were similar. Due to good pharmacokinetics, 68Ga-NOTA-MATBBN displayed higher tumor-to-tissue ratios compared with those of 68Ga-NOTA-ATBBN at the same time point, respectively.
Furthermore, the intestine uptakes of 68Ga-NOTA-MATBBN were below 1 %ID/g and significantly lower than the values of 68Ga-RM2 (0.18 ± 0.03 vs. 2.67 ± 1.14 %ID/g) in PC-3 tumor-bearing mice at 60 min postinjection, respectively (Roivainen 2013).
Mansi et al. (2013) demonstrated the values in excess of 30 % ID/g for pancreas uptake at 60 min postinjection for 68Ga-RM2. However, the pancreas uptake of 68Ga-NOTA-MATBBN was below 1 %ID/g in this study. It may be a result of rapid clearance of the tracer and detailed mechanisms required further investigation. The above results indicated that 68Ga-NOTA-MATBBN might be more suitable for imaging of abdominal cancer with high tumor uptake, low nontarget uptake, and predominantly renal excretion.
Conclusion
The BBN antagonist tracer modified with a peptide linker was successfully labeled with 68Ga via conjugated with NOTA. 68Ga-NOTA-MATBBN showed promising in vitro and in vivo pharmacokinetic performances that may improve diagnostic imaging of cancers overexpressing GRPR.
References
Ambrosini V, Fani M, Fanti S, Forrer F, Maecke HR (2011a) Radiopeptide imaging and therapy in Europe. J Nucl Med 52(Suppl 2):42S–55S
Ambrosini V, Fani M, Fanti S, Forrer F, Maecke HR (2011b) Radiopeptide imaging and therapy in Europe. J Nucl Med 52:42S–55S
Ananias HJ, Yu Z, Dierckx RA, van der Wiele C, Helfrich W, Wang F, Yan Y, Chen X, de Jong IJ, Elsinga PH (2011) (99 m)technetium-HYNIC(tricine/TPPTS)-Aca-bombesin(7-14) as a targeted imaging agent with microSPECT in a PC-3 prostate cancer xenograft model. Mol Pharm 8(4):1165–1173. doi:10.1021/mp200014h
Breeman WA, de Blois E, Sze Chan H, Konijnenberg M, Kwekkeboom DJ et al (2011) (68)Ga-labeled DOTA-peptides and (68)Ga-labeled radiopharmaceuticals for positron emission tomography: current status of research, clinical applications, and future perspectives. Semin Nucl Med 41:314–321
Carroll RE, Ostrovskiy D, Lee S, Danilkovich A, Benya RV (2000) Characterization of gastrin-releasing peptide and its receptor aberrantly expressed by human colon cancer cell lines. Mol Pharmacol 58:601–607
Chen X, Park R, Hou Y, Tohme M, Shahinian AH et al (2004) MicroPET and autoradiographic imaging of GRP receptor expression with 64Cu-DOTA-[Lys3]bombesin in human prostate adenocarcinoma xenografts. J Nucl Med 45:1390–1397
de Sá A, Matias AA, Prata MI, Geraldes CF, Ferreira PM et al (2010) Gallium labeled NOTA-based conjugates for peptide receptor-mediated medical imaging. Bioorgan Med Chem Lett 20:7345–7348
Dijkgraaf I, Franssen GM, McBride WJ, D’Souza CA, Laverman P et al (2012) PET of tumors expressing gastrin-releasing peptide receptor with an 18F-labeled bombesin analog. J Nucl Med 53:947–952
Dimitrakopoulou-Strauss A, Hohenberger P, Haberkorn U, Mäcke HR, Eisenhut M, Strauss LG (2007) 68Ga-labeled bombesin studies in patients with gastrointestinal stromal tumors: comparison with 18F-FDG. J Nucl Med 48(8):1245–1250
Dumont RA, Tamma M, Braun F, Borkowski S, Reubi JC, Maecke H, Weber WA, Mansi R (2013) Targeted radiotherapy of prostate cancer with a gastrin-releasing peptide receptor antagonist is effective as monotherapy and in combination with rapamycin. J Nucl Med 54:762–769
Fani M, Andre JP, Maecke HR (2008) 68 Ga-PET: a powerful generator based alternative to cyclotron-based PET radiopharmaceuticals. Contrast Media Mol Imaging 3:67–77
Fleischmann A, Waser B, Gebbers JO, Reubi JC (2005) Gastrin-releasing peptide receptors in normal and neoplastic human uterus: involvement of multiple tissue compartments. J Clin Endocrinol Metab 90:4722–4729
Graham MM, Menda Y (2011) Radiopeptide imaging and therapy in the United States. J Nucl Med 52(Suppl 2):56S–63S
Hoffman TJ, Gali H, Smith CJ, Sieckman GL, Hayes DL, Owen NK et al (2003) Novel series of 111In-labeled bombesin analogs as potential radiopharmaceuticals for specific targeting of gastrin-releasing peptide receptors expressed on human prostate cancer cells. J Nucl Med 44:823–831
Lang L, Li W, Guo N, Ma Y, Zhu L et al (2011) Comparison study of [18F]FAl-NOTA-PRGD2, [18F]FPPRGD2, and [68Ga]Ga-NOTA-PRGD2 for PET imaging of U87MG tumors in mice. Bioconjug Chem 22(12):2415–2422
Laverman P, Sosabowski JK, Boerman OC, Oyen WJ (2012) Radiolabelled peptides for oncological diagnosis. Eur J Nucl Med Mol Imaging 39:S78–S92
Lears KA, Ferdani R, Liang K, Zheleznyak A, Andrews R et al (2011) In vitro and in vivo evaluation of 64Cu-labeled SarAr-bombesin analogs in gastrin-releasing peptide receptor-expressing prostate cancer. J Nucl Med 52:470–477
Liu Z, Yan Y, Chin FT, Wang F, Chen X (2012) Dual integrin and gastrin-releasing peptide receptor targeted tumor imaging using 18F-labeled PEGylated RGD-bombesin heterodimer 18F-FB-PEG3-Glu-RGD-BBN. J Med Chem 18:11079–11087
Mansi R, Wang X, Forrer F, Waser B, Cescato R, Graham K, Borkowski S, Reubi JC, Maecke HR (2011) Development of a potent DOTA-conjugated bombesin antagonist for targeting GRPr-positive tumours. Eur J Nucl Med Mol Imaging 38(1):97–107
Nanda PK, Pandey U, Bottenus BN, Rold TL, Sieckman GL, Szczodroski AF, Hoffman TJ, Smith CJ (2012) Bombesin analogues for gastrin-releasing peptide receptor imaging. Nucl Med Biol 39(4):461–471
Reubi JC, Korner M, Waser B, Mazzucchelli L, Guillou L (2004) High expression of peptide receptors as a novel target in gastrointestinal stromal tumours. Eur J Nucl Med Mol Imaging 31:803–810
Reubi JC, Macke HR, Krenning EP (2005) Candidates for peptide receptor radiotherapy today and in the future. J Nucl Med 46:67S–75S
Roivainen A, Kähkönen E, Luoto P, Borkowski S, Hofmann B, Jambor I, Lehtiö K, Rantala T, Rottmann A, Sipilä H, Sparks R, Suilamo S, Tolvanen T, Valencia R, Minn H (2013) Plasma pharmacokinetics, whole-body distribution, metabolism, and radiation dosimetry of 68 Ga bombesin antagonist BAY86-7548 in healthy men. J Nucl Med 54:867–872
Sancho V, Di Florio A, Moody TW, Jensen RT (2011) Bombesin receptor-mediated imaging and cytotoxicity: review and current status. Curr Drug Deliv 8:79–134
Schuhmacher J, Zhang H, Doll J, Mäcke HR, Matys R et al (2005) GRP receptor-targeted PET of a rat pancreas carcinoma xenograft in nude mice with a 68 Ga-labeled bombesin (6–14) analog. J Nucl Med 46:691–699
Yang M, Gao HK, Zhou YR, Ma Y, Quan QM et al (2011) 18F-labeled GRPR agonists and antagonists: a comparative study in prostate cancer imaging. Theranostics 1:220–229
Zhang X, Cai W, Cao F, Schreibmann E, Wu Y, Wu JC et al (2006) 18F-labeled bombesin analogs for targeting GRP receptor-expressing prostate cancer. J Nucl Med 47:492–501
Acknowledgments
This work was partially supported by National Natural Science Foundation (81171399 and 81101077), CSC Foundation (2011832173), National Significant New Drugs Creation Program (2012ZX09505-001-001), Jiangsu Province Science and Technology Foundation (BE2012622, BK2011166 and BL2012031), Health Ministry of Jiangsu Province Fund (RC2011095 and H201028), Public Service Platform for Science and Technology Infrastructure Construction Project of Jiangsu Province (BM2012066), and University of Wisconsin-Madison Department of Medical Physics and Department of Radiology (Radiology R&D Award 1105-002).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Pan, D., Xu, Y.P., Yang, R.H. et al. A new 68Ga-labeled BBN peptide with a hydrophilic linker for GRPR-targeted tumor imaging. Amino Acids 46, 1481–1489 (2014). https://doi.org/10.1007/s00726-014-1718-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-014-1718-y