
Abstract
The lysine catabolism pathway differs in adult mammalian brain from that in extracerebral tissues. The saccharopine pathway is the predominant lysine degradative pathway in extracerebral tissues, whereas the pipecolate pathway predominates in adult brain. The two pathways converge at the level of ∆1-piperideine-6-carboxylate (P6C), which is in equilibrium with its open-chain aldehyde form, namely, α-aminoadipate δ-semialdehyde (AAS). A unique feature of the pipecolate pathway is the formation of the cyclic ketimine intermediate ∆1-piperideine-2-carboxylate (P2C) and its reduced metabolite l-pipecolate. A cerebral ketimine reductase (KR) has recently been identified that catalyzes the reduction of P2C to l-pipecolate. The discovery that this KR, which is capable of reducing not only P2C but also other cyclic imines, is identical to a previously well-described thyroid hormone-binding protein [μ-crystallin (CRYM)], may hold the key to understanding the biological relevance of the pipecolate pathway and its importance in the brain. The finding that the KR activity of CRYM is strongly inhibited by the thyroid hormone 3,5,3′-triiodothyronine (T3) has far-reaching biomedical and clinical implications. The inter-relationship between tryptophan and lysine catabolic pathways is discussed in the context of shared degradative enzymes and also potential regulation by thyroid hormones. This review traces the discoveries of enzymes involved in lysine metabolism in mammalian brain. However, there still remain unanswered questions as regards the importance of the pipecolate pathway in normal or diseased brain, including the nature of the first step in the pathway and the relationship of the pipecolate pathway to the tryptophan degradation pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
l-Lysine is an essential amino acid with the added importance that it is a limiting amino acid in the diet of many people around the world who rely on cereals as a staple in their diet (Papes et al. 2001). Our understanding of l-lysine catabolism in human tissues is therefore of paramount importance. Weissman and Schoenheimer (1941) demonstrated that l-lysine labeled with 15N in the α position and deuterium in the side chain is incorporated into proteins in the rat without a detectable change in the ratio of 15N to 2H. This finding was regarded as proof of the prior suggestion that l-lysine is unique among the common amino acids in that it cannot reversibly share its α-amino nitrogen with other common amino acids. Weissman and Schoenheimer (1941) therefore suggested that a pathway unique among the common amino acids exists for lysine catabolism. We now know that lysine catabolism is indeed unusual in that it proceeds via two distinct major pathways, the saccharopine pathway and the pipecolate pathway, both of which later converge into a common degradative pathway. The findings of Weissman and Schoenheimer (1941) may be explained by the fact that conversion of the α-amino group of lysine to an α-keto function (pipecolate pathway) or conversion of the ε-amino group of lysine to an aldehyde (saccharopine pathway) results in products that rapidly cyclize, essentially rendering unfavorable the formation of lysine by a transamination reaction. In the adult brain the pipecolate pathway predominates, whereas in extracerebral tissues the pipecolate pathway is a minor pathway for lysine degradation (Chang 1976, 1978). However, in the developing fetal brain the saccharopine pathway is highly active and predominates. During development, the capacity of the pipecolate pathway increases, becoming the major catabolic pathway for lysine degradation in adult brain (Rao et al. 1992). This relationship suggests a specific neuronal developmental role for the pipecolate pathway and its intermediate metabolites.
The two lysine catabolic pathways differ in that the saccharopine pathway is predominantly mitochondrial, whereas the pipecolate pathway is predominantly peroxisomal and cytosolic, as discussed in Sects. 3–7. Recently some of the key enzymes of the pipecolate pathway have been identified, yet questions still remain as to the relevance and importance of this pathway to the mammalian brain. A unique cyclic ketimine, which is not produced in the saccharopine pathway, is generated as an intermediate in the pipecolate pathway, namely, ∆1-piperideine-2-carboxylate (P2C). Perhaps P2C holds the key to elucidate the biological importance of the pipecolate pathway. This review traces the discoveries that highlight the importance of the pipecolate pathway in the brain and particularly the role of P2C in this pathway. The review also raises many unanswered questions that should provide the basis for future research, particularly in the areas designed to elucidate the neurochemical importance of lysine (and tryptophan) metabolism in normal and pathological states.
The saccharopine pathway: a major degradative pathway for lysine in extracerebral tissues and fetal brain but a minor pathway in adult mammalian brain
Higashino et al. (1965, 1967) were the first to identify saccharopine as a key intermediate in l-lysine degradation in mammalian tissues. These authors demonstrated that rat liver mitochondria in vitro convert l-lysine to saccharopine in the presence of α-ketoglutarate (α-KG), thus establishing the saccharopine pathway as a mitochondrial pathway (Fig. 1). The human enzyme that converts l-lysine to saccharopine in the presence of α-KG was investigated by Hutzler and Dancis (1968) and identified as an NADPH-dependent lysine-α-KG reductase (LKR). Studies by Dancis et al. (1969) on patients presenting with hyperlysinemia demonstrated that the accumulation of l-lysine is due to a deficiency of LKR. Although Higashino et al. (1971) found small amounts of free saccharopine in mouse liver, no detectable saccharopine was found in body fluids obtained from normal human volunteers (Carson et al. 1968) or in body fluids of rats that had been injected with 14C-labeled l-lysine. Thus, a rapid in vivo turnover of saccharopine was hypothesized by Carson et al. (1968). Evidence in support of this hypothesis was obtained by Fellows and Lewis (1973) who demonstrated much higher specific activity of saccharopine dehydrogenase (SDH) than of LKR in liver from various mammalian species. However, saccharopine can accumulate under certain pathological conditions. Thus, Carson et al. (1968) described the presence of saccharopine in physiological fluids of patients presenting with saccharopinuria. Fellows and Carson (1974) showed that saccharopinuria is caused by an absence of SDH, which catalyzes the NAD+-dependent oxidation of saccharopine to AAS and glutamate. Fellows and Lewis (1973) also demonstrated that a hepatic mammalian saccharopine oxidoreductase catalyzes the formation of l-lysine and α-KG from saccharopine in vitro, with highest specific activities in the livers of herbivores, and lowest levels in the livers of carnivores/omnivores. A cDNA clone that encodes a bifunctional mitochondrial LKR/SDH as well as a monofunctional SDH in mouse liver and kidney were isolated and characterized (Papes et al. 1999). Northern blot analyses indicated the existence of two mRNA species in mouse liver and kidney. The longer transcript, 3.4 kb in size, corresponds to the isolated cDNA and encodes the bifunctional mitochondrial enzyme. The shorter transcript, 2.4 kb in size, probably encodes a monofunctional dehydrogenase (Papes et al. 1999). A single gene encodes a bifunctional mitochondrial LKR/SDH protein in humans (Sacksteder et al. 2000).

The saccharopine pathway for the metabolism of l-lysine in mammals. In contrast to the pipecolate pathway (Fig. 2), which is predominantly cytosolic and peroxisomal, the saccharopine pathway occurs only in the mitochondria. The saccharopine pathway is the predominant pathway for l-lysine degradation in extracerebral tissues and developing brain. However, in the adult brain, the pipecolate pathway predominates. The two pathways converge at ∆1-piperideine-6-carboxylate/α-aminoadipate δ-semialdehyde (P6C/AAS). Enzymes: 1 the bifunctional enzyme l-lysine-α-ketoglutarate reductase/saccharopine dehydrogenase (LKR/SDH); 2 α-aminoadipate semialdehyde dehydrogenase (AASDH); 3 α-aminoadipate aminotransferase/kynurenine aminotransferase II (AADT/KAT II); 4 a series of mitochondrial enzymes common to l-lysine as well as tryptophan degradation (Fig. 4). Interconversion between AAS and P6C occurs spontaneously (non-enzymatically). Note that some of the enzyme steps shown here occur in other figures. The numbering of each enzyme-catalyzed step is maintained in the subsequent figures. Note also that some of the coenzymes and reactants/products in this pathway and other pathways (Figs. 2, 4) are omitted for simplicity
Rao et al. (1992) measured LKR/SDH activity in developing rat liver and brain. The authors found that enzyme activity is highly dependent on the stage of development in the rat brain, with high levels of activity in developing brain and low activity in adult brain (Rao et al. 1992). The decrease in activity of LKR/SDH in the brain contrasts with the increase in activity of this enzyme in the liver, where it becomes the major contributor to l-lysine catabolism in the liver of adult rats. Rao et al. (1992) also showed that glucagon induces the enzyme in both adult liver and brain. Hutzler and Dancis (1968, 1975) had previously shown that LKR/SDH enzyme activity is low in adult rat brain, and, as mentioned above, Chang (1976, 1978) had shown that the pipecolate pathway is the dominant l-lysine degradative pathway in the adult rat brain. These findings for rat brain have recently been corroborated by Sauer et al. (2011) for adult mouse brain. These authors found no detectable LKR/SDH activity, coupled with low LKR/SDH mRNA expression, in adult mouse brain (Sauer et al. 2011). Thus, the prior contention by Papes et al. (2001) that cortical and cerebellar expression of LKR/SDH mRNA in the mouse are almost as high as that found in the liver requires clarification. Papes et al. (2001) appear to have neglected to take into account the developmental changes in expression as noted by Rao et al. (1992). It appears that Papes et al. (2001) compared embryonic liver LKR/SDH mRNA expression (which is significantly lower than in adult liver) with adult cerebral/cerebellar LKR/SDH mRNA expression. Moreover, the conclusion by Papes et al. (2001) that LKR/SDH mRNA is comparable in rat brain to that in liver appears to contradict their previous findings of low LKR/SDH gene expression in adult mouse brain (Papes et al. 1999) and also the work of Sauer et al. (2011) mentioned above. The authors did, however, show that 15N label in the ε-amino position of l-lysine is transferred to glutamate in mouse cerebellar slices. The authors demonstrated that brain LKR/SDH expression is predominantly neuronal and suggested that the associated enzyme activities may contribute to a de novo neuronal glutamate and GABA biosynthetic route in neurons independent from the astrocytic glutamine-glutamate cycle, with particular importance in fetal brain development. This suggestion is further discussed in Sect. 9 in relation to tryptophan metabolism in the brain. In summary, the literature supports the notion that the saccharopine pathway exists in the adult rodent brain, but that it is of secondary importance to the pipecolate pathway for the metabolism of l-lysine.
The net reaction of LKR/SDH enzyme activity is the reductive amination of α-KG to glutamate balanced by the oxidative deamination of the ε-amino group of l-lysine (Fig. 1). The product of the oxidative deamination is AAS, which is in equilibrium with its cyclic ketimine form, namely ∆1-piperideine-6-carboxylate (P6C) (Soda et al. 1968). The equilibrium greatly favors P6C. It is at the level of P6C that the saccharopine and pipecolate pathways converge, and thereafter follow the same common degradative pathway. The pathway for catabolism of AAS/P6C is discussed in Sect. 8.
The nature of the first step of the pipecolate pathway in the brain remains unsolved
l-Pipecolate (L-PA, Fig. 2) is a natural metabolite in mammalian tissue including brain. Reported levels for adult mouse brain vary from a low of 3.4 nmol/g (w/w) for cerebrum to a high of 7.2 nmol/g (w/w) (or about 4.3 and 9.0 μM, respectively, assuming brain is 80 % water by weight) for the cerebellum (Kim and Giacobini 1985). Similar levels have been reported for adult rat brain (Nishio and Segawa 1983). Interestingly, l-pipecolate has been found to bind tightly to P2 fraction membranes of mouse brain. This binding is modulated by GABA (Gutierrez and Giacobini 1985). Given the neurochemical importance of l-pipecolate, it is somewhat surprising that its origin in brain is not well understood. One theoretical source of l-pipecolate is via the action of d-amino acid oxidases followed by reduction of the product (a cyclic ketimine).

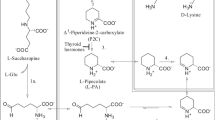
The pipecolate pathway for the metabolism of l-lysine in mammals. This pathway is the main degradative pathway for l-lysine in adult mammalian brain. Enzymes 1 and 2 are cytosolic. The enzymes 2 (AASDH) and 3 (AADT/KAT II) are the same as those in the saccharopine pathway. These enzyme activities are present in both mitochondria and cytosol. Nevertheless, the pathway for the conversion of l-lysine to AKA can be considered predominantly cytosolic with a peroxisomal component (as discussed in the text). Enzymes: 5 an enzyme that catalyzes conversion of the α-amino group of l-lysine to a keto function, possibly kynurenine aminotransferase III/glutamine transaminase L (KAT III/GTL); 6 d-amino acid oxidase (DAAO); 7 CRYM/ketimine reductase (CRYM/KR); 8 l-pipecolate oxidase (POX); 2–4 as per Fig. 1. Conversion between ∆1-piperideine-2-carboxylate (P2C) and α-keto-ε-aminocaproate (KAC) as well as between ∆1-piperideine-6-carboxylate (P6C) and α-aminoadipate δ-semialdehyde (AAS) is spontaneous (non-enzymatic). Thyroid hormones may play a major part in regulating this pathway as CRYM/KR (step 7) has been shown to be strongly regulated by T3, the active form of thyroxine
d-Lysine has been known for over 45 years to be metabolized in rats to l-pipecolate (Grove and Henderson 1968). The peroxisomal enzyme d-amino acid oxidase (DAAO) was suspected of initiating the metabolism of d-lysine. Indeed, Scannone et al. (1964) had previously shown that d-lysine is a substrate of mammalian DAAO. The product of d-lysine oxidative deamination is α-keto-ε-aminocaproate (KAC), which spontaneously cyclizes to the ketimine P2C (Fig. 2). This ketimine is subsequently reduced to l-pipecolate by a cytosolic P2C reductase (Meister 1962). Interestingly, P2C has been shown to form a stable complex with DAAO, and P2C is a potential competitive inhibitor of this enzyme with a very high enzyme affinity (Nishina et al. 1991). In rat brain, the specific activity of DAAO is highest in cerebellum and brainstem, with very low specific activity in the cerebral cortex (Horiike et al. 1994). Chang (1978) studied the metabolism in the rat brain of intraventricular administered 14C labeled l- and d-lysine and demonstrated that the major metabolite formed from both labeled compounds is l-[14C]pipecolate and that the two enantiomers are equally effective precursors of l-[14C]pipecolate. The conversion of d-[14C]lysine to l-[14C]pipecolate was presumably initiated by the action of DAAO, despite the possibility of strong product inhibition, followed by an enantiomeric reduction of labeled P2C. In the same study, Chang (1978) noted that some labeled α-aminoadipate (AAD) was also produced from both l- and d-[14C]lysine in the rat brain. In earlier work, Chang (1976) noted that [14C]pipecolate, but not [14C]AAD, was present in the urine of rats intraperitoneally injected with l-[14C]lysine. It was concluded that l-lysine metabolism in the brain occurs via a pathway involving pipecolate conversion to AAD (Chang 1976, 1978). As discussed above, this pathway (the pipecolate pathway) in the adult rat brain is distinct from the saccharopine pathway for lysine metabolism known to predominate in the liver and other tissues of the rat.
Given that the conversion of d-lysine to P2C in the brain is presumably catalyzed by a DAAO, how is l-lysine converted to P2C in that organ? Historically, Rothstein and Miller (1954) showed, using 15N and 14C as tracers and a “metabolite overloading” procedure, that l-lysine can be converted to l-pipecolate in the rat through loss of the α-amino group. But how is the α-amino group of lysine removed? The simplest explanation is that either an l-amino acid oxidase (LAAO) or an aminotransferase catalyzes the conversion of l-lysine to KAC. Because conversion of lysine to KAC is little understood, we provide here a detailed discussion of both possibilities. In both cases the product is KAC, which, as noted above, cyclizes to the ketimine P2C. Neuberger and Sanger (1943) suggested that acetylation of the ε-amino group may be the first step in the pathway leading to the oxidation of the α-amino group of l-lysine. In this regard, an acetyltransferase has been shown to be present in human tissues that acetylates thialysine (S-2-aminoethyl-l-cysteine) as well as l-lysine (to a lesser extent) at the N ε position (Coleman et al. 2004). However, work by Hernandez and Chang (1980) demonstrated that the l-pipecolate formed in vitro in rat kidney and brain from N ε-acetyl-l-lysine is derived from l-lysine. The study of Hernandez and Chang (1980) thus negates the possibility that N ε-acetyl-l-lysine as an obligatory intermediate in the pipecolate pathway of l-lysine metabolism.
Murthy and Janardanasarma (1999) provided an evidence for the presence of an LAAO/lysine oxidase in mouse brain. The authors, using labeled 14C-l-lysine as substrate, identified both P2C as well as its open-chain form (i.e., KAC) in mouse brain homogenates. However, the brain homogenate exhibited relatively low activity with lysine. Moreover, because a crude homogenate was used, the findings do not prove that an LAAO reaction is the major route for conversion of l-lysine to KAC in brain. Additionally, it is not clear what type of LAAO could catalyze such an oxidase reaction with l-lysine. A well-characterized mammalian (human) LAAO is an l-phenylalanine oxidase [interleukin-4-induced protein 1 (IL4I1)] (Chavan et al. 2002; Boulland et al. 2007). Two isoforms of the IL4I1 protein have been described. Isoform 1 is predominantly expressed in leukocytes, and isoform 2 is predominantly expressed in testis and brain (Wiemann et al. 2005). Isoform 2 may be identical to the enzyme described by Murthy and Janardanasarma. IL4I1 is lysosomal, exhibits an acidic pH optimum, and “prefers” aromatic amino acids such as phenylalanine as substrates. Thus, l-lysine is a relatively poor substrate (Mason et al. 2004). Moreover, IL4I1 is expressed at relatively low levels in the brain (Wiemann et al. 2005). l-Hydroxy acid oxidases are known to catalyze the oxidation of certain l-amino acids to a limited extent. However, l-lysine seems not to be a substrate, and l-hydroxy acid oxidases are not significantly expressed in brain (Miles and Holmes 1975; Duley and Holmes 1976; Urban et al. 1988). In summary, the contribution of an LAAO to the formation of KAC in brain remains uncertain.
Could an aminotransferase be responsible for the conversion of lysine to KAC in the brain? In contrast to most amino acids that are commonly found in proteins, l-lysine has traditionally been regarded as inert to the action of mammalian aminotransferases (Higashino et al. 1967). However, Papes et al. (2001) showed that when mouse brain cortex or cerebellar slices were incubated with l-[15N]glutamate, label could be detected in the α-amino group of l-lysine, but not in the ε-amino group (Papes et al. 2001). The simplest explanation for this finding is that despite the perceived notion that transamination reactions involving lysine are not favorable, the α-amino nitrogen can be transferred from glutamate to KAC in the brain, possibly as a result of the very high levels of glutamate in brain (10–12 mM). This finding suggests that transamination between glutamate and KAC is reversible—i.e., that transamination of l-lysine can occur in mouse brain such that the α-amino group is transferred to α-KG (or another suitable α-keto acid acceptor), thereby generating KAC (the first intermediate of the pipecolate pathway). If this reasoning is correct, what is the identity of this aminotransferase? Alanine:glyoxylate aminotransferase II (AGT II) is an enzyme with a relatively broad substrate specificity that can catalyze in vitro transamination of the α-amino group of l-lysine (Ogawa et al. 1990). However, lysine is a poor substrate compared to l-alanine, and AGT II is unlikely to catalyze this reaction to any great extent in vivo. Moreover, Ogawa et al. (1990) stated that AGT II activity is present in the rat only in liver and kidney homogenates.
Here, we consider the possibility that three other aminotransferases of broad α-keto acid and amino acid specificities may catalyze transamination of lysine at the α position. Cooper and Meister (1972, 1981, 1985) identified two glutamine transaminases in rat tissues, which they named glutamine transaminase K (GTK) and glutamine transaminase L (GTL). In the rat, GTK [synonyms: kynurenine aminotransferase I (KAT I); cysteine conjugate β-lyase isozyme 1 (CCBL 1)] activity is most abundant in kidney, but appreciable activity is also present in liver and other tissue. In the rat, GTL [synonyms: kynurenine aminotransferase III (KAT III); cysteine conjugate β-lyase isozyme 2 (CCBL 2)] activity is most abundant in liver, but activity can be detected in other tissues. Interestingly, GTK occurs in both the cytosolic and mitochondrial compartments of rat tissues (Cooper and Meister 1981). The same gene codes for both forms, but alternative splicing dictates whether or not a 32-amino acid mitochondrial targeting sequence at the N-terminus is present in the expressed protein (Malherbe et al. 1995; Tamburin et al. 1999). Cooper and Meister (1981, 1985) noted that GTL activity also occurs in cytosolic and mitochondrial compartments. It is possible that since the genes encoding KAT I/GTK and KAT III/GTL are highly conserved (including the N-terminus) (Yu et al. 2006), alternative gene splicing at the N-terminus may also dictate the relative distribution of KAT III/GTL between cytosol and mitochondria.
Both GTK and GTL exhibit very broad and somewhat overlapping amino acid and α-keto acid specificities. Thus, it is possible that one or both of these enzymes contribute to the formation of KAC from l-lysine in brain. In this regard, Pensa et al. (1989) purified an aminotransferasse from bovine brain that catalyzes transamination of thialysine at the α position. [The standard assay reaction mixture employed by these authors contained 10 mM l-thialysine and 0.5 mM α-keto-γ-methiolbutyrate in 20 mM sodium pyrophosphate buffer (pH 9.2). Formation of the product (i.e., AECK) derived from cyclization of the α-keto acid product was continuously measured spectrophotometrically.] Curiously, despite the similarity in structure between thialysine and lysine, the authors did not determine whether the enzyme also catalyzes transamination of l-lysine. An enzyme that catalyzes transamination of thialysine was also purified from bovine liver (Costa et al. 1986). The authors suggested that the enzyme responsible for transamination of thialysine is a glutamine transaminase.
Cooper and Meister (1981, 1985) showed that the glutamine analog l-albizziin is a good substrate of rat liver GTL, but a relatively poor substrate of rat kidney GTK. It is interesting to note that thialysine transaminase from bovine liver is also able to use albizziin as a substrate (Costa et al. 1986). Taken together, we considered the possibility that transamination of l-lysine at the α position may be catalyzed by KAT III/GTL. Similar to GTL purified from rat liver (Cooper and Meister 1972), recombinant mouse KAT III/GTL (mKAT III/GTL) has recently been shown to exhibit broad substrate l-amino acid and α-keto acid specificity (Han et al. 2009). l-Lysine was shown to be a substrate of mKAT III/GTL. At a concentration of 10 mM, l-lysine was about 25–40 % as effective as a substrate as the “best” amino acid substrates (conditions: 10 mM l-amino acid substrate; 2 mM glyoxylate as co-substrate; borate buffer pH 9.0, 45 °C) (Han et al. 2009).
We have now tested recombinant human KAT I/GTK, recombinant human KAT II/α-aminoadipate aminotransferase (AADT), another aminotransferase of broad substrate specificity, and recombinant mKAT III/GTL for their ability to catalyze transamination between 1 mM l-lysine and 1 mM α-keto-γ-methiolbutyrate (a good α-keto acid substrate of all three enzymes) in 100 mM potassium phosphate buffer (pH 7.4) at 37 °C (unpublished observations). These assay conditions are more physiological than those employed by Costa et al. (1986) and Pensa et al. (1989) for the measurement of thialysine transamination (see the previous paragraph). Both the disappearance of α-keto-γ-methiolbutyrate and appearance of methionine were followed by HPLC with CoulArray detection [e.g., (Lee et al. 2009)]. Under these conditions, the ability of KAT I/GTK and KAT III/GTL to catalyze transamination of l-lysine was <1 % that exhibited with glutamine. The ability of KAT II/α-aminoadipate aminotransferase to catalyze transamination between l-lysine and α-keto-γ-methiolbutyrate could not be detected. In a separate experiment (2-h endpoint determination; 37 °C), we investigated transamination between 10 mM l-lysine (or 10 mM l-glutamine) and 5 mM α-keto-γ-methiolbutyrate in 50 mM sodium pyrophosphate buffer (pH 9.2). As noted for the experiment carried out at pH 7.4, lysine was found to be an extremely poor substrate of KAT I/GTK at pH 9.2 (<0.1 % relative to that exhibited with glutamine) and activity with KAT II/á-aminoadipate aminotransferase could not be detected. However, although lysine is a less effective substrate than is glutamine of KAT III/GTL, the amount of lysine transaminated at pH 9.2 is substantial (about 5 % relative to that exhibited with glutamine). The data are thus in accord with the findings of Han et al. (2009) that KAT III can catalyze transamination of lysine at high pH values (≥9.0) at a moderate rate.
A consideration of the chemical properties of P6C (a double bond positional isomer of P2C) suggests a possible explanation for the apparent low level of activity of KAT III/GTL toward lysine at pH 7.4, but relatively higher activity at pH 9.2. Theoretically, P6C could arise through a one-step transamination of the ε-amino group with a suitable α-keto acid acceptor in an analogous fashion to that of the well-characterized transamination of the δ-amino group of ornithine. Instead, nature has devised a two-step process to convert lysine to AAS/P6C. Why? In Sect. 8 we discuss how P6C can react with PLP to form a Knoevenagel adduct. Perhaps the two-step conversion of l-lysine to AAS/P6C via saccharopine is designed by nature to avoid the problem of syncatalytic inactivation of a PLP-dependent aminotransferase by a Knoevenagel condensation of PLP cofactor with product P6C. We suggest that P2C may also inhibit an enzyme that catalyzes transamination of lysine at neutral pH due to a condensation with PLP cofactor. This inhibition is expected to be less of a problem at pH 9.2, because a greater proportion of P2C will be in the enamine form (Lu and Lewin 1998). It is possible that the imine reductases have evolved in part to rapidly reduce the double bond of P2C, preventing product inhibition of the parent aminotransferase.
In conclusion, of several enzymes considered as a possible source of KAC in the brain, KAT III/GTL appears to be the most likely candidate. KAT III/GTL is present in mammalian brain (Han et al. 2009 and references cited therein), but it is not clear how effective this enzyme is in transaminating lysine at physiological pH values. It is even conceivable that neither an LAAO nor an aminotransferase is mainly responsible for the generation of KAC, but rather a dual enzyme system analogous to the LKR/SDH for the production of P6C from lysine operates to generate P2C from lysine. Evidently, much more work is required to define the mechanism by which l-lysine is converted to KAC/P2C in the brain.
The discovery of specific cytosolic P2C reductases
In studies on ketimine reductase (KR) from bovine cerebellum, Nardini et al. (1988a) determined that, among potential substrates tested, this enzyme exhibits maximal activity with P2C (and NADPH rather than NADH as cofactor) and that it is also a cytosolic enzyme. P2C reductase activity was later found to be present mainly in the cytosolic supernatant fraction of rat cortex (Chang and Charles 1995). As it seems likely that P2C reductase activity occurs mainly in the cytosol, it is possible that the synthesis of KAC from l-lysine also occurs in the cytosol.
∆1-Piperideine-2-carboxylate formed from the spontaneous cyclization of KAC is reduced to l-pipecolate by specific reductases (Meister 1962; Meister and Buckley 1957; Aspen and Meister 1962a, b; Meister et al. 1957). Meister (1962), Meister and Buckley (1957) and Meister et al. (1957) partially purified an imine reductase from rat kidney and were the first to show that mammalian tissues contain reductase(s) that reduce the imine double bond of P2C and also that of its five-membered ring analog [i.e., ∆1-pyrroline-2-carboxylate (Pyr2C)], forming only the l-enantiomers of pipecolate and proline, respectively. The authors concluded that the same enzyme(s) catalyze(s) the reduction of both compounds, but are unable to catalyze the reduction of ∆1-pyrroline-5-carboxylate (Pyr5C) and P6C, which are isomers of P2C and Pyr2C, differing only in the position of the imine double bond. Pyr5C reductases that exist in various isoforms in mitochondria as well as in the cytosol were later characterized and shown to catalyze the reduction of both Pyr5C and P6C (Valle et al. 1973; Yeh et al. 1981). Note that enzymes that reduce Pyr5C/P6C and P2C/Pyr2C may be regarded as aldimine reductases and ketimine reductases (KRs), respectively.
Meister et al. (1957) showed that P2C/Pyr2C reductase is present in rat kidney, brain, cardiac muscle, spleen, and testis. With the exception of kidney, of all the organs investigated, rat brain exhibited the highest specific activity of P2C/Pyr2C reductase (Meister 1962; Meister et al. 1957). In later work, a P2C/Pyr2C reductase was partially purified from porcine kidney by Petrakis and Greenberg (1965) and these authors obtained a higher specific activity than that obtained previously from rat kidney by Meister et al. (1957). It is notable that the enzyme preparations obtained by both groups could use NADH as well as NADPH as cofactor. However, the results obtained by these authors do not preclude the possibility that mammalian tissues contain multiple KRs that differ in their affinities for NADH versus NADPH. In this regard, Meister et al. (1957) showed that NADH and NADPH were equally effective as cofactor for the KR purified from rat kidney, whereas Petrakis and Greenberg (1965) showed that the KR purified from porcine kidney exhibited a marked preference for NADPH over NADH. A cytosolic KR from porcine kidney was partially purified by Nardini et al. (1988b) and unlike the preparation obtained by Petrakis and Greenberg (1965), but similar to the preparation obtained by Meister et al. (1957), the preparation obtained by Nardini et al. (1988b) utilized NADH and NADPH equally well as cofactor when P2C was employed as substrate.
Garweg et al. (1980) demonstrated the presence of P2C/Pyr2C reductase activity and l-pipecolate in several regions of mouse, dog, and monkey brain. Both the level of l-pipecolate and P2C/Pyr2C reductase-specific activity differed considerably among the different brain regions. The authors found highest specific activities in the frontal and temporal cortex, with the striatum having the maximal specific activity. The enzyme in mouse brain exhibited an optimal pH of 5.2 with P2C and 5.4 with Pyr2C as ketimine substrates. These pH optima were similar to those determined by Meister et al. (1957) previously for a partially purified rat kidney KR. Among the various brain regions investigated, l-pipecolate levels were previously reported to be highest in the cerebellum (Kasé et al. 1973; Nishio and Segawa 1983), yet Garweg et al. (1980) found minimal P2C reductase activity in that region. A possible explanation for this apparent discrepancy is that NADH (and not NADPH) was used as a cofactor by Garweg et al. (1980). The cytosolic KR purified from bovine cerebellum by Nardini et al. (1988a) was found to have a marked preference for NADPH over NADH as a cofactor when P2C was used as the ketimine substrate. Nardini et al. also found maximal activity in the cerebellum and cerebral cortex among the brain regions investigated. The bovine cerebellar KR exhibited a similar pH optimum to that previously reported for other P2C reductases of ~5. The cerebellar enzyme was found to be a homodimer (monomer M r ~ 50,000), whereas a KR purified from porcine kidney was found to have a native M r of ~70,000 (Nardini et al. 1988b). However, no subunit composition data were presented for the porcine kidney enzyme. Both KRs purified by Nardini et al. were shown to reduce substrates other than P2C, namely several sulfur-containing cyclic ketimines. Perhaps not surprisingly, the sulfur-containing cyclic ketimine aminoethylcysteine ketimine (AECK), which is a thioether analog of P2C, was found to be a good substrate for both KRs.
In summary, the published findings strongly support the hypothesis that mammalian tissues contain at least two distinct KRs that are capable of reducing P2C to l-pipecolate. Moreover, in the mammalian brain there also appears to be regional differences in the expression of these two enzymes.
μ-Crystallin: the first P2C reductase identified in mammalian genome databases
In 2011, we purified a cytosolic lamb forebrain KR to apparent homogeneity and showed that this enzyme is similar to the previously reported purified porcine kidney KR (Hallen et al. 2011). Notably, the purified enzyme was able to use the same substrates [AECK, cystathionine ketimine (CysK) and P2C], as those previously described by Nardini et al. (1988b) for the porcine kidney enzyme. Moreover, both enzymes were found to use NADH and NADPH equally well as cofactor. Finally, the purified lamb brain KR was shown to be a homodimer (M r monomer, ~36,000). This value is consistent with the M r reported by Nardini et al. (1988b) for the porcine kidney native enzyme (~70,000). However, the size and substrate specificity of the lamb brain KR appear to be notably different from those of a KR previously purified from bovine cerebellum by Nardini et al. (1988a). As noted above, the bovine cerebellar KR was reported to be a homodimer (monomer M r ~ 50,000).
Mass spectrometric proteomic analysis of the purified lamb brain KR showed that the enzyme is an orthologue of a protein annotated in human and other mammalian genome databanks as μ-crystallin (CRYM). This finding suggested that human CRYM has a previously unsuspected biological role as a KR. Direct confirmation that human CRYM is indeed a KR was obtained by showing that highly purified recombinantly expressed human CRYM possesses substantial KR activity (Hallen et al. 2011). As noted for KRs purified from rat liver, porcine kidney and bovine brain, the lamb brain KR exhibits a pH optimum of ~5 with P2C as substrate (Hallen et al. 2011).
It is interesting to note that CRYM is now known to be a multifunctional protein with at least three disparate properties. In addition to our findings that CRYM is a KR (Hallen et al. 2011), the protein was previously well characterized as a structural protein of diurnal marsupial lens (Segovia et al. 1997). [For a recent review and characterization of crystallins (including μ-crystallin) in kangaroo lens see Augusteyn (2011).] Moreover, CRYM also strongly binds the active form of thyroxine, namely 3,5,3′-triiodothyronine (T3) in a redox-sensitive manner (Mori et al. 2002). The characterization of CRYM as a cytosolic thyroid hormone-binding protein (CTBP) is intriguing. The K d for binding of T3 in the presence of NADPH was reported to be 0.3 nM, which implies very strong binding of T3 to CRYM (Beslin et al. 1995). CTBP is considered the main cytosolic thyroid hormone-binding protein and it was originally purified and identified as CRYM on the basis of its extremely strong affinity for T3 in the presence of NADPH (Vie et al. 1997). Thyroid hormones have long been known to exert a major influence on brain development, and neonatal hypothyroidism in humans is known to cause mental retardation and neurological damage (Bernal 1999; Delange 2000). What role CRYM/KR has in brain development remains an unanswered question.
Lennon et al. (1980) demonstrated, from data obtained by Scatchard analysis of thyroid hormones binding to charcoal-extracted cytosol in the presence of 50 μM NADPH, that thyroid hormone binding is significantly lower in cerebellum compared to cerebrum at every stage of development. Suzuki et al. (1991) calculated the maximal binding capacity for cytosolic NADPH-dependent T3 binding in various rat tissues and also showed that levels of thyroid hormone binding are noticeably lower in the cerebellum than in the cerebrum of adult rat brain at all stages of development. The differences noted in NADPH-dependent T3 binding between cerebrum and cerebellum are reflected in the differences noted for mRNA CRYM/KR gene expression in adult mouse brain for the same regions, with very low expression in cerebellum compared to cerebrum (Tebbenkamp and Borchelt 2010). Among several tissues investigated in rats at various developmental stages, NADPH-dependent T3 binding was only detected in kidney, liver, heart and spleen after birth, increasing over the next 6 weeks (Suzuki et al. 1991). In contrast, NADPH-dependent T3 binding was detected in cerebrum and cerebellum 5 days before birth, increasing with a sharp transient spike at the time of birth, particularly in cerebrum. The NADPH-dependent T3 binding in cerebrum decreased after birth, but began to increase again 2 weeks after birth. The level in cerebellum did not show this increase. This spike in NADPH-dependent T3 binding at birth was not observed in other organs. In fetal rats, production of thyroid hormones is known to only occur 5–6 days before birth (Strait et al. 1990) with a surge at and after birth, reaching maximal levels 10 days after birth (Dubois and Dussault 1977). Prior to fetal production of thyroid hormones, the fetus is reliant on maternal thyroid hormones. The placenta, however, maintains a barrier to their supply and fetal thyroid hormone levels are known to be much lower than circulating maternal thyroid hormones prior to initiation of fetal thyroid hormone production (Calvo et al. 1992).
Studies on l-pipecolate levels in developing brain show a remarkable similarity to the observed changes in the concentrations of NADPH-dependent T3-binding protein. The highest levels were observed in mouse and rat brain during the perinatal period with levels 6–10 times higher than those in adult brain, and a subsequent rapid decrease after birth (Nishio and Segawa 1983; Kim and Giacobini 1985). This spike in l-pipecolate levels almost exactly mirrors the change in NADPH-dependent T3-binding protein concentrations noted at birth. Synthesis of l-pipecolate from l-lysine has been shown to be accelerated in the perinatal period in chick and mouse brain homogenates and this may also correlate with the observed changes in l-pipecolate levels (Nomura et al. 1978). It is interesting to note that by day 10 in rats, brain l-pipecolate is markedly decreased from its maximum just after birth (Nishio and Segawa 1983; Kim and Giacobini 1985). As noted previously, apart from the unexplained transient spike in NADPH-dependent T3-binding protein at birth, there is a gradual increase of NADPH-dependent T3 binding until adulthood, which also corresponds to the increases in thyroid hormone levels. It should be noted that the developmentally related expression of NADPH-dependent T3 binding (and by inference CRYM/KR expression) in the rat brain roughly correlates with the development of the pipecolate pathway. These results suggest that CRYM/KR and thyroid hormone levels may be major determining factors in the development of the pipecolate pathway in the brain. The relationships of thyroid hormones and the pipecolate pathway are further discussed in Sect. 9 in relation to tryptophan metabolism.
A complicating issue as regards T3 binding to its cognate receptor in the brain is the finding that there are at least two different CTBPs in cerebral tissues. As described above, CRYM/ketimine reductase (CRYM/KR) is a well-characterized CTBP. A distinguishing feature of human CRYM/KR is that it binds T3 in the presence of NADPH, but not in the presence of NADP+ (Vie et al. 1997). A 38-kDa protein (approximately the monomeric size of CRYM/KR) is present in rat liver cytosol and was found to bind T3 far better than do other cytosolic proteins (Moreno et al. 2003). As with the human CTBP (CRYM/KR), the rat liver 38-kDa protein strongly binds T3 in an NADPH-dependent fashion and may thus be identical to CRYM/KR. Interestingly, however, the protein also strongly binds 3,5-diiodothyronine (3,5-T2), but in an NADPH-independent fashion. 3,5-T2 is suspected of playing a non-genomic role in energy metabolism at the mitochondrial level independent of any protein synthesis (Goglia et al. 1994). Moreno et al. (2003) suggest that the NADPH/NADP+ ratio may regulate T3/3,5-T2 intracellular translocation. The effects on CRYM/KR enzyme activity by 3,5-T2 have yet to be evaluated.
Another CTBP has been isolated from rat brain that binds T3 tightly (K a 1.56 nM) (Lennon 1992). SDS-PAGE analysis of the purified protein suggested a monomeric M r of ~58,000. The binding capacity of this CTBP toward T3 increased two- to fourfold in the presence of NADPH with only a slight change in K a to 2.6 nM (Lennon 1992). NADP+ was found to bind to this protein, but only in the presence of the strong thiol reductant dithiothreitol. The protein purified by Lennon may be similar to the 58-kDa CTBP characterized and identified in Xenopus laevis liver as an aldehyde dehydrogenase (ALDH), which preferentially uses retinal as a substrate (Yamauchi and Tata 1994). However, T3 was shown to only weakly inhibit (at micromolar concentrations) the mitochondrial and cytosolic forms of human liver ALDH [competitively with respect to aldehyde substrate (propional) and uncompetitively with respect to NAD+] (Zhou and Weiner 1997). T3 was also shown to competitively inhibit the oxidation of octanal by recombinant mouse retinaldehyde dehydrogenase-3 at micromolar concentrations (Graham et al. 2006).
Motivated by the prior finding that T3 binds very strongly to CRYM in the presence of NADPH, we evaluated T3 as a potential inhibitor of KR and found that it is a strong reversible inhibitor when AECK (a sulfur-containing analog of P2C) is used as substrate at pH 5 (K i, 278 nM) (Hallen et al. 2011). A much stronger inhibition using low nM concentrations was found at neutral pH. Owing to technical difficulties in using AECK as a substrate at neutral pH a true K i could not be measured for T3. At neutral pH AECK exists predominantly in a tautomeric form (enamine) that is not a substrate of CRYM/KR (Nardini et al. 1988b). Thus, it was necessary to use an enzyme concentration greater than inhibitor concentration, resulting in difficulties in calculating an exact K i at neutral pH. Nevertheless, our results together with the low K d value determined by Beslin et al. (1995) at neutral pH, imply that enzyme activity would be tightly regulated in vivo by nM or even sub-nM concentrations of T3. Our kinetic data indicate that cyclic ketimines and T3 compete with each other in the active site of CRYM (Hallen et al. 2011). Therefore, we hypothesize that there will be a strong reciprocal relationship between the in vivo concentration of cyclic ketimines (and NADPH) and the concentration of free (i.e., unbound) T3. In this regard, it is interesting to note that CRYM had previously been implicated in regulating the bioavailability of T3 by a mechanism that involves redox-dependent translocation of T3 to the nucleus (Mori et al. 2002). Prior to our discovery that CRYM is a KR, this relationship was suggested to possibly be due to changes in the overall intracellular NADPH/NADP+ ratio (Beslin et al. 1995). However, while our findings are certainly consistent with this notion, they add an additional layer of complexity. Thus, the availability of KR substrates of CRYM may well be an even more important factor than the NADPH/NADP+ ratio in the regulation of T3 translocation to the nucleus. Inasmuch as CRYM effectively catalyzes the NADPH-dependent reduction of P2C to pipecolate (Hallen et al. 2011), our findings emphasize the important concept that T3 bioavailability is likely strongly dependent on the pipecolate pathway activity. Thus, any biological process that results in increased availability of the cyclic ketimine substrate P2C also has the potential to decrease the binding of T3 to CRYM and consequently to allow the translocation of free T3 to the nucleus. Conversely, any biological process that results in decreased concentration of P2C will promote increased tight binding of T3 to CRYM, reducing the translocation of free T3 to the nucleus. This is a novel concept in which the T3 bioavailability and its role as a transcription factor are regulated by a component of the pipecolate pathway (i.e., P2C reductase activity).
As the above discussion attests, the relationship between l-pipecolate levels and KR at the whole brain level is now well established. However, there are still some apparent inconsistencies that require an explanation. These include the observed high level of l-pipecolate in cerebellum yet low expression of CRYM/KR in that region, and the discovery by Nardini et al. (1988a) of a cerebellar KR with kinetic properties (and size) different from those reported by us for CRYM/KR. As we mentioned above, the findings suggest that the brain may contain at least two distinct P2C reductases/KRs, one of which is predominant in the forebrain (Hallen et al. 2011) and another that is prominent in the cerebellum (Nardini et al. 1988a). The forebrain enzyme is CRYM/KR that has been characterized in our laboratory. Although Nardini et al. (1988a) have partially characterized the cerebellar enzyme, its identity in mammalian genome data banks is not yet established. In future work, it will be important to determine not only the gene that codes for the cerebellar enzyme, but also to more fully characterize its kinetic and regulatory properties and to ascertain the relative importance of the two enzymes in the brain pipecolate pathway. It seems unlikely that the cerebellar enzyme will also be a thyroid hormone-binding protein. Thus, in future work it will be important to determine the mechanism involved in regulation of the cerebellar enzyme and why the brain requires two separate KRs.
At this point it is important to note that although our discussion has dealt mainly with the reduction of P2C, the KRs may also have additional important metabolic functions, particularly in relation to the sulfur-containing cyclic ketimines. The sulfur-containing ketimine substrates for CRYM/KR are thought to have neurotransmitter/neuromodulator roles. For example, Duprè et al. (1993) showed that [35S]lanthionine ketimine (LK) strongly binds to brain membranes with a resulting elevation of basal cAMP levels. [The structure of LK and some other sulfur-containing ketimines is shown in Fig. 3.] LK (and its ethyl ester derivative) has been shown by Hensley et al. (2010, 2011) to have neurotrophic properties, such as promoting neurite outgrowth in duck dorsal root ganglia as well as in motor neuron-like cells. As yet, no such relationships have been studied or noted for P2C or Pyr2C. The discovery that CRYM is a P2C reductase, and its enzyme activity regulated by T3 is consistent with the possibility that P2C and Pyr2C may also have neurotransmitter/neuromodulator roles. Thyroid hormones are known to affect neuronal differentiation and also to cause neurite outgrowth (Walter 1996). Further research is necessary to determine to what extent the inter-relationship between CRYM/KR, its cyclic ketimine substrates, as well as thyroid hormones, play in neuronal differentiation.

Structures of several known CRYM/KR substrates and one putative substrate [Pyr2C]. The known substrates include P2C and the sulfur-containing ketimines, aminoethylcysteine ketimine (AECK), lanthionine ketimine (LK), and cystathionine ketimine (CysK). LK in particular has been shown to have neurotrophic properties (see the text). The five-membered ring analog of P2C, namely, ∆1-pyrroline-2-carboxylate (Pyr2C), has been reported to be a substrate for semi-purified P2C/Pyr2C reductases (Meister et al. 1957). However, although it seems likely to be a substrate, Pyr2C has not yet been tested as a substrate of highly purified CRYM/KR
Northern blot analysis revealed that CRYM/KR mRNA in the mouse is most highly expressed in skin among the tissues evaluated, followed by brain and heart (Aoki et al. 2000). The expression of the CRYM transcript was found to follow the hair growth cycle with a significant increase in expression during mid- and late anagen growth phases. The exact function and importance of CRYM as regards hair growth is unknown. Aoki et al. (2000) suggested the possible involvement of CRYM in the development of mouse hair follicles during the anagen phase. The discovery of an enzyme function for CRYM as a KR (Hallen et al. 2011) places the suggestion of Aoki et al. in a new light. We suggest that enzyme substrates (e.g., sulfur-containing cyclic ketimines such as AECK and LK, Fig. 3) may play a role in regulating cell growth and/or cell differentiation.
Steroid hormones have a marked effect on the expression of CRYM/KR. For example, androgens have been shown to induce CRYM expression in human prostate cancer (Malinowska et al. 2009). Estrogen has also been shown to increase CRYM expression in bovine mammary glands (Li et al. 2006). There are marked seasonal differences in the expression levels of CRYM/KR in the hypothalamus of the song sparrow (Melospiza melodia) with highest expression in spring and lowest in autumn (Mukai et al. 2009). A similar seasonal variation of CRYM/KR gene expression also has been noted in the ground squirrel Ictidomys tridecemlineatus (Schwartz et al. 2013).
It has long been thought that owing to the predominance of the pipecolate pathway in the brain, the pathway must play a specific role in neuronal development (Chang 1976, 1978). That role is still poorly understood. However, the findings that (1) CRYM/KR is a crucial enzyme in the pipecolate pathway, (2) the CRYM/KR activity is potently regulated by T3, and (3) CRYM/KR substrates are potentially neuroactive and are highly informative. These findings are beginning to not only help in the elucidation of the biological role of the pipecolate pathway (and its associated enzyme activities), but also to emphasize the previously overlooked role of the pipecolate pathway in brain physiology and function.
Our discovery that T3 regulates CRYM/KR activity (and enzyme substrate levels) raises the question as to whether at least some of the biological actions of T3 are due to the interaction of T3 with CRYM/KR. In our opinion, KRs and their cyclic ketimine substrates (including sulfur-containing cyclic ketimines) are important topics for future neurochemical studies.
The significance of CRYM/KR in psychiatric and neurological disease
There is typically a delay of a few weeks before tricyclic antidepressants elicit a therapeutic response. This delay suggests that long-term neuroadaptive changes underlie the antidepressant actions rather than inhibition of neurotransmitter reuptake and signaling. This suggestion was the motivation for research performed on mice that were treated chronically for 28 days with the tricyclic antidepressant amitriptyline at dosages, corrected for body weight, known to have a therapeutic effect in humans (Böhm et al. 2006). The nucleus accumbens is an area of the forebrain that has been suggested to be affected in schizophrenia (Chronister and DeFrance 1982; Gray et al. 1995) as well as in depression (Shirayama and Chaki 2006; Bewernick et al. 2010). Böhm et al. (2006) microdissected dopaminergic cells from the nucleus accumbens of control mice and of mice treated with amitriptyline, followed by RNA amplification and DNA microarray analysis. Among the 7,523 transcripts analyzed 64 were found to be significantly up-regulated and 31 significantly down-regulated. Interestingly, among those genes significantly up-regulated, CRYM/KR mRNA levels showed the greatest difference compared to control mice (24.3-fold increase) (Böhm et al. 2006). This finding suggests that amitriptyline may exert its effect, not only through its well-documented inhibition of the serotonin and norepinephrine transporters in the brain, but also possibly in part by stimulating the increased reduction of cyclic ketimines. This significant change in gene expression must be further evaluated in the context of the now known enzyme role of CRYM/KR.
d-Serine is an important co-agonist of the NMDA glutamate receptor. Endogenous levels of d-serine are regulated in part by DAAO (Snyder and Kim 2000). Hypo-stimulation of glutamate NMDA excitatory receptors is viewed as an important component of schizophrenia (Olney et al. 1999). Increased brain DAAO activity has been found in schizophrenia (Madeira et al. 2008) as well as decreased d-serine levels (Hashimoto et al. 2003). Beneficial effects of d-serine administration had previously been noted in schizophrenia (Tsai et al. 1998; Kantrowitz et al. 2010). The hypo-function of the NMDA receptor is commonly attributed to increased kynurenate (KynA) levels (as discussed in Sect. 9). However, we propose an additional contributory effect that is related to CRYM/KR of the pipecolate pathway in brain. As noted in Sect. 3, P2C is known to inhibit DAAO, but the sulfur-containing analog AECK is a far more potent DAAO inhibitor (Ricci et al. 1983). We suggest that levels of cyclic ketimine substrates of CRYM/KR (such as AECK and P2C) in the brain regulate DAAO activity and d-serine levels, and therefore also play a role in schizophrenia. The role of the pipecolate pathway in schizophrenia is further discussed in Sect. 9 in relation to tryptophan metabolism.
Gene expression profiling of schizophrenia pre-frontal cortex has revealed the CRYM/KR gene expression to be consistently decreased compared to that in controls (Middleton et al. 2002). In addition, CRYM/KR gene expression was shown to be down-regulated in pre-frontal cortices from a subset of schizophrenia patients (Akbarian et al. 2005). Moreover, low levels of the CRYM/KR protein have been found in schizophrenia pre-frontal cortex (Martins-de-Souza et al. 2009). CRYM/KR protein expression has, however, been found to be up-regulated in the corpus callosum of schizophrenia patients (Sivagnanasundaram et al. 2007). Whether these differences are a primary cause or a secondary compensatory result of the pathology remains to be determined. Given the possible involvement of DAAO in schizophrenia and the potential role of cyclic ketimines such as AECK, in regulating activity of this enzyme, down-regulation of CRYM/KR gene/protein expression may represent a secondary compensatory mechanism to maintain glutamate NMDA receptor tone.
Dysregulation of CRYM/KR gene expression, with subsequent alterations in cyclic ketimine substrate levels, may be involved in the pathological or secondary compensatory mechanisms of a number of neurodegenerative and neuromuscular diseases. Abnormal expression of CRYM/KR has been reported in facioscapulohumeral muscular dystrophy (FSHD) with large increases of CRYM/KR protein detected in muscle biopsies from affected patients (Reed et al. 2007). No such increase was noted in biopsies of several other myopathies (Reed et al. 2007). In future work, the effects of resultant lower ketimine substrate levels should be evaluated as possibly being causal or contributory in the pathology. The CRYM/KR gene is also up-regulated in a murine model of familial amyotrophic lateral sclerosis (ALS) (Fukada et al. 2007). Fukada et al. (2007) suggested that this up-regulation may be related to the pathogenesis of familial ALS. In this regards, a member of the cyclic ketimine family, namely, LK, administered as its ethyl ester, has been shown to substantially alleviate ALS symptoms, and prevent weight loss in an ALS murine model (Hensley 2007). CRYM/KR gene expression has also been shown to be significantly up-regulated in a murine model of Alzheimer disease (George et al. 2010).
Finally, there are two known point mutations of human CRYM, both of which are associated with nonsyndromic deafness (Abe et al. 2003). The effects of these mutations on enzyme activity must now be evaluated. The inference is that levels of CRYM/KR substrates are important determinants in hearing as CRYM mRNA is highly expressed in human inner ear (Aoki et al. 2000).
The peroxisomal connection: oxidation of l-pipecolate
Patients with hyperlysinemia are known to excrete increased amounts of l-pipecolate in their urine relative to unaffected controls (Woody and Pupene 1970). The accumulation of l-lysine in these patients is now known to be due to a block in LKR/SDH (also known as AAS synthase) (Sacksteder et al. 2000). As noted above, the saccharopine pathway is thought to be the main route for extracerebral l-lysine degradation in adult mammals. However, Woody and Pupene (1970) suggested that in patients with hyperlysinemia the increased level of l-pipecolate is indicative of an alternative pathway for lysine degradation i.e., via the l-pipecolate pathway. Zellweger syndrome is the result of a severe genetic disorder characterized by the failure to develop normal peroxisomes in affected individuals. The disease is characterized by impaired brain developmental and mental retardation (Versmold et al. 1977). Plasma l-pipecolate levels were found to be near normal in two newborn infants with Zellweger syndrome, but the level rose markedly within 2 months to pathological levels (Dancis and Hutzler 1986). The increase in plasma l-pipecolate in patients with Zellweger syndrome suggests that most, if not all, l-pipecolate oxidation occurs in peroxisomes in humans. Indeed, Wanders et al. (1988, 1989) showed that the oxidation of l-pipecolate is localized to peroxisomes in human liver and that this oxidation is deficient in liver from patients with Zellweger syndrome. These findings were subsequently confirmed by experiments showing that radiolabeled l-pipecolate is oxidized by a peroxisomal fraction of human liver and that this activity is deficient in patients with Zellweger syndrome (Mihalik et al. 1989). Rao and Chang (1990) also confirmed that l-pipecolate is oxidized in human liver peroxisomes and rigorously confirmed the identity of the product formed to be the cyclic ketimine P6C in equilibrium with its open-chain form AAS.
Early studies on the oxidation of l-pipecolate by Pseudomonas putida demonstrated that l-pipecolate is oxidized to P6C/AAS by an FAD-requiring dehydrogenase (Baginsky and Rodwell 1967). Subsequent studies showed that l-pipecolate oxidase (POX) is widespread in nature (Mihalik and Rhead 1989; 1991). Interestingly, however, differences occur in the subcellular location of the enzyme among various mammalian species. In rabbit, dog, guinea pig, pig, and sheep oxidation of l-pipecolate occurs in the mitochondrial fraction (Mihalik and Rhead 1991), whereas in rats oxidation of l-pipecolate occurs in both mitochondrial and peroxisomal fractions (Rao et al. 1993). In the rat brain, POX activity in the mitochondria is located in the matrix, whereas the activity in the peroxisomes is membrane-associated (Rao et al. 1993). POX activity in brain and liver was observed to occur early in rat fetal development, reaching maximum specific activity in the perinatal period immediately before birth, followed by a noticeable decline in specific activity with age (Rao et al. 1993). These changes are similar to those observed for CRYM/KR expression (Lennon et al. 1980) and also for l-pipecolate levels in brain (Kim and Giacobini 1985).
In contrast to the findings observed with tissues from other mammalian species (rabbit, dog, guinea pig, pig, sheep, rat), POX in the kidneys of the Rhesus monkey is only present in the peroxisomal fraction (Mihalik and Rhead 1989). POX was subsequently purified to apparent homogeneity from Rhesus monkey liver and shown to be monomeric, with a M r of ~46,000 and to possess a covalently bound FAD prosthetic group (Mihalik et al. 1991). Purified monkey liver POX was also shown to oxidize l-proline to a limited extent (Mihalik et al. 1991). Through peptide sequencing of the purified monkey liver POX, a reverse genetic approach was used to identify the cDNA for human POX (Dodt et al. 2000). The human POX possesses a C terminal AHL peroxisomal targeting sequence and is expressed in cultured human fibroblasts in a fashion consistent with its location in peroxisomes (Dodt et al. 2000). Previously, it was shown that d-pipecolate is an effective substrate of the peroxisomal enzyme DAAO (Zaar et al. 1986). Inasmuch as POX carries out an identical reaction to that catalyzed by DAAO (except that l-picolinate is the substrate rather than d-picolinate) it was considered that POX may be phylogenetically related to DAAO. However, comparative sequence analysis showed that human POX has no discernible relationship to DAAO (Dodt et al. 2000). Rather, human POX was found to be most similar to various monomeric sarcosine oxidases and dehydrogenases. The recombinantly expressed human enzyme was shown to oxidize both sarcosine (N-methyl glycine) and l-pipecolate equally effectively. [Both sarcosine and pipecolate may be regarded as N-substituted amino acids.] However, in patients with peroxisomal deficiency disorders, plasma pipecolate levels were found to be greatly elevated (95–126 μM; control 1.8 μM), whereas plasma sarcosine levels were undetectable in both controls and patients. The authors suggested that POX mainly oxidizes l-pipecolate in vivo in humans (Dodt et al. 2000). As was found for the monkey POX, the human enzyme contains a covalently bound FAD prosthetic group. This covalent binding of the cofactor in human and Rhesus POX is in contrast to DAAO, where the cofactor is non-covalently bound (Dodt et al. 2000).
The markedly elevated levels of l-pipecolate in patients with Zellweger syndrome are thought to contribute to the neuropathological symptoms (Arneson et al. 1982). l-Pipecolate has been shown to reduce neuronal cell viability and cause neuronal apoptosis (Matsumoto et al. 2003). Matsumoto et al. (2003) suggested that l-pipecolate may cause encephalopathy by apoptosis rather than through depression of neurotransmission. The mechanism involved has yet to be elucidated. However, it may involve activation of POX and subsequent formation of reactive oxygen species (ROS) as a related enzyme, namely proline oxidase, has been implicated in contributing to apoptosis through generation of ROS (Liu et al. 2006). Significant apoptosis takes place during normal brain development (de Graaf-Peters and Hadders-Algra 2006) and we suggest that formation of l-pipecolate and its subsequent oxidation by POX may be a contributing factor in this apoptosis.
The convergence of pathways and the mystery of B6-dependent epilepsy solved
As noted previously in Sect. 2, the saccharopine and pipecolate pathways (Figs. 1, 2, respectively) for the metabolism of l-lysine both converge at the step in which AAS is oxidized to AAD. An NAD+-requiring oxidoreductase [namely, α-aminoadipate semialdehyde dehydrogenase (AASDH)] responsible for the oxidation of AAS to AAD was first isolated from P. putida and characterized (Calvert and Rodwell 1966). Subsequently, an AASDH was shown to be present in Streptomyces clavuligerus and its highly conserved gene was successfully sequenced (Pérez-Llarena et al. 1998). Rao and Chang (1990) followed their successful isolation and characterization of AAS as the oxidation product of POX in tissues of several mammalian species with the characterization of a cytosolic AASDH enzyme in human liver (Chang et al. 1990). However, it was not until 16 years later that the human AASDH gene was identified.
The identification of the human AASDH gene came about as a result of a curious series of events. An inexplicable (at the time) severe intractable form of early onset epilepsy was first described in 1954 in which the patients exhibited resistance to conventional pharmaceutical interventions and yet showed an abrupt therapeutic response upon administration of pyridoxine (Hunt et al. 1954). [Pyridoxine is a B6 vitamer that is converted in vivo to the most biologically active form of vitamin B6, namely, pyridoxal 5′-phosphate (PLP)] The mechanism for the therapeutic effect of pyridoxine, however, remained unexplained for more than 50 years. Glutamate decarboxylase (GAD) is a PLP-dependent enzyme that converts the excitatory neurotransmitter glutamate to the inhibitory neurotransmitter GABA. Therefore, it was reasonable to suggest that a possible cause of seizures alleviated by vitamin B6 is due to an endogenous compound that reacts with PLP and/or a deficiency of GAD (Gospe et al. 1994). However, no linkage was found between the two brain GAD isoforms and pyridoxine-dependent seizures (Battaglioli et al. 2000). Moreover, the gene defect associated with pyridoxine-dependent seizures was mapped to chromosome 5q31 (Cormier-Daire et al. 2000), a region not associated with GAD genes.
The first important clue as to the origin of pyridoxine-dependent seizures came from the observation that a patient with a defect in Pyr5C dehydrogenase (hyperprolinemia type II) presented with both a vitamin B6 deficiency and seizures (Walker et al. 2000). It was suspected by Walker et al. (2000) that accumulation of Pyr5C resulting from this enzyme defect was in some way inactivating vitamin B6. Farrant et al. (2001) confirmed that this is indeed the case and showed that Pyr5C reacts with PLP through a Knoevenagel condensation, thus effectively preventing it from being used as an enzyme cofactor. In effect, Pyr5C acts as an anti-vitamin by covalently removing PLP. Thus, a defect in Pyr5C dehydrogenase accounts for one form of epilepsy responsive to vitamin B6 therapy. However, the majority of epileptic patients responsive to vitamin B6 therapy do not have this enzyme defect. As emphasized previously in Sect. 2, AAS is in equilibrium with its cyclic form P6C, and P6C is an analog of Pyr5C. Thus, it was rationalized that a defect in the human homologue of the previously described microbial AASDH gene (Pérez-Llarena et al. 1998) may be responsible for the disorder. By using a highly conserved region of the microbial AASDH gene, the sequence in the human genome analogous to this microbial enzyme was identified as the gene coding for aldehyde dehydrogenase 7A1 (ALDH7A1) and this gene mapped to chromosome 5q31 previously identified through linkage analysis (Mills et al. 2006). Mutations in ALDH7A1 were shown by Mills et al. (2006) and Plecko et al. (2007) to be present in several patients presenting with pyridoxine-responsive seizures. Interestingly, the AASDH encoded by the ALDH7A1 gene was already well known as antiquitin, a highly conserved protein with 60 % identity to that, for example, of a protein present in the garden pea (Lee et al. 1994). The identity of human aldehyde dehydrogenase AASDH as antiquitin/ALDH7A1 was conclusively established by Mills et al. (2006). Thus, a defect in antiquitin/ALDH7A1/AASDH will result in accumulation of P6C. It was shown that P6C, in a similar manner to Pyr5C, also forms a Knoevenagel condensation product with PLP (Mills et al. 2006; Plecko et al. 2007). [It is not currently known whether P2C, which is a double bond positional isomer of P6C, can also react with PLP in a similar manner to that exhibited by P6C, but a condensation product between PLP and P2C seems highly likely. Thus, an important function of KRs may be to maintain levels of P2C at a low level to avoid compromising PLP-dependent enzymes.]
Folinic acid-responsive seizures also result from mutations in antiquitin/AASDH/ALDH7A1 (Hyland et al. 1995; Nicolai et al. 2006). However, the biochemical mechanism involved is not currently understood (Hyland et al. 1995; Nicolai et al. 2006). The implication is that the biochemical repercussions resulting from accumulated P6C may be more complex than can be explained solely on the basis of PLP removal. Moreover, even though increased urinary AAS is an important indicator of antiquitin/ALDH7A1/AASDH deficiency (pyridoxine-dependent epilepsy), this compound is increased in the urine of patients with other disorders Mills et al. 2012). For example, sulfite was also shown in vitro with human liver homogenates to be a strong specific inhibitor of AASDH, with an accompanying increase in AAS (Mills et al. 2012). This finding may explain the marked accumulation of AAS in sulfite oxidase deficiency as well as in a deficiency of molybdenum, a sulfite oxidase cofactor (Mills et al. 2012).
At this point it is worth emphasizing the different subcellular localizations of the pipecolate and saccharopine pathways. The first step in the pipecolate pathway (Fig. 2), as discussed in Sect. 3, involves a conversion of l-lysine to KAC/P2C in the cytosol. As also discussed, this step is possibly catalyzed by GTL/KAT III, which is present in the cytosolic fraction of mammalian tissues. The second step of the pipecolate pathway, namely, the reduction of P2C by CRYM/KR also takes place in the cytosol. The next enzyme in the pipecolate pathway (POX, responsible for oxidation of pipecolate) occurs in the peroxisomes as discussed previously in Sect. 7. Another enzyme further downstream in the pathway (AASDH) is also present in the cytosolic compartment (Chang et al. 1990). It is thus reasonable to suggest that the pipecolate pathway is predominantly a cytosolic pathway with a peroxisomal component. The saccharopine pathway on the other hand is a well-established mitochondrial pathway. However, there is one caveat that requires discussion. Recently, AASDH was definitively established to be present in the cytosol and mitochondria of various mammalian tissues (Wong et al. 2010; Chan et al. 2011). This distribution results from two isoforms of the enzyme—a cytosolic isoform (ALDH7A1_v2) that lacks a mitochondrial targeting sequence, and a mitochondrial isoform that possesses an additional 28 amino acid mitochondrial signaling peptide at the N-terminus (ALDH7A1_v1) (Brocker et al. 2010). The mitochondrial ALDH7A1 was found, by Brocker et al. (2010), to be the predominant form in mouse kidney and liver in which the saccharopine pathway is the major lysine degradative pathway. Both cytosolic and mitochondrial ALDHA71 isoforms are present in adult brain where the pipecolate pathway is predominant (Brocker et al. 2010). These findings suggest that even though the pipecolate and saccharopine pathways merge at the metabolite AAS, further catabolism of AAS may well be separated in different compartments depending on the pathway i.e., cytosolic/peroxisomal and mitochondrial. Fibroblasts normally metabolize l-lysine through the saccharopine pathway and not through the pipecolate pathway. Nevertheless, Struys and Jakobs (2010) demonstrated formation of l-pipecolate from l-lysine in AASDH-deficient fibroblasts. The authors presented an evidence that l-pipecolate is produced in these cells via saccharopine and AAS/P6C (Struys and Jakobs 2010). Struys and Jakobs (2010) suggested that the enzyme responsible for this alternative pathway for the formation of l-pipecolate may be Pyr5C reductase.
AASDH is a member of the aldehyde dehydrogenase superfamily, and besides its role in the reduction of AAS it also is involved in the detoxification of other reactive aldehydes such as betaine aldehyde, converting it to the osmoprotectant glycine betaine (N,N,N-trimethylglycine) (Brocker et al. 2010). The enzyme can also act on longer chain aldehydes derived from the oxidation of lipids (Brocker et al. 2010). AASDH was first purified from Atlantic seabream (Mylio macrocephalus) and characterized as an aldehyde dehydrogenase based on its activity with acetaldehyde (Tang et al. 2002). The role of AASDH in lysine metabolism was unknown at that time. Reactive aldehydes are potentially toxic and can readily form adducts and condensation products with small molecules (e.g., amines) as well as with proteins and DNA (Marnett 2002; Brooks and Theruvathu 2005). In addition to its mitochondrial and cytosolic subcellular localization, AASDH is present in the nucleus (Brocker et al. 2010), where it is suggested to protect against aldehyde-induced DNA damage (Chan et al. 2011). In this regard, AASDH/ALDHA71 is similar to aldehyde dehydrogenase 3A1 (ALDH3A1), which is also localized to both mitochondria and nucleus (Pappa et al. 2005). Interestingly, AASDH accumulates maximally in the nucleus during the G1-S phase of replication, which is a time when oxidative stress is known to be most deleterious (Havens et al. 2006). However, its role in detoxification of aldehydes may not be its only function in the nucleus (Chan et al. 2011). We suggest that AASDH may also function in a similar manner to nuclear lactate dehydrogenase and glyceraldehyde-3-phosphate dehydrogenase, both of which are known to be part of the histone 2B transcription activation complex (Dai et al. 2008).
In summary, mutations in AASDH can give rise to pyridoxine-dependent and folinic acid-dependent seizures. Moreover, AASDH is a multifunctional enzyme with roles in: (1) lysine degradation; (2) protection against oxidative stress by converting reactive aldehydes derived from lipid peroxidation (and other metabolic processes) to less reactive/toxic carboxylic acids; (3) conversion of betaine aldehyde to the osmoprotectant betaine; and (4) possible protection against DNA damage by reactive aldehydes and/or functioning to regulate DNA transcription.
Interconnections between lysine and tryptophan pathways: developmental and regulatory aspects
AAD formed by AASDH is subsequently transaminated to form α-ketoadipate (AKA). Historically, Broquist et al. (1961), in experiments on Saccharomyces cerivisiae, first described a transaminase reaction involving AAD and AKA that was suspected to play a role in lysine biosynthesis. However, it was not clear whether AAD aminotransferase activity is catalyzed by a discreet enzyme or whether the activity is catalyzed by an already characterized aminotransferase. Subsequently, Matsuda and Ogur (1969) showed that yeast contains two aminotransferases that catalyze the reversible transamination between AAD and α-KG to form AKA and glutamate. The authors named these aminotransferases glutamate-AKA transaminase I and II (GKAT I and GKAT II). These GKAT isozymes were shown to be distinct from aspartate aminotransferase and two forms of alanine aminotransferase (Matsuda and Ogur 1969).
The first indication of the presence of a mammalian AADT (Figs. 4, 5, AADT) came from the discovery in rat liver mitochondria of an enzyme capable of catalyzing transamination between AAD and α-KG (Nakatani et al. 1970). The purified enzyme was found to possess a loosely bound PLP cofactor. Even though the first indications were that AADT is a mitochondrial enzyme, enzyme activity was later demonstrated to be present in both the cytosolic and mitochondrial fractions of rat liver and kidney (Tobes and Mason 1975, 1977). Cytosolic activity is consistent with a role in the pipecolate pathway and mitochondrial activity is consistent with a role in the saccharopine pathway.

The interconnection of l-lysine and l-tryptophan degradative pathways in mammals. Enzymes: 2–8 as per Figs. 1 and 2; 1a indoleamine dioxygenase/tryptophan dioxygenase (IDO/TDO); 2a kynurenine formylase; 3a kynurenine monooxygenase (depicted as occurring on the mitochondrial outer membrane); 4a enzymes catalyzing the conversion of 3-hydroxykynurenate to α-ketoadipate (AKA). Note that as discussed in the text, the conversion of l-lysine to KAC in the pipecolate pathway may be catalyzed by KAT III/GTL. The most important aminotransferase that catalyzes the conversion of α-aminoadipate to α-ketoadipate in the pipecolate pathway for l-lysine degradation is the same enzyme that catalyzes the conversion of kynurenine to kynurenate [i.e., α-aminoadipate aminotransferase/kynurenine aminotransferase II (AADT/KAT II); enzyme 3 in this figure and Figs. 1, 2]. Thus, any perturbation in one pathway will influence the other. This interconnection between lysine and tryptophan degradative pathways has implications in psychiatric as well as neurological illnesses as discussed in the text

The remarkable amino acid substrate versatility of α-aminoadipate aminotransferase/kynurenine aminotransferase II (AADT/KAT II). Not only is the enzyme active with AAD and Kyn, but it can also transaminate thyroid hormones (e.g., T3) and halogenated tyrosines as discussed in the text. The thyroid hormone T3 is a strong inhibitor of CRYM/KR, and AADT/KAT II activity is also potentially regulated by circulating thyroid hormones. In the text, we postulate that thyroid hormones play a pivotal role in the development of the pipecolate pathway in mammalian brain. The biological importance of T3 transamination is unknown although it has been suggested that KAT (i.e., KAT II) may be important for the metabolism of thyroid hormones (Tobes and Mason 1978)
Aminotransferases often exhibit wider specificity than first realized, leading to some confusion in the literature regarding nomenclature, and AADT is an example (Figs. 4, 5). An enzyme designated kynurenine aminotransferase (KAT) that catalyzes transamination between l-kynurenine (Kyn) and α-KG was shown to be present in Pseudomonas fluorescens by Miller et al. in 1953 and in animal tissues by Mason in 1954. However, Tobes and Mason (1975, 1977) convincingly showed that AADT and KAT are identical enzymes in the rat. [This enzyme is now designated as KAT II. As noted above, KAT I is identical to GTK and KAT III is identical to GTL. An additional KAT (KAT IV) is identical to mitochondrial aspartate aminotransferase (Guidetti et al. 2007).] Tobes and Mason (1975, 1977) found that rat liver possesses mostly mitochondrial KAT activity, whereas in rat kidney the supernatant fraction is more active. AAD was demonstrated to be a better substrate than Kyn. Takeuchi et al. (1983) also confirmed that AADT from rat liver mitochondria is identical to KAT. These authors showed that enzyme activity is strongly inhibited by Hg++ as well as by p-chloromercuribenzoate, suggesting that a crucial cysteine is in or near the active site. Once again AAD was found to be a better substrate than Kyn.
The relationship between AADT and KAT, however, may be more complicated than the above discussion suggests. Deshmukh and Mungre (1989) reported that bovine kidney possesses appreciable AADT activity but minimal KAT activity, suggesting that different enzymes catalyze the two reactions. In spite of evidence noted above suggesting that AADT and KAT are identical enzymes, Mawal and Deshmukh (1991) separated two enzymes chromatographically from rat liver supernatant fractions and claimed that AAD and Kyn transamination are due to two distinctly different enzymes, seemingly throwing into doubt the work of Tobes and Mason (1975, 1977). However, a soluble rat kidney enzyme with dual AADT/KAT activities was identified, cloned, and functionally expressed by Buchli et al. (1995). The predicted amino acid sequence showed no evidence of a mitochondrial signaling sequence. Among the α-keto acids tested when Kyn was employed as the amino acid substrate, the recombinant enzyme was shown to have highest specific activity with α-KG and relatively poor activity with pyruvate. The authors also showed that this Kyn-α-KG aminotransferase is distinct from KAT I/GTK. Previously, two AADT isoenzymes were purified from human liver (AADT I and II) by Okuno et al. (1993). AADTI was shown to be localized to the supernatant (cytosol) fraction and AADTII to the mitochondria. AADTI was shown to be highly specific for AAD and glutamate as amino acid substrates and α-KG and AKA as α-keto acid substrates. On the other hand, AADTII was shown to possess KAT activity. AADTI was shown to have a high K m (20 mM) for AAD and a low K m (1.4 mM) for glutamate. AADTII, on the other hand, was found to have a low K m (0.25 mM) for AAD and a high K m (12.5 mM) for glutamate. AADTII was also shown to have a low K m (0.95 mM) for Kyn. The human gene (AADT) encoding AADT was subsequently identified and localized to chromosome 4q32.2 (Goh et al. 2002). Human AADT was found to be 72 and 73 % identical to the mouse and rat KAT II orthologues, respectively. The gene is suspected to code for AADTII and possesses a mitochondrial signaling peptide. Recombinant human AADT/KAT II, as noted above, has a broad amino acid and α-keto acid specificity (Han et al. 2008). Among the amino acids tested as substrates, the highest catalytic efficiency (K cat/K m) was noted with AAD (196 min−1 mM−1) followed by Kyn (126 min−1 mM−1) (Han et al. 2008). The predicted mitochondrial signal sequence is the first N-terminus 22 amino acid, but a recombinant enzyme that lacked this sequence showed no enzyme activity (Han et al. 2010). Further research is required to ascertain how the enzyme activity is apportioned between mitochondria and cytosol. A cytosolic subcellular localization is consistent with its role in the pipecolate pathway and a mitochondrial location with the saccharopine pathway.
The initiation of the Kyn metabolic pathway, through the action of indoleamine 2,3-dioxygenase (IDO) or tryptophan 2,3-dioxygenase (TDO), is known to suppress the immune response and in particular to prevent rejection of the developing fetus (Gonzalez et al. 2008). The N-formylkynurenine formed by the action of either IDO or TDO is rapidly degraded to form Kyn by the action of kynurenine formylase, an enzyme that is abundant in most mammalian tissues (Mehler and Knox 1950). The Kyn pathway undergoes marked developmental changes in fetal brain. Kyn and KynA levels in rat and primate fetal brain are markedly high in comparison to post-natal brain, with a gradual decrease in Kyn as well as KynA levels during the course of gestation and a subsequent rapid decrease after birth (Beal et al. 1992). The high level of Kyn pathway activity in fetal brain and subsequent decrease during development are similar to the changes noted for the high levels and subsequent decrease in expression of LKR/SDH (Rao et al. 1992) (the first enzyme activities in the lysine saccharopine pathway) as discussed in Sect. 2. The decrease in Kyn and KynA levels are also inversely related to the developing ascendancy of the pipecolate pathway as the dominant lysine degradation pathway in adult mammalian brain (Chang 1976). KynA is known to inhibit neurite outgrowth by NMDA receptor stimulation in granule cells in culture (Pearce et al. 1987). We suggest that the high levels of LKR/SDH observed in fetal brain neurons may represent a compensatory mechanism to overcome this inhibition by supplying a de novo source of glutamate.
A day before birth in rat fetal brain there is a spike in Kyn and KynA levels (Beal et al. 1992) similar to that observed for rat fetal brain pipecolate (Kim and Giacobini 1985; Nishio and Segawa 1983) as well as NADPH-dependent cytosolic thyroid hormone binding (Suzuki et al. 1991). At day 1 post-delivery in rat brain, KynA levels decrease to adult levels (Beal et al. 1992). This finding is in marked contrast with the formation of l-pipecolate (as discussed in Sect. 5), where expression of NADPH-dependent T3-binding protein (and by inference CRYM/KR) only reaches detectable levels a few days before birth in fetal rat brain (Suzuki et al. 1991). There is a rapid increase in neurite outgrowth and development of excitatory synapses after birth (Lin et al. 2002), which corresponds to the period of rapid decreases in KynA levels. Beal et al. (1992) also showed that brain levels of norepinephrine, dopamine, and DOPAC increase at a time that correlates with the spikes seen with CRYM/KR and also Kyn and KynA.
In Sect. 3, we presented arguments suggesting that the first enzyme(s) in the pipecolate pathway for lysine degradation may be KAT III/GTL. On the other hand, KAT II/AADT is well documented to be involved downstream in the catabolism of both tryptophan and lysine. GTK/GTL are also believed to be responsible for the biosynthesis of the CRYM/KR sulfur-containing cyclic ketimine enzyme substrates (such as AECK and LK) (Pensa et al. 1989). One of the cyclic ketimines, namely, LK (as mentioned in Sects. 5 and 6), has been shown to cause neurite extension in duck dorsal root ganglia and motor neuron-like cells (Hensley et al. 2010, 2011). We hypothesize that a neuro-differentiation effect may be a generalized characteristic of all the cyclic ketimines and that activation of the pipecolate pathway may be a critical factor in this rapid post-natal neuronal development.
Thyroid hormones play an important regulatory role in the pipecolate pathway as discussed in Sect. 5. They may also play a role in regulation of KAT II/AADT enzyme regulation. Levels of metabolites in the tryptophan/kynurenine pathway are markedly altered in thyroxine-treated rat liver, with a threefold increase in Kyn and KynA levels (Okamoto et al. 1971). KAT activity increased proportionally. Okamoto et al. (1971) suggested that this effect is due to an increase in de novo KAT synthesis. Tobes and Mason (1978) showed that KAT (i.e., KAT II) not only functions to transaminate both AAS and Kyn, but also effectively transaminates halogenated tyrosines (such as 3,5-diiodotyrosine) as well as, if not better than, Kyn or AAS, and suggested that halogenated tyrosine transaminase and KAT (KAT II) are identical enzymes based on their kinetic observations (Tobes and Mason 1978). It had earlier been suggested that halogenated tyrosine transaminase is identical to thyroid transaminase (Nakano 1967; Nakano and Danowski 1964; Nakano et al. 1964). The remarkable activity of KAT II as a KYN/AAD/T3 aminotransferase is shown in Fig. 5. The marked increase in fetal thyroid hormone production before and after birth in rats, thus, may be causally related to the spikes observed in KynA and pipecolate formation at this time through induction of CRYM/KR expression.
The activity of KAT II/AADT is important in preventing neurotoxicity and neurodegeneration as it regulates the levels of AAD, KynA, and possibly thyroid hormones in the brain. AAD is a 6-carbon dicarboxylic acid homologue of l-glutamate, which is known to be toxic to glial cells and is taken up by astrocytes prior to exerting its gliotoxic effects (Huck et al. 1984). AAD is also thought to interfere with glutamate uptake into astrocytes (Tsai et al. 1996). In astrocyte cultures, KynA synthesis is inhibited by AAD as well as by l-pipecolate (Chang et al. 1997). In contrast to the toxicity demonstrated by AAD, KynA is a neuroprotectant and an endogenous inhibitor of excitatory glutamate NMDA receptors (Stone 1993). Therefore, the biosynthesis of KynA in the brain and the regulation of KAT enzymes are of utmost importance. In this regard, l-cysteine sulfinate, an endogenous intermediate in sulfur-containing amino acid metabolism, has been found to be a selective AADT/KAT II enzyme inhibitor and is suspected of modulating KAT II activity as well as AAD levels in the brain (Kocki et al. 2003).
Changes in brain KynA levels have been noted in various neurological and psychiatric conditions. For example, in Alzheimer’s disease brain, a significant increase in AAD synthesis was found coupled with a decrease in KAT activity (Yang et al. 1995). An intriguing finding is that KynA levels are increased in the cerebrospinal fluid and cortex of schizophrenia patients (Erhardt et al. 2001; Schwarcz et al. 2001). As noted elsewhere in this review, AKA formed by KAT II/AADT is a common intermediate in l-tryptophan as well as in l-lysine catabolic pathways, and it is reasonable to suggest that the metabolism of the one amino acid will influence the metabolism of the other. This relationship may explain the observed positive therapeutic response observed with l-lysine supplementation in a cohort of schizophrenia patients (Wass et al. 2011). It is possible that a decrease in the activity of the pipecolate pathway for lysine metabolism plays a role in schizophrenia. A decrease in this pathway will result in lower amounts of AAD and hence less inhibition of KAT II, leading to increased KynA. Thus, the positive effect of lysine in schizophrenic patients may be due to stimulation of the pipecolate pathway in brain leading to decreased KynA. The CRYM/KR substrates P2C and AECK have been noted in Sect. 6 as also of possible importance in schizophrenia by virtue of their strong inhibition of DAAO.
Thus far our discussion of l-lysine degradation has been restricted to biological processes in the pipecolate and saccharopine pathways that terminate at AKA formation. Further degradation occurs only in the mitochondria, namely, subsequent formation of glutaryl-CoA and eventual catabolism to CO2 in the TCA cycle. These steps are not discussed in the present review. Detailed reviews of this pathway may be found, for example, in Benevenga and Blemings (2007) and Sauer et al. (2011).
Summary
The pipecolate pathway has evolved as the main l-lysine degradative pathway in adult mammalian brain, whereas in the developing brain and extracerebral tissues the saccharopine pathway predominates. The reason for this dichotomy is not fully understood, but we are slowly beginning to understand the importance of the pipecolate pathway in brain metabolism and function. We predict that the discovery of an enzyme in mammalian forebrain (namely CRYM/KR) that is capable of effectively reducing the intermediate metabolite P2C to l-pipecolate will be a significant step in advancing our knowledge of the pipecolate pathway and its neurological/neurochemical importance. P2C (as well as other cyclic ketimine substrates of KR) may play a crucial role in brain development, metabolism, and electrophysiology/neurotransmitter regulation. The finding that T3 strongly regulates CRYM/KR enzyme activity suggests a previously unsuspected inter-relationship between amino acid metabolism and the endocrine system. With the completion of the human genome project, scientists were at first astounded by the relatively low number of genes coding for proteins (~21,000). However, it is now apparent that many gene-encoded proteins exist in multiple forms, and many possess multiple and often disparate roles. Two pivotal enzymes in the lysine degradation pathways, namely, CRYM/KR and AASDH, are examples of “moonlighting” enzymes that have evolved to possess two or more disparate functions.
The mechanism by which l-lysine is oxidized to KAC/P2C is unknown. One possibility is that the reaction is catalyzed by KAT III/GTL. If verified, inasmuch as Kyn is an important metabolite in l-tryptophan metabolism, the involvement of an aminotransferase that catalyzes transamination of both l-lysine and l-kynurenine would suggest a close relationship between lysine degradation and tryptophan degradation. Changes in the concentration of the product of Kyn transamination, namely, KynA, are implicated in many neurological and psychiatric disorders. We suggest that any discussion as to the relevance of the tryptophan-Kyn degradation pathway in neurodegeneration and illness should also take into account its “close relation”, namely, the lysine degradation pathway.
Abbreviations
- AAD:
-
α-Aminoadipate
- AADT:
-
α-Aminoadipate transaminase (aminotransferase)
- AAS:
-
α-Aminoadipate δ-semialdehyde
- AASDH:
-
α-Aminoadipate δ-semialdehyde dehydrogenase
- α-KG:
-
α-Ketoglutarate
- AKA:
-
α-Ketoadipate
- ALDH:
-
Aldehyde dehydrogenase
- ALS:
-
Amyotrophic lateral sclerosis
- CTBP:
-
Cytosolic thyroid hormone-binding protein
- CRYM:
-
μ-Crystallin
- DAAO:
-
d-Amino acid oxidase
- GAD:
-
Glutamate decarboxylase
- GTK:
-
Glutamine transaminase K
- GTL:
-
Glutamine transaminase L
- KAC:
-
α-Keto-ε-aminocaproate
- KAT:
-
Kynurenine aminotransferase
- KR:
-
Ketimine reductase
- Kyn:
-
l-Kynurenine
- KynA:
-
Kynurenate
- IL4I1:
-
Interleukin-4-induced protein 1
- LAAO:
-
l-Amino acid oxidase
- LKR/SDH:
-
Lysine-α-ketoglutarate reductase/saccharopine dehydrogenase
- P2C:
-
∆1-Piperideine-2-carboxylate
- P6C:
-
∆1-Piperideine-6-carboxylate
- PLP:
-
Pyridoxal 5′-phosphate
- POX:
-
Pipecolate oxidase
- Pyr2C:
-
∆1-Pyroline-2-carboxylate
- Pyr5C:
-
∆1-Pyroline-5-carboxylate
- T3 :
-
3,5,3′-Triiodothyronine
References
Abe S, Katagiri T, Saito-Hisaminato A, Usami S, Inoue Y, Tsunoda T, Nakamura Y (2003) Identification of CRYM as a candidate responsible for nonsyndromic deafness, through cDNA microarray analysis of human cochlear and vestibular tissues. Am J Hum Genet 72:73–82
Akbarian S, Ruehl MG, Bliven E, Luiz LA, Peranelli AC, Baker SP, Roberts RC, Bunney WE Jr, Conley RC, Jones EG, Tamminga CA, Guo Y (2005) Chromatin alterations associated with down-regulated metabolic gene expression in the prefrontal cortex of subjects with schizophrenia. Arch Gen Psychiatry 62:829–840
Aoki N, Ito K, Ito M (2000) μ-Crystallin, thyroid hormone-binding protein, is expressed abundantly in the murine inner root sheath cells. J Invest Dermatol 115:402–405
Arneson DW, Tipton RE, Ward JC (1982) Hyperpipecolic acidemia. Occurrence in an infant with clinical findings of the cerebrohepatorenal (Zellweger) syndrome. Arch Neurol 39:713–716
Aspen AJ, Meister A (1962a) The preparation and some properties of α-aminoadipic-δ-semialdehyde (∆1-piperideine-6-carboxylic acid). Biochemistry 1:600–605
Aspen AJ, Meister A (1962b) Conversion of α-aminoadipic acid to l-pipecolic acid by Aspergillus nidulans. Biochemistry 1:606–612
Augusteyn RC (2011) Lens growth and protein changes in the eastern grey kangaroo. Mol Vis 17:3234–3242
Baginsky ML, Rodwell VW (1967) Metabolism of pipecolic acid in a Pseudomonas species. V. Pipecolate oxidase and dehydrogenase. J Bacteriol 94:1034–1039
Battaglioli G, Rosen DR, Gospe SM Jr, Martin DL (2000) Glutamate decarboxylase is not genetically linked to pyridoxine-dependent seizures. Neurology 55:309–311
Beal MF, Swartz KJ, Isacson O (1992) Developmental changes in brain kynurenic acid concentrations. Brain Res Dev Brain Res 68:136–139
Benevenga NJ, Blemings KP (2007) Unique aspects of lysine nutrition and metabolism. J Nutr 137(6 Suppl 2):1610S–1615S
Bernal J (1999) Iodine and brain development. BioFactors 10(2–3):271–276
Beslin A, Vie MP, Blondeau JP, Francon J (1995) Identification by photoaffinity labelling of a pyridine nucleotide-dependent tri-iodothyronine-binding protein in the cytosol of cultured astroglial cells. Biochem J 305:729–737
Bewernick BH, Hurlemann R, Matusch A, Kayser S, Grubert C, Hadrysiewicz B, Axmacher N, Lemke M, Cooper-Mahkorn D, Cohen MX, Brockmann H, Lenartz D, Sturm V, Schlaepfer TE (2010) Nucleus accumbens deep brain stimulation decreases ratings of depression and anxiety in treatment-resistant depression. Biol Psychiatry 67:110–116
Böhm C, Newrzella D, Herberger S, Schramm N, Eisenhardt G, Schenk V, Sonntag-Buck V, Sorgenfrei O (2006) Effects of antidepressant treatment on gene expression profile in mouse brain: cell type-specific transcription profiling using laser microdissection and microarray analysis. J Neurochem 97(Suppl 1):44–49
Boulland ML, Marquet J, Molinier-Frenkel V, Moller P, Guiter C, Lasoudris F, Copie-Bergman C, Baia M, Gaulard P, Leroy K, Castellano F (2007) Human IL4I1 is a secreted l-phenylalanine oxidase expressed by mature dendritic cells that inhibits T-lymphocyte proliferation. Blood 110:220–227
Brocker C, Lassen N, Estey T, Pappa A, Cantore M, Orlova VV, Chavakis T, Kavanagh KL, Oppermann U, Vasiliou V (2010) Aldehyde dehydrogenase 7A1 (ALDH7A1) is a novel enzyme involved in cellular defense against hyperosmotic stress. J Biol Chem 285:18452–18463
Brooks PJ, Theruvathu JA (2005) DNA adducts from acetaldehyde: implications for alcohol-related carcinogenesis. Alcohol 35:187–193
Broquist HP, Stiffey AV, Albrecht AM (1961) Biosynthesis of lysine from α-ketoadipic acid and α-aminoadipic acid in yeast. Appl Microbiol 9:1–5
Buchli R, Alberati-Giani D, Malherbe P, Kohler C, Broger C, Cesura AM (1995) Cloning and functional expression of a soluble form of kynurenine/α-aminoadipate aminotransferase from rat kidney. J Biol Chem 270:29330–29335
Calvert AF, Rodwell VW (1966) Metabolism of pipecolic acid in a Pseudomonas species. 3. L-α-aminoadipate δ-semialdehyde:nicotinamide adenine dinucleotide oxidoreductase. J Biol Chem 241:409–414
Calvo R, Obregon MJ, Escobar del Rey F, Morreale de Escobar G (1992) The rat placenta and the transfer of thyroid hormones from the mother to the fetus. Effects of maternal thyroid status. Endocrinology 131:357–365
Carson NA, Scally BG, Neill DW, Carre LJ (1968) Saccharopinuria: a new inborn error of lysine metabolism. Nature 218:679
Chan CL, Wong JW, Wong CP, Chan MK, Fong WP (2011) Human antiquitin: structural and functional studies. Chem Biol Interact 191:165–170
Chang YF (1976) Pipecolic acid pathway: the major lysine metabolic route in the rat brain. Biochem Biophys Res Commun 69:174–180
Chang YF (1978) Lysine metabolism in the rat brain: the pipecolic acid-forming pathway. J Neurochem 30:347–354
Chang YF, Charles AK (1995) Uptake and metabolism of ∆1-piperidine-2-carboxylic acid by synaptosomes from rat cerebral cortex. Biochim Biophys Acta 1238:29–33
Chang YF, Ghosh P, Rao VV (1990) l-Pipecolic acid metabolism in human liver: l-α-aminoadipate δ-semialdehyde oxidoreductase. Biochim Biophys Acta 1038:300–305
Chang YF, Cauley RK, Chang JD, Rao VV (1997) l-α-Aminoadipate inhibits kynurenate synthesis in rat brain hippocampus and tissue culture. Neurochem Res 22:825–829
Chavan SS, Tian W, Hsueh K, Jawaheer D, Gregersen PK, Chu CC (2002) Characterization of the human homolog of the IL-4 induced gene-1 (Fig. 1). Biochim Biophys Acta 1576:70–80
Chronister RB, DeFrance JF (1982) Implications from studies of the nucleus accumbens: toward a multi-factor theory of schizophrenia. Med Hypotheses 9:115–123
Coleman CS, Stanley BA, Jones AD, Pegg AE (2004) Spermidine/spermine-N 1-acetyltransferase-2 (SSAT2) acetylates thialysine and is not involved in polyamine metabolism. Biochem J 384:139–148
Cooper AJL, Meister A (1972) Isolation and properties of highly purified glutamine transaminase. Biochemistry 11:661–671
Cooper AJL, Meister A (1981) Comparative studies of glutamine transaminases from rat tissues. Comp Biochem Phys B Comp Biochem 69:137–145
Cooper AJL, Meister A (1985) Glutamine transaminase L from rat liver. Methods Enzymol 113:338–343
Cormier-Daire V, Dagoneau N, Nabbout R, Burglen L, Penet C, Soufflet C, Desguerre I, Munnich A, Dulac O (2000) A gene for pyridoxine-dependent epilepsy maps to chromosome 5q31. Am J Hum Genet 67:991–993
Costa M, Pensa B, Fontana M, Foppoli C, Cavallini D (1986) Transamination of l-cystathionine and related compounds by a bovine liver enzyme. Possible identification with glutamine transaminase. Biochim Biophys Acta 881:314–320
Dai RP, Yu FX, Goh SR, Chng HW, Tan YL, Fu JL, Zheng L, Luo Y (2008) Histone 2B (H2B) expression is confined to a proper NAD+/NADH redox status. J Biol Chem 283:26894–26901
Dancis J, Hutzler J (1986) The significance of hyperpipecolatemia in Zellweger syndrome. Am J Hum Genet 38:707–711
Dancis J, Hutzler J, Cox RP, Woody NC (1969) Familial hyperlysinemia with lysine-ketoglutarate reductase insufficiency. J Clin Invest 48:1447–1452
de Graaf-Peters VB, Hadders-Algra M (2006) Ontogeny of the human central nervous system: what is happening when? Early Hum Dev 82:257–266
Delange F (2000) The role of iodine in brain development. Proc Nutr Soc 59:75–79
Deshmukh DR, Mungre SM (1989) Purification and properties of 2-aminoadipate: 2-oxoglutarate aminotransferase from bovine kidney. Biochem J 261:761–768
Dodt G, Kim DG, Reimann SA, Reuber BE, McCabe K, Gould SJ, Mihalik SJ (2000) l-Pipecolic acid oxidase, a human enzyme essential for the degradation of l-pipecolic acid, is most similar to the monomeric sarcosine oxidases. Biochem J 345:487–494
Dubois JD, Dussault JH (1977) Ontogenesis of thyroid function in the neonatal rat. Thyroxine (T4) and triiodothyronine (T3) production rates. Endocrinology 101:435–441
Duley JA, Holmes RS (1976) l-α-Hydroxyacid oxidase isozymes. Purification and molecular properties. Eur J Biochem 63:163–173
Duprè S, Fontana M, Costa M, Pecci L, Ricci G, Cavallini D (1993) Characterization of [35S]lanthionine ketimine specific binding to bovine brain membranes. Biochem Biophys Res Commun 195:673–677
Erhardt S, Blennow K, Nordin C, Skogh E, Lindstrom LH, Engberg G (2001) Kynurenic acid levels are elevated in the cerebrospinal fluid of patients with schizophrenia. Neurosci Lett 313:96–98
Farrant RD, Walker V, Mills GA, Mellor JM, Langley GJ (2001) Pyridoxal phosphate de-activation by ∆1-pyrroline-5-carboxylic acid. Increased risk of vitamin B6 deficiency and seizures in hyperprolinemia type II. J Biol Chem 276:15107–15116
Fellows FC, Carson NA (1974) Enzyme studies in a patient with saccharopinuria: a defect of lysine metabolism. Pediatr Res 8:42–49
Fellows FC, Lewis MH (1973) Lysine metabolism in mammals. Biochem J 136:329–334
Fukada Y, Yasui K, Kitayama M, Doi K, Nakano T, Watanabe Y, Nakashima K (2007) Gene expression analysis of the murine model of amyotrophic lateral sclerosis: studies of the Leu126delTT mutation in SOD1. Brain Res 1160:1–10
Garweg G, von Rehren D, Hintze U (1980) l-Pipecolate formation in the mammalian brain. Regional distribution of ∆1-pyrroline-2-carboxylate reductase activity. J Neurochem 35:616–621
George AJ, Gordon L, Beissbarth T, Koukoulas I, Holsinger RM, Perreau V, Cappai R, Tan SS, Masters CL, Scott HS, Li QX (2010) A serial analysis of gene expression profile of the Alzheimer’s disease Tg2576 mouse model. Neurotox Res 17:360–379
Goglia F, Lanni A, Horst C, Moreno M, Thoma R (1994) In vitro binding of 3,5-di-iodo-l-thyronine to rat liver mitochondria. J Mol Endocrinol 13:275–282
Goh DL, Patel A, Thomas GH, Salomons GS, Schor DS, Jakobs C, Geraghty MT (2002) Characterization of the human gene encoding α-aminoadipate aminotransferase (AADAT). Mol Genet Metab 76:172–180
Gonzalez A, Varo N, Alegre E, Diaz A, Melero I (2008) Immunosuppression routed via the kynurenine pathway: a biochemical and pathophysiologic approach. Adv Clin Chem 45:155–197
Gospe SM Jr, Olin KL, Keen CL (1994) Reduced GABA synthesis in pyridoxine-dependent seizures. Lancet 343:1133–1134
Graham CE, Brocklehurst K, Pickersgill RW, Warren MJ (2006) Characterization of retinaldehyde dehydrogenase 3. Biochem J 394:67–75
Gray JA, Joseph MH, Hemsley DR, Young AM, Warburton EC, Boulenguez P, Grigoryan GA, Peters SL, Rawlins JN, Taib CT et al (1995) The role of mesolimbic dopaminergic and retrohippocampal afferents to the nucleus accumbens in latent inhibition: implications for schizophrenia. Behav Brain Res 71:19–31
Grove J, Henderson LM (1968) The metabolism of d- and l-lysine in the intact rat, perfused liver and liver mitochondria. Biochim Biophys Acta 165:113–120
Guidetti P, Amori L, Sapko MT, Okuno E, Schwarcz R (2007) Mitochondrial aspartate aminotransferase: a third kynurenate-producing enzyme in the mammalian brain. J Neurochem 102:103–111
Gutierrez MD, Giacobini E (1985) Identification and characterization of pipecolic acid binding sites in mouse brain. Neurochem Res 10:691–702
Hallen A, Cooper AJL, Jamie JF, Haynes PA, Willows RD (2011) Mammalian forebrain ketimine reductase identified as μ-crystallin; potential regulation by thyroid hormones. J Neurochem 118:379–387
Han Q, Cai T, Tagle DA, Robinson H, Li J (2008) Substrate specificity and structure of human aminoadipate aminotransferase/kynurenine aminotransferase II. Biosci Rep 28:205–215
Han Q, Robinson H, Cai T, Tagle DA, Li J (2009) Biochemical and structural properties of mouse kynurenine aminotransferase III. Mol Cell Biol 29:784–793
Han Q, Cai T, Tagle DA, Li J (2010) Structure, expression, and function of kynurenine aminotransferases in human and rodent brains. Cell Mol Life Sci 67:353–368
Hashimoto K, Fukushima T, Shimizu E, Komatsu N, Watanabe H, Shinoda N, Nakazato M, Kumakiri C, Okada S, Hasegawa H, Imai K, Iyo M (2003) Decreased serum levels of d-serine in patients with schizophrenia: evidence in support of the N-methyl-d-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry 60:572–576
Havens CG, Ho A, Yoshioka N, Dowdy SF (2006) Regulation of late G1/S phase transition and APCCdh1 by reactive oxygen species. Mol Cell Biol 26(12):4701–4711
Hensley K (2007) Lanthionine-related compounds for the treatment of inflammatory diseases. US Patent 2007/0197515A1 (US 2007/0197515 A1)
Hensley K, Christov A, Kamat S, Zhang XC, Jackson KW, Snow S, Post J (2010) Proteomic identification of binding partners for the brain metabolite lanthionine ketimine (LK) and documentation of LK effects on microglia and motoneuron cell cultures. J Neurosci 30:2979–2988
Hensley K, Venkova K, Christov A, Gunning W, Park J (2011) Collapsin response mediator protein-2: an emerging pathologic feature and therapeutic target for neurodisease indications. Mol Neurobiol 43:180–191
Hernandez MF, Chang YF (1980) In vitro synthesis of l-pipecolate from l-lysine: inconsistent with ε-N-acetyl-l-lysine as an obligatory intermediate. Biochem Biophys Res Commun 93:762–769
Higashino K, Tsukada K, Lieberman I (1965) Saccharopine, a product of lysine breakdown by mammalian liver. Biochem Biophys Res Commun 20:285–290
Higashino K, Fujioka M, Aoki T, Yamamura T (1967) Metabolism of lysine in rat liver. Biochem Biophys Res Comm 29:95–100
Higashino K, Fujioka M, Yamamura Y (1971) The conversion of l-lysine to saccharopine and α-aminoadipate in mouse. Arch Biochem Biophys 142(2):606–614
Horiike K, Tojo H, Arai R, Nozaki M, Maeda T (1994) d-Amino-acid oxidase is confined to the lower brain stem and cerebellum in rat brain: regional differentiation of astrocytes. Brain Res 652:297–303
Huck S, Grass F, Hortnagl H (1984) The glutamate analog α-aminoadipic acid is taken up by astrocytes before exerting its gliotoxic effect in vitro. J Neurosci 4(10):2650–2657
Hunt AD Jr, Stokes J Jr, Mc CW, Stroud HH (1954) Pyridoxine dependency: report of a case of intractable convulsions in an infant controlled by pyridoxine. Pediatrics 13:140–145
Hutzler J, Dancis J (1968) Conversion of lysine to saccharopine by human tissues. Biochim Biophys Acta 158:62–69
Hutzler J, Dancis J (1975) Lysine-ketoglutarate reductase in human tissues. Biochim Biophys Acta 377:42–51
Hyland K, Buist NR, Powell BR, Hoffman GF, Rating D, McGrath J, Acworth IN (1995) Folinic acid responsive seizures: a new syndrome? J Inherit Metab Dis 18:177–181
Kantrowitz JT, Malhotra AK, Cornblatt B, Silipo G, Balla A, Suckow RF, D’Souza C, Saksa J, Woods SW, Javitt DC (2010) High dose d-serine in the treatment of schizophrenia. Schizophr Res 121:125–130
Kasé Y, Kataoka M, Miyata T, Okano Y (1973) Pipecolic acid in the dog brain. Life Sci 13:867–873
Kim JS, Giacobini E (1985) Pipecolic acid levels and transport in developing mouse brain. Brain Res 354:181–186
Kocki T, Luchowski P, Luchowska E, Wielosz M, Turski WA, Urbanska EM (2003) l-Cysteine sulphinate, endogenous sulphur-containing amino acid, inhibits rat brain kynurenic acid production via selective interference with kynurenine aminotransferase II. Neurosci Lett 346:97–100
Lee P, Kuhl W, Gelbart T, Kamimura T, West C, Beutler E (1994) Homology between a human protein and a protein of the green garden pea. Genomics 21:371–378
Lee JI, Nian H, Cooper AJL, Sinha R, Dai J, Bisson WH, Dashwood RH, Pinto JT (2009) α-Keto acid metabolites of naturally occurring organoselenium compounds as inhibitors of histone deacetylase in human prostate cancer cells. Cancer Prev Res 2:683–693
Lennon AM (1992) Purification and characterization of rat brain cytosolic 3,5,3′-triiodo-l-thyronine-binding protein. Evidence for binding activity dependent on NADPH, NADP and thioredoxin. Eur J Biochem 210:79–85
Lennon AM, Osty J, Nunez J (1980) Cytosolic thyroxine-binding protein and brain development. Mol Cell Endocrinol 18:201–214
Li RW, Meyer MJ, Van Tassell CP, Sonstegard TS, Connor EE, Van Amburgh ME, Boisclair YR, Capuco AV (2006) Identification of estrogen-responsive genes in the parenchyma and fat pad of the bovine mammary gland by microarray analysis. Physiol Genomics 27:42–53
Lin YC, Huang ZH, Jan IS, Yeh CC, Wu HJ, Chou YC, Chang YC (2002) Development of excitatory synapses in cultured neurons dissociated from the cortices of rat embryos and rat pups at birth. J Neurosci Res 67:484–493
Liu Y, Borchert GL, Surazynski A, Hu CA, Phang JM (2006) Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: the role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene 25:5640–5647
Lu SP, Lewin AH (1998) Enamine/imine tautomerism in α, β-unsaturated α-amino acids. Tetrahedron 54:15097–15104
Madeira C, Freitas ME, Vargas-Lopes C, Wolosker H, Panizzutti R (2008) Increased brain d-amino acid oxidase (DAAO) activity in schizophrenia. Schizophr Res 101:76–83
Malherbe P, Alberati-Giani D, Kohler C, Cesura AM (1995) Identification of a mitochondrial form of kynurenine aminotransferase/glutamine transaminase K from rat brain. FEBS Lett 367:141–144
Malinowska K, Cavarretta IT, Susani M, Wrulich OA, Uberall F, Kenner L, Culig Z (2009) Identification of μ-crystallin as an androgen-regulated gene in human prostate cancer. Prostate 69:1109–1118
Marnett LJ (2002) Oxy radicals, lipid peroxidation and DNA damage. Toxicology 181–182:219–222
Martins-de-Souza D, Gattaz WF, Schmitt A, Maccarrone G, Hunyadi-Gulyas E, Eberlin MN, Souza GH, Marangoni S, Novello JC, Turck CW, Dias-Neto E (2009) Proteomic analysis of dorsolateral prefrontal cortex indicates the involvement of cytoskeleton, oligodendrocyte, energy metabolism and new potential markers in schizophrenia. J Psychiatr Res 43:978–986
Mason M (1954) The kynurenine transaminase of rat kidney. J Biol Chem 211:839–844
Mason JM, Naidu MD, Barcia M, Porti D, Chavan SS, Chu CC (2004) IL-4-induced gene-1 is a leukocyte l-amino acid oxidase with an unusual acidic pH preference and lysosomal localization. J Immunol 173:4561–4567
Matsuda M, Ogur M (1969) Separation and specificity of the yeast glutamate-α-ketoadipate transaminase. J Biol Chem 244:3352–3358
Matsumoto S, Yamamoto S, Sai K, Maruo K, Adachi M, Saitoh M, Nishizaki T (2003) Pipecolic acid induces apoptosis in neuronal cells. Brain Res 980:179–184
Mawal MR, Deshmukh DR (1991) α-Aminoadipate and kynurenine aminotransferase activities from rat kidney. Evidence for separate identity. J Biol Chem 266:2573–2575
Mehler AH, Knox WE (1950) The conversion of tryptophan to kynurenine in liver. II. The enzymatic hydrolysis of formylkynurenine. J Biol Chem 187:431–438
Meister A (1962) ∆1-Piperideine-2-carboxylate and ∆1-Pyrroline-2-carboxylate Reductase. Methods Enzymol 5:878–882
Meister A, Buckley SD (1957) Pyridine nucleotide-dependent reduction of the α-keto acid analog of lysine to l-pipecolic acid. Biochim Biophys Acta 23:202–203
Meister A, Radhakrishnan AN, Buckley SD (1957) Enzymatic synthesis of l-pipecolic acid and l-proline. J Biol Chem 229:789–800
Middleton FA, Mirnics K, Pierri JN, Lewis DA, Levitt P (2002) Gene expression profiling reveals alterations of specific metabolic pathways in schizophrenia. J Neurosci 22:2718–2729
Mihalik SJ, Rhead WJ (1989) l-Pipecolic acid oxidation in the rabbit and cynomolgus monkey. Evidence for differing organellar locations and cofactor requirements in each species. J Biol Chem 264:2509–2517
Mihalik SJ, Rhead WJ (1991) Species variation in organellar location and activity of l-pipecolic acid oxidation in mammals. J Comp Physiol B 160:671–676
Mihalik SJ, Moser HW, Watkins PA, Danks DM, Poulos A, Rhead WJ (1989) Peroxisomal l-pipecolic acid oxidation is deficient in liver from Zellweger syndrome patients. Pediatr Res 25:548–552
Mihalik SJ, McGuinness M, Watkins PA (1991) Purification and characterization of peroxisomal l-pipecolic acid oxidase from monkey liver. J Biol Chem 266:4822–4830
Miles JL, Holmes RS (1975) The ontogeny of l-α-hydroxyacid oxidase isozymes in the mouse. J Exp Zool 192:119–125
Miller IL, Tsuchida M, Adelberg EA (1953) The transamination of kynurenine. J Biol Chem 203:205–211
Mills PB, Struys E, Jakobs C, Plecko B, Baxter P, Baumgartner M, Willemsen MA, Omran H, Tacke U, Uhlenberg B, Weschke B, Clayton PT (2006) Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med 12:307–309
Mills PB, Footitt EJ, Ceyhan S, Waters PJ, Jakobs C, Clayton PT, Struys EA (2012) Urinary AASA excretion is elevated in patients with molybdenum cofactor deficiency and isolated sulphite oxidase deficiency. J Inherit Metab Dis 35:1031–1036
Moreno M, Silvestri E, Lombardi A, Visser TJ, Goglia F, Lanni A (2003) Identification of 3,5-diiodo-l-thyronine-binding proteins in rat liver cytosol by photoaffinity labeling. Endocrinology 144:2297–2303
Mori J, Suzuki S, Kobayashi M, Inagaki T, Komatsu A, Takeda T, Miyamoto T, Ichikawa K, Hashizume K (2002) Nicotinamide adenine dinucleotide phosphate-dependent cytosolic T3 binding protein as a regulator for T3-mediated transactivation. Endocrinology 143:1538–1544
Mukai M, Replogle K, Drnevich J, Wang G, Wacker D, Band M, Clayton DF, Wingfield JC (2009) Seasonal differences of gene expression profiles in song sparrow (Melospiza melodia) hypothalamus in relation to territorial aggression. PLoS One 4(12):e8182
Murthy SN, Janardanasarma MK (1999) Identification of l-amino acid/l-lysine α-amino oxidase in mouse brain. Mol Cell Biochem 197:13–23
Nakano M (1967) Purification and properties of halogenated tyrosine and thyroid hormone transaminase from rat kidney mitochondria. J Biol Chem 242:73–81
Nakano M, Danowski TS (1964) Thyroid-hormone transaminase and oxidase in rat-kidney mitochondria. Biochim Biophys Acta 85:18–28
Nakano M, Tsuchiya S, Danowski TS (1964) Purification of thyroid hormone-transaminase from rat kidney mitochondria. Proc Soc Exp Biol Med 115:16–18
Nakatani Y, Fujioka M, Higashino K (1970) α-Aminoadipate aminotransferase of rat liver mitochondria. Biochim Biophys Acta 198:219–228
Nardini M, Ricci G, Vesci L, Pecci L, Cavallini D (1988a) Bovine brain ketimine reductase. Biochim Biophys Acta 957:286–292
Nardini M, Ricci G, Caccuri AM, Solinas SP, Vesci L, Cavallini D (1988b) Purification and characterization of a ketimine-reducing enzyme. Eur J Biochem 173:689–694
Neuberger A, Sanger F (1943) The availability of the acetyl derivatives of lysine for growth. Biochem J 37:515–518
Nicolai J, van Kranen-Mastenbroek VH, Wevers RA, Hurkx WA, Vles JS (2006) Folinic acid-responsive seizures initially responsive to pyridoxine. Pediatr Neurol 34:164–167
Nishina Y, Sato K, Shiga K (1991) Isomerization of ∆1-piperideine-2-carboxylate to ∆2-piperideine-2-carboxylate on complexation with flavoprotein d-amino acid oxidase. J Biochem 109:705–710
Nishio H, Segawa T (1983) Determination of pipecolic acid in rat brain areas by high-performance liquid chromatography of dansyl derivatives with fluorimetric detection. Anal Biochem 135:312–317
Nomura Y, Schmidt-Glenewinkel T, Giacobini E (1978) In vitro formation of piperidine, cadaverine and pipecolic acid in chick and mouse brain during development. Dev Neurosci 1:239–249
Ogawa T, Kimoto M, Sasaoka K (1990) Dimethylarginine:pyruvate aminotransferase in rats. Purification, properties, and identity with alanine:glyoxylate aminotransferase 2. J Biol Chem 265:20938–20945
Okamoto H, Okada F, Hayaishi O (1971) Kynurenine metabolism in hyperthyroidism. A biochemical basis for the low NAD(P) level in hyperthyroid rat liver. J Biol Chem 246:7759–7763
Okuno E, Tsujimoto M, Nakamura M, Kido R (1993) 2-Aminoadipate-2-oxoglutarate aminotransferase isoenzymes in human liver: a plausible physiological role in lysine and tryptophan metabolism. Enzyme Protein 47:136–148
Olney JW, Newcomer JW, Farber NB (1999) NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res 33:523–533
Papes F, Kemper EL, Cord-Neto G, Langone F, Arruda P (1999) Lysine degradation through the saccharopine pathway in mammals: involvement of both bifunctional and monofunctional lysine-degrading enzymes in mouse. Biochem J 344:555–563
Papes F, Surpili MJ, Langone F, Trigo JR, Arruda P (2001) The essential amino acid lysine acts as precursor of glutamate in the mammalian central nervous system. FEBS Lett 488:34–38
Pappa A, Brown D, Koutalos Y, DeGregori J, White C, Vasiliou V (2005) Human aldehyde dehydrogenase 3A1 inhibits proliferation and promotes survival of human corneal epithelial cells. J Biol Chem 280:27998–28006
Pearce IA, Cambray-Deakin MA, Burgoyne RD (1987) Glutamate acting on NMDA receptors stimulates neurite outgrowth from cerebellar granule cells. FEBS Lett 223:143–147
Pensa B, Achilli M, Fontana M, Caccuri AM, Cavallini D (1989) S-Aminoethyl-l-cysteine transaminase from bovine brain: purification to homogeneity and assay of activity in different regions of the brain. Neurochem Int 15:285–291
Pérez-Llarena FJ, Rodríguez-García A, Enguita FJ, Martín JF, Liras P (1998) The pcd gene encoding piperideine-6-carboxylate dehydrogenase involved in biosynthesis of α-aminoadipic acid is located in the cephamycin cluster of Streptomyces clavuligerus. J Bacteriol 180:4753–4756
Petrakis PL, Greenberg DM (1965) Studies on l-proline:NAD(P)+-oxidoreductase of hog kidney. Biochim Biophys Acta 99:78–95
Plecko B, Paul K, Paschke E, Stoeckler-Ipsiroglu S, Struys E, Jakobs C, Hartmann H, Luecke T, di Capua M, Korenke C, Hikel C, Reutershahn E, Freilinger M, Baumeister F, Bosch F, Erwa W (2007) Biochemical and molecular characterization of 18 patients with pyridoxine-dependent epilepsy and mutations of the antiquitin (ALDH7A1) gene. Hum Mutat 28:19–26
Rao VV, Chang YF (1990) l-Pipecolic acid metabolism in human liver: detection of l-pipecolate oxidase and identification of its reaction product. Biochim Biophys Acta 1038:295–299
Rao VV, Pan X, Chang YF (1992) Developmental changes of l-lysine-ketoglutarate reductase in rat brain and liver. Comp Biochem Physiol B: Comp Biochem 103:221–224
Rao VV, Tsai MJ, Pan X, Chang YF (1993) l-Pipecolic acid oxidation in rat: subcellular localization and developmental study. Biochim Biophys Acta 1164:29–35
Reed PW, Corse AM, Porter NC, Flanigan KM, Bloch RJ (2007) Abnormal expression of μ-crystallin in facioscapulohumeral muscular dystrophy. Exp Neurol 205:583–586
Ricci G, Nardini M, Caccuri AM, Federici G (1983) Interaction between 1,4-thiazine derivatives and d-amino-acid oxidase. Biochim Biophys Acta 748:40–47
Rothstein M, Miller LL (1954) The conversion of lysine to pipecolic acid in the rat. J Biol Chem 211:851–858
Sacksteder KA, Biery BJ, Morrell JC, Goodman BK, Geisbrecht BV, Cox RP, Gould SJ, Geraghty MT (2000) Identification of the α-aminoadipic semialdehyde synthase gene, which is defective in familial hyperlysinemia. Am J Hum Genet 66:1736–1743
Sauer SW, Opp S, Hoffmann GF, Koeller DM, Okun JG, Kolker S (2011) Therapeutic modulation of cerebral l-lysine metabolism in a mouse model for glutaric aciduria type I. Brain 134:157–170
Scannone H, Wellner D, Novogrodsky A (1964) A study of amino acid oxidase specificity, using a new sensitive assay. Biochemistry 3:1742–1745
Schwarcz R, Rassoulpour A, Wu HQ, Medoff D, Tamminga CA, Roberts RC (2001) Increased cortical kynurenate content in schizophrenia. Biol Psychiatry 50:521–530
Schwartz C, Hampton M, Andrews MT (2013) Seasonal and regional differences in gene expression in the brain of a hibernating mammal. PLoS One 8(3):e58427
Segovia L, Horwitz J, Gasser R, Wistow G (1997) Two roles for μ-crystallin: a lens structural protein in diurnal marsupials and a possible enzyme in mammalian retinas. Mol Vis 3:9
Shirayama Y, Chaki S (2006) Neurochemistry of the nucleus accumbens and its relevance to depression and antidepressant action in rodents. Curr Neuropharmacol 4:277–291
Sivagnanasundaram S, Crossett B, Dedova I, Cordwell S, Matsumoto I (2007) Abnormal pathways in the genu of the corpus callosum in schizophrenia pathogenesis: a proteome study. Proteomics Clin Appl 1:1291–1305
Snyder SH, Kim PM (2000) d-Amino acids as putative neurotransmitters: focus on d-serine. Neurochem Res 25:553–560
Soda K, Misono H, Yamamoto T (1968) l-Lysine:α-ketoglutarate aminotransferase. I. Identification of a product, ∆1-piperideine-6-carboxylic acid. Biochemistry 7:4102–4109
Stone TW (1993) Neuropharmacology of quinolinic and kynurenic acids. Pharmacol Rev 45:309–379
Strait KA, Schwartz HL, Perez-Castillo A, Oppenheimer JH (1990) Relationship of c-erbA mRNA content to tissue triiodothyronine nuclear binding capacity and function in developing and adult rats. J Biol Chem 265:10514–10521
Struys EA, Jakobs C (2010) Metabolism of lysine in α-aminoadipic semialdehyde dehydrogenase-deficient fibroblasts: evidence for an alternative pathway of pipecolic acid formation. FEBS Lett 584:181–186
Suzuki S, Hashizume K, Ichikawa K, Takeda T (1991) Ontogenesis of the high affinity NADPH-dependent cytosolic 3,5,3′-triiodo-l-thyronine-binding protein in rat. Endocrinology 129:2571–2574
Takeuchi F, Otsuka H, Shibata Y (1983) Purification, characterization and identification of rat liver mitochondrial kynurenine aminotransferase with α-aminoadipate aminotransferase. Biochim Biophys Acta 743:323–330
Tamburin M, Mostardini M, Benatti L (1999) Kynurenine aminotransferase I (KATI) isoform gene expression in the rat brain: an in situ hybridization study. NeuroReport 10:61–65
Tang WK, Cheng CH, Fong WP (2002) First purification of the antiquitin protein and demonstration of its enzymatic activity. FEBS Lett 516:183–186
Tebbenkamp AT, Borchelt DR (2010) Analysis of chaperone mRNA expression in the adult mouse brain by meta analysis of the Allen Brain Atlas. PLoS One 5:e13675
Tobes MC, Mason M (1975) l-kynurenine aminotransferase and l-α-aminoadipate aminotransferase. I. Evidence for identity. Biochem Biophys Res Commun 62:390–397
Tobes MC, Mason M (1977) α-Aminoadipate aminotransferase and kynurenine aminotransferase. Purification, characterization, and further evidence for identity. J Biol Chem 252:4591–4599
Tobes MC, Mason M (1978) Kynurenine aminotransferase and α-aminoadipate aminotransferase: III evidence for identity with hologenated tyrosine aminotransferase. Life Sci 22:793–802
Tsai MJ, Chang YF, Schwarcz R, Brookes N (1996) Characterization of l-α-aminoadipic acid transport in cultured rat astrocytes. Brain Res 741:166–173
Tsai G, Yang P, Chung LC, Lange N, Coyle JT (1998) d-Serine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry 44:1081–1089
Urban P, Chirat I, Lederer F (1988) Rat kidney l-2-hydroxyacid oxidase. Structural and mechanistic comparison with flavocytochrome b2 from baker’s yeast. Biochemistry 27:7365–7371
Valle D, Downing SJ, Phang JM (1973) Proline inhibition of pyrroline-5-carboxylate reductase: differences in enzymes obtained from animal and tissue culture sources. Biochem Biophys Res Commun 54:1418–1424
Versmold HT, Bremer HJ, Herzog V, Siegel G, Bassewitz DB, Irle U, Voss H, Lombeck I, Brauser B (1977) A metabolic disorder similar to Zellweger syndrome with hepatic acatalasia and absence of peroxisomes, altered content and redox state of cytochromes, and infantile cirrhosis with hemosiderosis. Eur J Pediatr 124:261–275
Vie MP, Evrard C, Osty J, Breton-Gilet A, Blanchet P, Pomerance M, Rouget P, Francon J, Blondeau JP (1997) Purification, molecular cloning, and functional expression of the human nicotinamide-adenine dinucleotide phosphate-regulated thyroid hormone-binding protein. Mol Endocrinol 11:1728–1736
Walker V, Mills GA, Peters SA, Merton WL (2000) Fits, pyridoxine, and hyperprolinaemia type II. Arch Dis Child 82:236–237
Walter IB (1996) Triiodothyronine exerts a trophic action on rat sensory neuron survival and neurite outgrowth through different pathways. Eur J Neurosci 8:455–466
Wanders RJ, Romeyn GJ, van Roermund CW, Schutgens RB, van den Bosch H, Tager JM (1988) Identification of l-pipecolate oxidase in human liver and its deficiency in the Zellweger syndrome. Biochem Biophys Res Commun 154:33–38
Wanders RJ, Romeyn GJ, Schutgens RB, Tager JM (1989) l-Pipecolate oxidase: a distinct peroxisomal enzyme in man. Biochem Biophys Res Commun 164:550–555
Wass C, Klamer D, Katsarogiannis E, Palsson E, Svensson L, Fejgin K, Bogren IB, Engel JA, Rembeck B (2011) l-Lysine as adjunctive treatment in patients with schizophrenia: a single-blinded, randomized, cross-over pilot study. BMC Med 9:40
Weissman N, Schoenheimer RJ (1941) The relative stability of l(+)-lysine in rats studied with deuterium and heavy nitrogen. J Biol Chem 140:779–795
Wiemann S, Kolb-Kokocinski A, Poustka A (2005) Alternative pre-mRNA processing regulates cell-type specific expression of the IL4l1 and NUP62 genes. BMC Biol 3:16
Wong JW, Chan CL, Tang WK, Cheng CH, Fong WP (2010) Is antiquitin a mitochondrial Enzyme? J Cell Biochem 109(1):74–81
Woody NC, Pupene MB (1970) Excretion of pipecolic acid by infants and by patients with hyperlysinemia. Pediatr Res 4:89–95
Yamauchi K, Tata JR (1994) Purification and characterization of a cytosolic thyroid-hormone-binding protein (CTBP) in Xenopus liver. Eur J Biochem 225:1105–1112
Yang Y, Cauley RK, Zhao W, Chang JD, Chang CK, Chang YF (1995) Demographic study of old age and Alzheimer’s disease on enzymes forming kynurenate and aminoadipate. Amino Acids 9:40 (Abstract)
Yeh GC, Harris SC, Phang JM (1981) Pyrroline-5-carboxylate reductase in human erythrocytes. J Clin Invest 67:1042–1046
Yu P, Li Z, Zhang L, Tagle DA, Cai T (2006) Characterization of kynurenine aminotransferase III, a novel member of a phylogenetically conserved KAT family. Gene 365:111–118
Zaar K, Angermüller S, Völkl A, Fahimi HD (1986) Pipecolic acid is oxidized by renal and hepatic peroxisomes. Implications for Zellweger’s cerebro-hepato-renal syndrome (CHRS). Exp Cell Res 164:267–271
Zhou J, Weiner H (1997) Binding of thyroxine analogs to human liver aldehyde dehydrogenases. Eur J Biochem 245:123–128
Acknowledgments
Part of the work cited from the authors’ laboratories was supported by NIH grants RO1 ES8421 (to AJLC), and Macquarie University Research Excellence Scholarship (to AH). We thank Dr. John Pinto for determining whether KAT I, KAT II or KAT III can catalyze transamination between l-lysine and α-keto-γ-methiolbutyrate. We also thank Dr. Jianyong Li (Virginia Tech) for the generous gift of the three KAT enzymes.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hallen, A., Jamie, J.F. & Cooper, A.J.L. Lysine metabolism in mammalian brain: an update on the importance of recent discoveries. Amino Acids 45, 1249–1272 (2013). https://doi.org/10.1007/s00726-013-1590-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-013-1590-1