
Abstract
Alkylations of chiral or achiral Ni(II) complexes of glycine Schiff bases constitute a landmark in the development of practical methodology for asymmetric synthesis of α-amino acids. Straightforward, easy preparation as well as high reactivity of these Ni(II) complexes render them ready available and inexpensive glycine equivalents for preparing a wide variety of α-amino acids, in particular on a relatively large scale. In the case of Ni(II) complexes containing benzylproline moiety as a chiral auxiliary, their alkylation proceeds with high thermodynamically controlled diastereoselectivity. Similar type of Ni(II) complexes derived from alanine can also be used for alkylation providing convenient access to quaternary, α,α-disubstituted α-amino acids. Achiral type of Ni(II) complexes can be prepared from picolinic acid or via recently developed modular approach using simple secondary or primary amines. These Ni(II) complexes can be easily mono/bis-alkylated under homogeneous or phase-transfer catalysis conditions. Origin of diastereo-/enantioselectivity in the alkylations reactions, aspects of practicality, generality and limitations of this methodology is critically discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The asymmetric synthesis of α-amino acids (α-AAs) continues to be a subject of intense current research interest and ever increasing technological importance in major areas of chemical industries dealing with health care and food production. In particular, pharmaceutical and agrochemical industries heavily depend on the progress in amino acids availability and large-scale production. It would not be overstatement to say that the price of pharmaceuticals and the rate of the development of new drug candidates practically reflect the current methodological state in the area of asymmetric synthesis of amino acids (Izumi et al. 1978; Coppola and Schuster 1987).
Although a significant progress in this area has been already achieved (Duthaler 1994; Maruoka and Ooi 2003; Ma 2003; Nájera and Sansano 2007; Soloshonok and Sorochinsky 2010; Aceña et al. 2012), a general and cost-effective access to structurally varied α-AAs, on relatively large scale, has not been reached thus far. To name just a few of the most relevant synthetic strategies, the asymmetric hydrogenation of α,β-dehydro-α-amino acid derivatives (Etayo and Vidal-Ferrán 2013) or the Strecker reaction (Wang et al. 2011a) has produced outstanding results in the last decades. However, both methods cannot be successfully applied to the synthesis of all types of α-AAs.
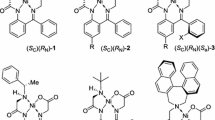
In this respect, the alkylation of nucleophilic equivalents of glycine and higher amino acids is probably the most practical and direct method for the synthesis of structurally varied and/or conformationally restricted, sterically constrained α-AAs. One of the most useful types of achiral nucleophilic glycine equivalents is imines derived from glycine esters 1 and 2 (Fig. 1) introduced for homologation of glycine into various higher amino acids (Stork et al. 1976). This area was revolutionized by O’Donnell who demonstrated that imine 3, derived from benzophenone and tert-butyl ester of glycine, can be alkylated with high enantioselectivity using cinchona alkaloids-derived phase-transfer catalysts (O’Donnell and Eckrich 1978; O’Donnell et al. 1988, 1989; O’Donnell 2001, 2004). Subsequent work by many research groups (Corey et al. 1997; Chinchilla et al. 2000; Park et al. 2002; Shibuguchi et al. 2002), most notably by Lygo (Lygo and Wainwright 1997; Lygo et al. 2001, 2002, Lygo and Andrews 2004) and Maruoka (Ooi et al. 1999, 2003; Hashimoto and Maruoka 2007; Ooi and Maruoka 2007; Shirakawa and Maruoka 2013) have expanded the synthetic utility of this glycine equivalent through the design of new and more efficient catalysts, mostly quaternary ammonium salts, leading to enantioselectivities usually higher than 99 %. One problem still associated with this methodology is the inherent hydrolytical instability of Schiff base and products of its homologation, resulting in incomplete chemical yields.

Achiral nucleophilic glycine equivalents
Another milestone in the asymmetric synthesis of α-AAs is the application of chiral derivatives of glycine. The most remarkable examples are Schöllkopf bislactims 4 (Schöllkopf et al. 1981; Schöllkopf 1983a, b; Undheim 2008), Williams diarylmorpholinones 5 (Sinclair et al. 1986; Williams et al. 1988; Williams and Im 1991; Sebahar and Williams 2000) and Seebach imidazo- and oxazolidinones 6 (Fitzi and Seebach 1988; Seebach et al. 1996) (Fig. 2). In these cyclic templates, one of the faces of the corresponding in situ prepared enolate is effectively sterically shielded providing for virtually complete stereoselectivity. Acyclic nucleophilic glycine equivalents, such as Myers glycinamide 7 (Myers et al. 1997) and Evans oxazolidinone 8 (Evans and Weber 1986), have also been successfully applied for the synthesis of α-amino acids. However, the main disadvantage of compounds 4–8 is the low C–H acidity of the glycine methylene moiety, which requires the use of strong bases, such as n-BuLi, low temperatures, usually −78 °C, and anhydrous conditions to form the corresponding enolates. Furthermore, preparation of these derivatives requires multi-step sequences, rendering them relatively expensive, in particular for application on a large scale.

Chiral nucleophilic glycine equivalents
Transition metal complexes derived from glycine Schiff bases constitute a convenient template for an easy functionalization of the glycine moiety. Belokon et al. (1983) described Cu(II) complexes of glycine and other amino acids using (S)-2-[N-(N′-benzylprolyl)amino]-benzophenone (BPB, (S)-11) as chiral ligand and studied their reactivity, in particular aldol addition reactions. Further development of this approach revealed that the corresponding Ni(II) complex (S)-9 is more practical due to its higher stability and overall reactivity (Belokon et al. 1985a). In addition, the paramagnetic character of the Cu(II) complexes impedes their analysis using NMR techniques. The most synthetically appealing features of Ni(II) complex (S)-9 include its easy preparation from available and inexpensive starting materials and generally highly diastereoselective functionalization of the glycine moiety. In particular, the homologation of the glycine complex (S)-9 can be conducted via the following major types of reactions: alkyl halide alkylations, the subject of this review, Michael (Soloshonok et al. 1999, 2000a, b, 2005a), aldol (Soloshonok et al. 1993, 1995, 1996a, b) and Mannich (Soloshonok et al. 1997) addition reactions (Scheme 1). All these reactions can be conducted under operationally convenient reaction conditions (Boyall et al. 2002; Soloshonok and Berbasov 2004; Moore et al. 2005; Soloshonok et al. 2006a, b; Yasumoto et al. 2007), at ambient temperature or moderate heating, commercial grade solvents and, in most of cases, just in the open air. The decomposition or disassembly of the homologation products 10 is also operationally convenient, requiring heating of products 10 in a mixture of methanol and 3 N HCl. Finally, isolation of the target amino acids can be achieved using in situ derivatization or by use of ion-exchange chromatography on Dowex-type resins. It is very important to emphasize that at the disassembly step, the chiral ligand (S)-11 can be quantitatively recovered and, in most cases, reused to produce new portions of the glycine complex (S)-9.

Strategies for the homologation of the glycine complex (S)-9
Complex (S)-9 is a red, crystalline compound and can be purified by crystallization or chromatography on SiO2. Crystal structure analysis of (S)-9 reveals that the complex is square planar, neutral with two positive charges at the central Ni ion neutralized by two negative charges (CON− and COO−) of the tetradentate ligand (Kožíšek et al. 2004). Crystallographic data and theoretical calculations suggest that in one of the low-energy conformations, the benzyl group is located above the metal in the coordination plane. It is assumed that possible weak interaction between the Ni atom and the benzyl group can additionally stabilize the conformation of the complex in which the benzyl group is fixed above the central Ni atom and reduces the distance between the plane of the benzyl group and the nickel atom (Popkov et al. 2003). Consequently, much work has been focused on the preparation of analogs of (S)-9 at the benzyl moiety with the aim of improving the diastereoselective outcome of the alkylation reactions.
A second type of Ni(II) complexes derived from glycine are based on achiral ligands. For instance, Belokon et al. (1997) and Soloshonok et al. reported the preparation of Ni(II) complexes 12 and 13 derived from 2-(N-(α-picolyl)amino)benzophenone (PABP) and -acetophenone, respectively (Fig. 3). Complexes 12 and, in particular, 13 were found to be excellent achiral glycine equivalents in the Michael addition reactions with chiral oxazolidinone-derived Michael acceptors (Soloshonok et al. 2000c, d, 2004; Cai et al. 2004), providing for very efficient synthesis of various sterically constrained β-substituted amino acids (Qiu et al. 2001; Soloshonok 2002) used for total synthesis of oxazolomicine antibiotics (Yamada et al. 2008) and new thalidomide derivatives (Yamada et al. 2006; Soloshonok et al. 2009a). Another type of achiral Ni(II) complexes 14 and 15 was recently introduced by Soloshonok et al. (2005b, c, 2009b). The major advantage of complexes 14 and 15 is their easy modular assembly and structural flexibility allowing to tailor virtually any desired reactivity and physicochemical properties. In addition, the cost of preparing complexes 14 and 15 is much lower compared to those derived from N-benzylproline or picolinic acid. Both types of achiral complexes 12–15 are suitable for diastereo- or enantioselective homologation reactions, using chiral reagents or catalysts, respectively.

Achiral Ni(II) complexes of glycine Schiff bases
Discussing general features of Ni(II) complexes 9, 12–15 reactivity, it should be mentioned that when their synthesis or their homologation is conducted under strongly basic conditions and in the presence of oxygen, one unusual oxidative reaction takes place (Scheme 2). It is assumed that in situ generated enolate react with molecular oxygen to trigger a cascade of transformations resulting in the formation of binuclear Ni(II) complexes 16 (Soloshonok and Ueki 2007). These products possess an inherent helical chirality and their chiroptical properties can be applied in the area of chiral nano-technology (Soloshonok et al. 2010).

Synthesis of binuclear Ni(II) complex with inherent helical chirality
Despite the large number of contributions reported thus far on the synthetic applications of Ni(II) complexes of glycine and other α-AAs, it is surprising the lack of review articles covering this important area. In fact, the only general review published by Belokon (1992) was a result of a symposium presentation. Later on, Soloshonok reported the progress in Michael additions to Ni(II) complexes of glycine (Soloshonok 2002; Soloshonok et al. 2009b), and very recently Popkov accounted for their applications in the synthesis of radiolabelled compounds (Popkov and De Spiegeleer 2012). Accordingly, the general aim of this review is to cover the most direct homologation reactions on these glycine equivalents, i.e. alkylations, providing a comprehensive outlook of this important transformation leading to α-AAs after disassembling the corresponding alkylated Ni(II) complexes. The specific aspects shown are the synthesis of the starting nickel complexes, their differences in reactivity and the differences in alkylation conditions (homogeneous vs. heterogeneous), as well as those topics related with the diastereo- and/or enantioselectivity of the reactions.
Synthesis of Ni(II) complexes of Schiff’s bases derived from o-amino-benzo-/aceto-phenones and α-amino acids
The Ni(II) complexes of the Schiff bases derived from (S)-2-[N-(N′-benzylprolyl)amino]benzophenone (BPB) (S)-11 and glycine or higher α-amino acids can be prepared on large scale by a simple sequence of reactions starting from N-benzylproline (BP) (S)-17 (Scheme 3). In the original procedure published by Belokon et al. (1988, 1998) (S)-17 is converted in situ to the corresponding acyl chloride using excess of SOCl2 at low temperature followed by the reaction with o-aminobenzophenone to afford the ligand (S)-11 in 81 % yield. However, the use of almost 50 % excess of (S)-17 on the amidation stage renders this procedure unattractive for large-scale preparation of the target Ni(II) complexes. An improved procedure, described by Soloshonok et al. included the in situ preparation of a mixed anhydride by addition of MsCl and N-MeIm to (S)-17 in a CH2Cl2 solution. The reaction of thus prepared mixed anhydride with o-aminobenzophenone was completed at 40–50 °C affording higher than 90 % conversion of the phenone to the target ligand (S)-11 which was isolated in 94 % yield (Ueki et al. 2003a). The ligand (S)-11 can be purified by crystallization of the crude product from ethanol as well as by precipitation of the corresponding hydrochloride salt from a solution of the reaction mixture in acetone. Further formation of the Ni(II) complexes (S)-9 and (S,S)-18a–f easily takes place on heating (S)-11 in methanol in the presence of base, the corresponding α-amino acids and a source of Ni(II) ions (Belokon et al. 1988, 1998; Nádvorník and Popkov 2002; Ueki et al. 2003a; Soloshonok et al. 2008a; Nádvorník et al. 2008). Excess of aliphatic α-amino acids and Ni(NO3)2·6H2O was used in a standard protocol in order to obtain Ni(II) complexes (S)-9 and (S,S)-18a–c in >95 % yields. Using near stoichiometric amounts of nickel salt and aromatic α-amino acids led to lower yields of the Ni(II) complexes (S,S)-18d–f. In the case of the Ni(II) complex (S,S)-18a the use of enantiomerically pure (S)- or racemic (R/S)-alanine had no influence on the final diastereomeric purity (>90 %) of the isolated complex. Since the complex was synthesized in highly alkaline media at elevated temperatures, racemization of the amino acid, via enolate formation, proceeds fast and the ratio of the diastereomeric complexes reflects the thermodynamic equilibrium between them. The Ni(II) complexes (S,S)-18b,c containing bulkier substituents were obtained in diastereomerically pure form while the diastereomeric excess of (S,S)-18d–f in all cases was >95 %. It should be noted that the N-atom of the proline residue is chiral; however, it creates no problem in terms of possible diastereomers formation as the absolute configuration of the N-stereogenic center is completely controlled by the proline’s C-chirality.

Synthesis of Ni(II) complexes of Schiff bases derived from 2-[N-(N′-benzylprolyl)amino]benzophenone (S)-11 and α-amino acids
These methods were also successfully used for preparing Ni complexes 19–27 (De and Thomas 1997; Ueki et al. 2003a; Belokon et al. 2002; Saghiyan et al. 2006, 2010; Popkov et al. 2002), containing ligands with various substituents on the o-amino-benzo/aceto-phenone moiety as well as the proline chiral auxiliary (Fig. 4).

Modified chiral Ni(II) complexes of Schiff bases of α-amino acids
Ni(II) glycine-derived achiral complexes 12 and 13 as well as alanine-derived racemic complex 30 were prepared by condensation of the acid chloride derived from picolinic acid 28 with o-amino-benzo/aceto-phenones followed by heating ligands 29 with glycine or (S/R)-alanine and Ni(NO3)2 in the presence of KOH or MeONa in methanol (Belokon et al. 1997, 2001, 2003a) (Scheme 4). The structures of complexes 12, 13 and 30 were confirmed by X-ray analysis. It should be noted that application of excess of SOCl2 for the preparation of the acid chloride of 28 led to substantial formation of byproducts and laborious purification of ligands 29. On the other hand, when activation of 28 was carried out by the mixed anhydride method using ethyl chloroformate, complete conversion of o-aminobenzophenone as well as o-aminoacetophenone was observed during the condensation step. The amidation proceeded smoothly giving rise, in quantitative chemical yield, to the target ligands 29, which could be used without any additional purification for preparing the corresponding Ni(II) complexes 12, 13 and 30 (Ellis et al. 2003a; Ueki et al. 2003b).

Synthesis of the Ni(II)-complexes of glycine and alanine Schiff bases with 2-[N-(α-picolyl)amino]aceto/benzophenones
On the basis of the mixed anhydride method, a one-pot, two-step protocol, which did not require isolation of ligand 29a as a separate step, has been developed for preparation of Ni(II) complexes derived from picolinic acid (Deng et al. 2007). Optimization of the one-pot procedure conditions using sterically hindered isobutyl chloroformate, mixed bases NaH/KOH and THF as solvent allowed to obtain the Ni(II) complexes 12, 30 and 31 in 84–98 % yield (Scheme 5).

One-pot synthesis of Ni(II) complexes derived from picolinic acid
Most recently, the need for a more flexible methodology and inexpensive achiral Ni(II) complexes led to the development of the new generation of achiral glycine equivalents 14 (Ellis and Soloshonok 2006; Ellis et al. 2006) and 15 (Scheme 6). The synthesis of glycine derivatives 14 and 15 is based on the combination of three different modules: phenone module, acid module 32, and amine module 34. The nature of these modules allowed for a rational control of geometry, physical properties and reactivity of derivatives 14 and 15. Further advantage of this new generation of ligands/Ni(II)-complexes is that the reaction of phenones and commercially available bromoacetyl bromide 32 was usually accomplished in MeCN solution in the presence of K2CO3 furnishing acetamide intermediate 33 with high chemical purity. Next, the reaction of 33 with secondary or primary amines 34 afforded ligands 35 and 36. This reaction was also accomplished in MeCN solution with K2CO3. Formation of the Ni(II) complexes 14 and 15 readily took place on moderate heating of MeOH solution of ligands 35 and 36, glycine and Ni(II) in the presence of KOH. Potassium hydroxide was utilized to catalyze the imine formation as well as to neutralize the corresponding acid formed from the reaction. All three synthetic steps usually proceeded with excellent yields (>95 %) and purification of the intermediate products was not required. This new generation approach has a very attractive cost structure as the average cost of these complexes is less than $1 per gram. To illustrate how easily the physicochemical properties, in particular solubility, of the corresponding Ni(II) complexes can be rationally controlled one can consider a series of derivatives 14a–d. Thus, the complex 14a is soluble in DMF or chlorinated solvents such as CHCl3 and CH2Cl2. Piperidine-derived 14b can be dissolved in acetone or methanol, while the N,N-di-n-butylamine derivative 14c can be used for phase-transfer reactions using benzene and toluene. Finally, N,N-di-n-octylamine containing complex 14d is soluble in n-hexane. In principle, this approach allows preparation of the corresponding Ni(II) complexes soluble in virtually any known solvents including water or a perfluorinated (fluorous) phase.

Synthesis of new generation of nucleophilic glycine equivalents
Besides the synthesis of amino acids via homologation of the corresponding glycine derivatives, this type of new generation of glycine equivalents, containing residues of chiral amines, can also be used for deracemization and (S) to (R) interconversion of higher amino acids (Soloshonok et al. 2009c; Sorochinsky et al. 2013).
Alkylation of the Ni(II) complexes of the Schiff bases of (S)-2-[N-(N′-benzylprolyl)-amino]benzophenone and α-amino acids
The diastereoselective α-alkylation of chiral Ni(II) complex (S)-9 with alkyl halides is based on the high acidity of the α-protons. In this context, the corresponding enolate can undergo alkylation reactions under different operationally convenient basic conditions and in high chemical yields. When the enolate was generated using n-BuLi in THF at −70 °C followed by treatment with alkyl halides under kinetically controlled conditions the alkylation reactions afforded the corresponding diastereomeric Ni(II) complexes 18 with good chemical yields and 41–42 % diastereoisomeric excess (Belokon et al. 1988) (Scheme 7). Thus, the kinetic stereoselectivity for the monoalkylation is not very high and in the case of sterically small electrophiles is not favored at all. The (S) absolute configuration of the N-benzylproline residue in (S)-9 induced the (S) stereochemistry of the newly formed stereogenic center in the major diastereomers. Alkylation of Ni(II) complex (S)-9 is usually conducted in aprotic solvents such as DMF or MeCN in the presence of NaOH at ambient temperature affording diastereomeric alkylation products which under the experimental conditions undergo α-epimerization leading to thermodynamic control. Thus, in the presence of 2.5 equivalents of NaOH all alkyl halides tested gave thermodynamically favored monoalkylated complex with (S)-amino acid residue in up to 98 % de and with yields in the 70–96 % range (Belokon et al. 1988; Collet et al. 1998, 2000; Gu et al. 2002; Le Chevalier Isaad et al. 2008; Kawamoto et al. 2012). While activated benzyl, allyl and propargyl halides can be used as both bromides and chlorides, introduction of non-activated alkyl groups required application of the corresponding bromides or iodides. In order to avoid dialkylation, all active halides were used in slightly less than 1 equiv. Enantiomerically pure α-amino acids with different side chains can be obtained after separation of alkylated diastereoisomeric complexes by fractional recrystallization or column chromatography on SiO2. Disassembly of the diastereomerically pure alkylation products is usually conducted via hydrolysis with aqueous HCl in MeOH and isolation of the target amino acids by ion-exchange chromatography. The Ni(II) ions remain on the column and can be regenerated with 1 N HCl solution. This procedure also allowed recovery of the chiral ligand (S)-11 which can be reused for the preparation of starting glycine complex (S)-9.

Alkylation of the glycine Ni(II) complex (S)-9
Successful application of asymmetric alkylation reactions between the glycine equivalent (S)-9 and ω-trifluoromethyl alkyl iodides 37 allowed for an efficient access to enantiomerically pure, linear ω-(trifluoromethyl)-containing α-amino acids (Wang et al. 2011b) (Scheme 8). After optimization of bases, solvents and reaction temperature, the alkylation was performed in the presence of NaOH at ambient temperature in DMF for half an hour to afford the alkylation adducts (S,S)-38 with high diastereoselectivity. However, the chemical yields of Ni(II) complexes gradually decreased with the alkyl chain length, indicating the increasing electronic effect of the trifluoromethyl group (Soloshonok et al. 1994a, b). The standard procedure for hydrolysis was performed by heating (S,S)-38c in methanol/6 N HCl to afford 2-amino-6,6,6-trifluorohexanoic acid (S)-39 in 96 % yield.

Alkylation of the glycine Ni(II) complex (S)-9 with ω-trifluoromethyl alkyl iodides 37
Alkylation of chiral glycine equivalent (S)-9 with (3-trifluoromethyl)phenyldiazirinyl bromides 40 has been developed for efficient synthesis of photoreactive l-phenylalanine derivatives (Fishwick et al. 1994; Hashimoto et al. 2002) (Scheme 9). The reactions proceeded in the presence of powdered NaOH in MeCN at room temperature with virtually complete diastereoselectivity to afford alkylated Ni(II) complexes (S,S)-41 in high yield. Hydrolysis of the Ni(II) complexes (S,S)-41a,b occurred rapidly in refluxing HCl/MeOH, without destruction of the diazirine ring, to give the amino acids (S)-42 after cation-exchange chromatography. On the other hand treatment of the Ni(II) complexes (S,S)-41c,d with 50 % TFA–CH2Cl2 at room temperature selectively deprotected the N-Boc groups affording the amino containing Ni(II) complexes (S,S)-43. Subsequent reactions of (S,S)-43 with FmocOSu and biotin N-hydroxysuccinimide under standard conditions afforded N-protected Ni(II) complexes (S,S)-44 and (S,S)-45. Finally, acidic hydrolysis allowed quantitative disassembly of the Ni(II) complexes (S,S)-44 and (S,S)-45 giving rise to Fmoc-protected and biotinylated amino acid derivatives (S)-46 and (S)-47, respectively. These amino acids are suitably protected for incorporation into bioactive peptides to investigate their properties and functions.

Synthesis of 3-(trifluoromethyl)phenyldiazirine based alanine derivatives
An approach to 3-(trans-2-nitrocyclopropyl)alanine (2S,1′R,2′S)-50 was developed based on the alkylation of the Ni(II) complex (S)-9 with enantiomerically enriched trans 1-(iodomethyl)-2-nitrocyclopropane (1S,2S)-48. The fast and highly diastereoselective alkylation of (S)-9 in a DMF/MeCN mixture using NaH as a base yielded the alkylation product (S,S)-49 as a single diastereomer (Larionov et al. 2003; Zlatopolskiy et al. 2004) (Scheme 10). Only trace amounts of other diastereomers of 49 were observed by 1H NMR analysis of the reaction mixture. The Ni(II) complex (S,S)-49 was decomposed by treatment with 6 N HCl to give the amino acid (2S,1′R,2′S)-50 with 95 % ee and 67 % yield after ion-exchange chromatography and crystallization. Following this protocol the deuterated compound (S)-[D]2-9, which was prepared by a hydrogen/deuterium exchange with a very high degree of deuteration (>97 % of the incorporation of two deuterium atoms per molecule), was alkylated with the racemic iodide 48 in a mixture of CD3CN and DMF to give deuterated Ni(II) complex (S)-[D2]-49 in 70 % yield. The subsequent hydrolysis of the Ni(II) complex (S)-[D2]-49 gave the corresponding deuterated amino acid (2S)-[D2]-50 isolated in 60 % yield by ion-exchange chromatography.

Synthesis of 3-(trans-2-nitrocyclopropyl)alanine (2S,1′R,2′S)-50 and the corresponding deuterated amino acid (2S)-[D2]-50
A simple synthesis of the potent arginase inhibitor 2-amino-6-boronohexanoic acid (S)-53 included alkylation of the Ni(II) complex (S)-9 with pinacol 4-bromobutylboronate 51 (Vadon-Legoff et al. 2005) (Scheme 11). When the reaction was performed using 3.0 equiv of t-BuOK as a base in THF at room temperature alkylated Ni(II) complex (S,S)-52 was formed with 80 % de and 83 % yield. Application of other solvents or bases resulted in lower yields and stereochemical outcome of alkylated Ni(II) complex (S,S)-52. Further recrystallization and hydrolysis of the alkylated Ni(II) complex (S,S)-52 by heating in HCI/MeOH with simultaneous deprotection of the boronic acid led to 2-amino-6-boronohexanoic acid (S)-53 with 97 % ee.

Synthesis of 2-amino-6-boronohexanoic acid (S)-53
Taking into account the biological importance of phosphorus-functionalized amino acids (Kukhar et al. 1994), a highly selective procedure for the synthesis of ω-phosphino- and phosphono-α-amino acids through the alkylation of the Ni(II) complex (S)-9 has been developed. Alkylation of (S)-9 with the halogenoalkylphosphinate 54a and halogenoalkylphosphonates 54b,c in MeCN solution at ambient temperature in the presence of powdered KOH favored the formation of (S,S)-diastereoisomers 55a–c in a 86–90 % de (Soloshonok et al. 1992a, b) (Scheme 12). On the other hand alkylation of the Ni(II) complex (S)-9 with diisopropyl iodomethylphosphonate 54d gave the corresponding diastereoisomerically pure complex (S,S)-55d. Decomposition of complexes (S,S)-55 under mild conditions with 2 N HCl furnished amino acids (S)-56, which had free carboxyl groups and esterified phosphinic and phosphonic groups. Enantiomerically pure α-amino-ω-phosphinic and -phosphonic acids (S)-57 can be obtained from the corresponding esters after hydrolysis with HCl under reflux and treatment with propylene oxide.

Synthesis of α-amino-ω-phosphinic- and phosphonic acids (S)-57
Alkylation of the Ni(II) complex (S)-9 with the phosphine-containing reagents 58 and 59, followed by sulfurization in the case of 55, afforded alkylated Ni(II) complexes (S,S)-60 and (S,S)-61 as single diastereomers (Burck et al. 2009) (Scheme 13). After column chromatography the complexes (S,S)-60 and (S,S)-61 were isolated as red powders in yields of about 90 %. The (S) configuration at the α-carbon atom of the glycine fragment was confirmed for both products by X-ray analysis. Ni(II) complexes (S,S)-60 and (S,S)-61 were hydrolyzed by heating in a mixture of MeOH and 2 M HCl and after simple work-up, including the recovery of the free ligand (S)-11, methyl esters (S)-62 and (S)-63 were isolated in 93 and 82 % yield, respectively.

Synthesis of phosphine substituted amino acid derivatives (S)-62 and (S)-63
Alkylation of the Ni(II) complex (S)-9 conducted under thermodynamically controlled conditions at room temperature in the presence of KOH or NaOH was efficiently carried out with sterically hindered 2′,2′-dimethylbenzyl halides 64 (Tang et al. 2000; Soloshonok, et al. 2001a). The benzylation occurred at temperature from 25 to 50 °C furnishing products (S,S)-65 in 95 % chemical yield and 94 % de (Scheme 14). Investigation of the chiroptical properties of major complexes allowed assigning the absolute configuration of the α-stereogenic carbons as (S). It could be mentioned that the corresponding dibenzylation of Ni(II) complex (S)-9 did not take place in these cases at all. After isolation via column chromatography diastereomerically pure complexes (S,S)-65 were decomposed following the standard procedure to afford free amino acids (S)-66 which were isolated in high chemical yield by cation-exchange chromatography along with quantitative recovery of the chiral ligand (S)-11. Hydrogenation of benzyl ethers (S)-66a in a solution of conc. HCl/MeOH afforded enantiomerically pure 2′,6′-dimethyltyrosine (S)-67 which is used as a key structural unit in the design of numerous bioactive peptides, some of which have potential medicinal applications.

Synthesis of 2′,6′-dimethyltyrosine (S)-67
The Ni(II) complex (S)-9 has been effectively alkylated with α,ω-dibromide reagents under homogeneous conditions (Wang et al. 2013). Optimized protocol using NaOH as base in MeCN at 60 °C and 0.5 equiv of dihalogenated reagents such as para- and meta-bis(bromomethyl)benzenes 68a and 68b produced the corresponding bis-alkylated products 69a–b in 79–82 % yield (Scheme 15). It is worth mentioning that this bis-alkylation proceeded with excellent diastereoselectivity, as only the (S,S,S′,S′)-diastereomers were observed by NMR analysis of the crude reactions mixtures. Similarly, meta-substituted pyridine 68c led to the corresponding bis-alkylation product 69c in about 68 % yield. The method also enabled to use non-activated alkyl dibromides, and the reaction of (S)-9 with 1,3-dibromopropane 68d produced 69d in similar yield to those obtained with benzylic reagents. All bis-alkylation reactions demonstrated complete stereochemical control giving rise to diastereomerically pure products 69. The disassembly of the Ni(II) complex 69d was carried out using standard conditions (6 N HCl, MeOH) and was followed by isolation of free (2S,6S)-diaminopimelic acid 70 using a cation-exchange resin.

Alkylation of the Ni(II) complex (S)-9 with α,ω-dibromide reagents. Synthesis of diaminopimelic acid (2S,6S)-70
Depending on the reaction conditions the alkylation of the Ni(II) complex (S)-9 with α,α’-dibromo-o-xylene 71 allowed to obtain both mono- and bis-alkylation complexes 72 and 73 (Belokon et al. 2003b) (Scheme 16). Monoalkylation product (S,S)-72 was formed under standard alkylation conditions at ambient temperature with ratio of reagents 1:1 in 72 % yield. Decrease of the quantity of the alkylating agent 71 up to 0.5 equiv and heating of the reaction mixture to 50 °C resulted in producing bis-alkylated complex (S,S,S′,S′)-73 in 62 % yield. The decomposition of complex (S,S,S′,S′)-73 by aqueous HCl gave bis-amino acid (S,S′)-75. On the other hand decomposition of monomeric complex (S,S)-72 with aqueous HCl led to the intramolecular alkylation of the free amino group with the formation of cyclic amino acid (S)-74.

Alkylation of the Ni(II) complex (S)-9 with α,α′-dibromo-o-xylene 71
Of particular interest is the preparation of enantiomerically pure α,β-dialkyl substituted α-amino acids via alkylation of the Ni(II) complex (S)-9 with racemic sec-alkyl bromide 76. The reaction, conducted in DMF in the presence of KOH, generated a mixture of three diastereomers 77, out of four theoretically possible products (Gu et al. 2003, 2004) (Scheme 17). The reaction was conducted at low temperatures and the corresponding alkylation products were obtained in good combined yield (96 %) and diastereomeric purity (80 % de) of the major product. Relative stereochemistry of the products 77 was confirmed by X-ray crystallography. While the major product S(2S,3S)-77 was kinetically controlled, the authors could not explain the origin of this relatively high S(2S,3S)-77/S(2S,3R)-77 selectivity.

Alkylation of the Ni(II) complex (S)-9 with racemic sec-alkyl bromide 76
Further investigation of reactions between the Ni(II) complex (S)-9 and racemic sec-alkyl bromide 78a under standard conditions using DMF as a solvent and powdered NaOH as a base has shown that lowering the temperature in this case decreased the rate of alkylation but not markedly improved the diastereoselectivity. For example, reaction conducted at −15 °C resulted in formation of diastereomers (S)(2S,3R)-79 and (S)(2S,3S)-79 in a ratio of 1.9:1 and combined yield 89 % (Soloshonok et al. 2008a) (Scheme 18). Complex (S)(2S,3R)-79 was isolated in diastereomerically pure form by column chromatography on silica gel and disassembled to afford enantiomerically pure β-methylphenylalanine (2S,3R)-80. To explain the stereochemical outcome of this reaction, theoretically possible transition states representing the interactions between the si-face of the enolate derived from the Ni(II) complex (S)-9 and the (S)- and (R)-enantiomers of the sec-alkyl bromide 78a have been discussed.

Synthesis of β-methylphenylalanine (2S,3R)-80
The synthetic approach based on the alkylation of glycine equivalent (S)-9 was applied for the preparation of β,β-diphenylalanine and its fluorinated analog (S)-2-amino-3,3-bis-(4-fluorophenyl)propanoic acid as key intermediates for the synthesis of dipeptidyl peptidase IV inhibitors (Scheme 19). Fast alkylation of Ni(II) complex (S)-9 with Ph2CHBr in DMF using sodium hydroxide as a base led to formation of two diastereomeric complexes in ratio 58:42 and 95:5 after 5 and 60 min, respectively (Tararov et al. 1997). The initial ratio of the diastereomers reflected the kinetic diastereoselectivity of the reaction. When thermodynamic equilibrium was established after 1 h the major diastereomer (S,S)-81 was isolated from the reaction mixture by crystallization. β,β-Diphenylalanine (S)-82 was obtained in enantiomerically pure form after hydrolysis of Ni(II) complex (S,S)-81 and precipitation with ethylene diamine tetraacetic acid (EDTA) in 60 % overall yield. Diphenylalanine (R)-82 was successfully synthesized according to the same synthetic protocol starting from Ni(II) complex (R)-9. On the other hand, alkylation of Ni(II) complex (S)-9 with chlorides Ph2CHCl and (4-F-C6H4)2CHCl in the presence of NaOH or KOH as a base led to relatively slow reaction rates and formation of undesired byproducts. Careful study of the reaction with (4-F-C6H4)2CHCl including solvent, base and temperature has shown that alkylation runs smoothly with 3 equiv of NaH as a base, DMF as a solvent at −20 °C for 2.5 h yielding the diastereoisomeric complex (S)-83 in 86 % yield and excellent diastereoselectivity (Deng et al. 2008). More recently, an operationally convenient procedure for the alkylation of Ni(II) complex (S)-9 with (4-F-C6H4)2CHCl employing well-soluble in MeCN potassium tert-butoxide as a base provided nearly quantitative yield of product (2S,2′S)-83 with excellent stereochemical outcomes (>95 % de) (Soloshonok and Ono 2009). Diastereomerically pure product (2S,2′S)-83 was obtained by column chromatography and hydrolyzed by standard procedure. The target amino acid (S)-84 was isolated as hydrochloric salt by reversed-phase chromatography in high isolated yield (>95 %).

Synthesis of β,β-diphenylalanine (S)-82 and its fluorinated analog (S)-84
Although alkylations of glycine Ni(II) complex (S)-9 are typically carried out under homogeneous conditions to achieve the best stereoselectivity of the corresponding homologated products, heterogeneous phase-transfer catalysis (PTC) conditions are usually milder and never lead to formation of double alkylation products. It should be mentioned that quite low solubility of (S)-9 in usual PTC solvents such as benzene or toluene limited the synthetic potential of these reactions. Thus, in early experiments on phase-transfer benzylation and allylation of glycine Ni(II) complex (S)-9 CH2Cl2 was used as organic solvent affording products with low stereochemical outcome (Belokon et al. 1988). However, it was recently demonstrated that CH2Cl2 could be used as a practical reagent for the methylene dimerization of Ni(II) complexes under PTC producing diastereoisomeric dimers. The first asymmetric version of “methylene dimerization” was reported by Belokon et al. by treatment of Ni(II) complex (S)-9 with 0.5 equiv of CH2Br2 and powdered NaOH in dry MeCN resulting in the formation of Ni(II) complex (S,S,S′,S′)-85 as a major product, which was converted into 4-aminoglutamic acid (2S,4S)-86 (Belokon et al. 1987) (Scheme 20). The main disadvantages of these reaction conditions were the formation of the diastereomeric product as well as substantial amount of decomposition products. As a result the Ni(II) complex (S,S,S′,S′)-85 could be obtained in about 50 % yield after painstaking column purification. Optimization of the reaction conditions for methylene dimerization of Ni(II) complex (S)-9 by screening various bases, solvents, phase-transfer catalysts, and concentration of the reagents showed that application of tetra-n-butylammonium bromide (TBAB), aqueous NaOH, and a solution of Ni(II) complex (S)-9 in CH2Cl2 gave within 1 h the best outcome of products (S,S,S′,S′)-85 and (S,S,S′,R′)-85 in a ratio of 1:1 and 95 % combined yield (Taylor et al. 2004; Soloshonok et al. 2006c). The initial 1:1 ratio of the two products was gradually changed under the thermodynamic control, leading to complete disappearance of the complex (S,S,S′,R′)-85 after 24 h, leaving complex (S,S,S′,S′)-85 as a sole diastereomer. This result suggested that (S,S,S′,S′)-85 was the thermodynamically controlled product and (S,S,S′,R′)-85 was the kinetically controlled product. Fast epimerization for 1 h of diastereomer (S,S,S′,R′)-85 to diastereomer (S,S,S′,S′)-85 was achieved by treatment of the corresponding 1:1 mixture with strong bases such as guanidine, producing Ni(II) complex (S,S,S′,S′)-85 as a sole product in 80 % isolated yield. Thus, prepared product (S,S,S′,S′)-85 without any chromatographic purification was disassembled under the standard conditions to give 4-aminoglutamic acid (2S,4S)-86.

Methylene dimerization of Ni(II) complex (S)-9 under phase-transfer conditions
The reaction most likely proceeds through the formation of an intermediate mono-alkylated Ni(II) complex 87, which can undergo dehydrohalogenation, leading to the formation of an unsaturated Ni(II) complex 88 (Scheme 21). This intermediate complex can react as Michael acceptor with the starting glycine complex (S)-9 furnishing the reaction products (S,S,S′,S′)-85 and (S,S,S′,R′)-85. The intermediate Michael acceptor 88 was isolated in a reaction conducted in benzene with CH2Br2 as an alkylating reagent. The Michael addition between complexes 88 and (S)-9 under the standard PTC conditions successfully gave rise to a mixture of products (S,S,S′,S′)-85 and (S,S,S′,R′)-85 in a ratio similar to that observed in the direct methylene dimerization of (S)-9 in CH2Cl2.

Synthesis of intermediate Michael acceptor 88
Further experiments with halogenated solvents have shown that benzylation of (S)-9 in the presence of tetrabutylammonium iodide (TBAI) in a 1:1 mixture of ClCH2CH2Cl and 30 % aqueous NaOH afforded diastereoisomeric complexes (S,S)-89 and (S,R)-89 in a 85:15 ratio and good overall yield without any trace of possible dimerization products (Houck et al. 2012) (Scheme 22). The minor isomers (S,R)-89 could be epimerized to the most stable (S,S)-89 by treatment with MeONa without purification of the initial mixture. In this manner the single isomer (S,S)-89 was obtained in excellent overall yield. Reactions with substituted benzyl bromides, allyl bromides and propargyl bromides proceeded in good overall yields and diastereoselectivities after MeONa-mediated equilibration. Ethyl bromoacetate was also successfully used for alkylation. On the other hand, PTC alkylations of Ni(II) complex (S)-9 with non-activated alkyl halides such as MeI and BuI were less successful and the reaction did not proceed at all with sterically demanding alkyl iodides.

Alkylation of glycine Ni(II) complex (S)-9 under phase-transfer conditions
Alkylation of alanine Ni(II) complex (S,S)-18a with benzyl and allyl halides was successfully carried out using 1.1–5 equiv. excess of the alkylating reagent producing the major diastereoisomer (S,S)-90 in >80 % de (Belokon et al. 1985b, 1988) (Scheme 23). The ratio of the diastereoisomers was not influenced by the diastereoisomeric purity of the starting material and the experimental conditions. Since new complexes 90 contain no α-proton, the stereochemistry of the amino acid moiety is controlled kinetically. Sterically constrained α-methyl-α-amino acids can be prepared in enantiomerically pure form by chromatography separation of the corresponding diastereomeric Ni(II) complexes followed by their hydrolytic disassembly and isolation of the target amino acid.

Alkylation of the alanine Ni(II) complex (S,S)-18a
It is interesting to note that alkylation of alanine Ni(II) complex (S,S)-18a with trans-cinnamyl bromide (Qiu et al. 2000) or fluorine-containing benzyl chlorides (Soloshonok et al. 1990; Kukhar et al. 1993) and KOH as a base afforded the corresponding products (S,S)-91 and (S,S)-92 in high chemical yields, and the diastereoselectivity of these reactions (>90 % de) was better than the usually observed for the benzylation or allylation of alanine Ni(II) complex (S,S)-18a (Scheme 24). After separation of the alkylated diastereomers on silica gel, decomposition of products (S,S)-91 and (S,S)-92 led to enantiomerically pure α-trans-cinnamyl-α-alanine (S)-93 and α-methyl(fluorophenyl)alanines (S)-94, respectively, in good overall yields.

Benzylation and allylation of alanine Ni(II) complex (S,S)-18a
Application of NaOH as a base in DMF at ambient temperature for the alkylation of the alanine Ni(II) complex (S,S)-18a with 1,1,1-trifluoro-4-iodobutane 37c did not result in formation of any desirable product. In this case the alkylation could be successfully performed using t-BuOK as a strong and non-nucleophilic base affording the product (S,S)-95 in 74 % yield and with complete diastereoselectivity (Wang et al. 2011b) (Scheme 25). Unfortunately, application of 1,1,1-trifluoro-3-iodopropane 37b and 1,1,1-trifluoro-2-iodoethane 37a, containing a shorter alkyl chain, did not allow for preparation of the corresponding products, indicating limitations of this method. Disassembly of the diastereomerically pure complex (S,S)-95 under the standard conditions afforded the target 2-amino-6,6,6-trifluoro-2-methylhexanoic acid (S)-96 in 96 % yield.

Synthesis of 2-amino-6,6,6-trifluoro-2-methylhexanoic acid (S)-96
The reactions between alanine Ni(II) complex (S,S)-18a and sterically constrained benzyl bromides 66 in DMF using powdered NaOH as base were studied under different temperatures. It turned out that regardless of the reaction temperature the benzylation occurred with diastereoselectivities in range 70–74 % de affording Ni(II) complexes (S,S)-97 as major reactions products (Soloshonok et al. 2001a) (Scheme 26). This is in good agreement with the kinetically controlled stereochemical outcome of the reactions of alanine Ni(II) complex (S,S)-18a with benzyl bromides. Products (S,S)-97 were easily isolated by column chromatography in diastereomerically pure form and their decomposition gave α-amino acids (S)-98 isolated by ion-exchange chromatography. O-benzyl protected α-amino acid (S)-98a was hydrogenated under standard conditions to afford enantiomerically pure α,2′,6′-trimethyltyrosine (S)-99.

Synthesis of α,2′,6′-trimethyltyrosine (S)-99
The alkylation of the alanine Ni(II) complex (S,S)-18a with racemic sec-alkyl bromides 78a-c, in contrast to the corresponding reactions of the glycine Ni(II) complex (S)-9, occurred under standard conditions at −15 °C with high stereochemical outcome of >84 % allowing isolation of the major products (S)(2S,3S)-100a–c in good chemical yield (Soloshonok et al. 2001b, 2008a) (Scheme 27). Further decomposition of the complex (S)(2S,3S)-100a afforded the enantiomerically pure α,β-dimethylphenylalanine (2S,3S)-101. Because of the simplicity of the experimental procedure and high chemical and stereochemical outcome, this direct alkylation of alanine Ni(II) complex (S,S)-18a with racemic 78 is a synthetically useful approach for preparing α,β-dialkyl-substituted α-amino acids.

Alkylation of the alanine Ni(II) complex (S,S)-18a with racemic sec-alkyl bromides 78
Surprisingly, the alkylation of the ethyl-containing Ni(II) complex (S,S)-18b with 78a occurred at a noticeably slower rate at ambient temperature giving rise to a mixture of (S)(2S,3R)-102 and (S)(2S,3S)-102 in a low ratio of 1:1.6 (Scheme 28). Attempts to improve the stereochemical outcome by lowering the reaction temperature were unsuccessful. Thus, the reaction conducted at −10 °C was too sluggish to produce reliable amounts of product. Even slower reaction rates and the same stereochemical outcome were observed in the alkylation of the leucine-derived complex (S,S)-18c indicating that an increase in the steric bulk of the amino acid residue interferes with the reactivity of the Ni(II) complexes and stereoselectivity of the alkylation.

Alkylation of the Ni(II) complexes (S,S)-18b,c with (1-bromoethyl)benzene 78a
Several attempts have been made to increase the diastereoselectivity of the alkylation reactions using derivatives of Ni(II) complex (S)-9 with various substituents on the benzyl group of the proline chiral auxiliary. For example, Saghiyan et al. reported that 2-chloro-, 3,4-dimethyl-, 3,4-dichloro- and 2-fluoro-substituted glycine Ni(II) complexes (S)-22a–d gave better diastereoselectivity of alkylation with BnBr and MeI conducted in DMF and NaOH or NaH as a base at ambient temperature than the original Ni(II) complex (S)-9 (Belokon et al. 2002; Saghiyan et al. 2006, 2010) (Scheme 29). The best results both in terms of stereoselectivities and the rate of reactions were obtained for 2-chloro-, 3,4-dichloro- and 2-fluoro-substituted Ni(II) complexes (S)-22a, (S)-22c and (S)-22d affording the alkylated products (S,S)-103 in 95–97 % de. The 3,4-dimethyl-substituted Ni(II) complex (S)-22b was less efficient with the 93 % de of alkylated product (S,S)-103. In these cases the stereochemical outcome of the alkylation reactions reflected the position of thermodynamic equilibrium between the diastereoisomeric complexes (S,R)/(S,S).

Alkylation of modified glycine Ni(II) complexes (S)-22
The best kinetic stereoselectivity of alkylation of alanine-derived Ni(II) complexes with allyl and benzyl bromides was observed in the case of 2-chloro- and 2-fluoro-substituted complexes (S,S)-23a and (S,S)-23d ranging from 94 to 99 % de (Scheme 30). In general, modified Ni(II) complexes (S,S)-23a,d were found efficient for highly stereoselective syntheses of α-methyl-α-amino acids with high enantiomeric purity (ee > 95 %).

Alkylation of modified alanine Ni(II) complexes (S,S)-23
In contrast to the above discussed results, the methylation of alanine-derived Ni(II) complex (S,S)-25, containing ortho- and para-methyl groups which provide both steric hindrance and supposedly donation of the electron density to nickel orbitals, with 13CH3I led to the corresponding Ni(II) complex (S,S)-105 in only 66 % de (Popkov et al. 2002) (Scheme 31). After chromatographic purification on silica gel, which did not affect the ratio of the diastereomers, complex (S,S)-105 was used for preparation of [13C]-α-aminoisobutyric acid 106.

Methylation of alanine Ni(II) complex (S,S)-25
Due to the generality and simplicity of the experimental procedures as well as high stereochemical outcome of the alkylation reactions of the Ni(II) complexes of the Schiff bases of glycine and alanine, derived from (S)- or (R)-BPB, this approach has been used to provide fast access to isotopically enriched valine 107 (Chaykovski et al. 2003), δ-silanediol amino acid 108 (Kim and Sieburth 2012), linear backbone of (2S,4R)-4-methylpipecolic acid 109 (Hung et al. 2010) as well as pyrenylalanine 110 (Alves et al. 2004; Chen et al. 2012) (Fig. 5).

α-Amino acids prepared by alkylation of the chiral glycine and alanine Ni(II) complexes
Alkylation of the Ni(II)-complexes of glycine Schiff bases of 2-[N-(α-picolyl)-amino]benzophenone
The achiral Ni(II) complex of glycine Schiff base of 2-[N-(α-picolyl)-amino]benzophenone 12 was found to a be stable yet highly reactive nucleophilic glycine equivalent and its alkylation could be carried out at ambient temperature without inert atmosphere or dried and degassed solvent. The corresponding acetophenone-derived complex 13, useful for Michael addition reactions (Ellis et al. 2009), was rather unsuitable for the alkylation reactions due to the relatively high C–H acidity of the methyl group. On the other hand, the reactions of Ni(II)-complex 12 with an excess of alkyl halides, conducted under homogeneous conditions in DMF resulted in complete (>99 %) α,α-dialkylation offering a convenient method for preparation of sym-α,α-AAs 112 (Ellis et al. 2003a, b) (Scheme 32). Generation of the corresponding enolate from Ni(II)-complex 12 could be effectively achieved using simple sodium and potassium hydroxides or alkoxides. However, NaO-t-Bu was significantly more efficient than KOH as a base allowing conducting the reactions under homogeneous conditions as only 3.5 equivalents of base was required to complete the dialkylation process. Activated alkyl halides, such as allyl, benzyl and cinnamyl, could be used as bromides, while introduction of usual alkyl groups required application of the corresponding iodides. It should be noted that use of only 3.5 equivalents of alkyl halide was enough for complete, fast, and clean dialkylation of 12 to afford compounds 111 in high isolated yields. By contrast, alkylation of complex 12 with iso-propyl and iso-butyl bromides or iodides, even under high temperatures and long reaction times resulted in quantitative formation of only monoalkylated products 113. Dialkylation products 112 were isolated simply by pouring the reaction mixture into ice water followed by filtration of the precipitate. Procedure for isolation of the target symmetrically α,α-disubstituted α-amino acids 112 included heating of the corresponding products 111 in methanol in the presence of 1 N hydrochloric acid followed by neutralization of the resultant mixture with aqueous ammonia. The ligand can be extracted and recycled to produce back the glycine complex 12 and symmetrically α,α-disubstituted α-amino acids 112a and 112b were isolated in free form in 91 and 93 % yield using cation-exchange resin.

Alkylation of the glycine Ni(II)-complex 12 under homogeneous conditions
Achiral Ni(II) complex 12 was found to undergo alkylation with alkyl halides under phase-transfer conditions in the presence of benzyltriethylammonium bromide as catalyst and 30 % aqueous NaOH as the base. The reactions were extremely fast and clean, proceeding to completion with an equimolar ratio (1:1) of substrate and alkylating agent to give the mono-alkylated products 113 quantitatively (Belokon et al. 2003a) (Scheme 33). When the reaction of Ni(II) complex 12 was carried out using 4 equiv of propargyl bromide at room temperature under phase-transfer conditions in dichloromethane in the presence of tetrapropylammonium iodide, quantitative conversion of Ni(II) complex 12 to the mono-substituted product was observed within 45 min with further clean conversion of 113 to the target disubstituted complex 114 in >98 % yield after running the reaction overnight.

Alkylation of the glycine Ni(II)-complex 12 under phase-transfer conditions
Application of glycine Ni(II) complex 12 for preparation of cyclic amino acids was also demonstrated. For example, a method for preparing 2-aminoindane-2-carboxylic acid 117 included a two-step alkylation of Ni(II)-complex 12 with o-dibromoxylylene 71 (Ellis et al. 2003c) (Scheme 34). The first step, monoalkylation of 12 with 71, conducted under the phase-transfer conditions, gave the corresponding complex 115 in excellent chemical yield. Without any purification the intermediate 115 was cyclized under homogeneous conditions in DMF with NaO-t-Bu as a base to give the product 116 in high chemical yield. Finally, decomposition of Ni(II) complex 116 was conducted under standard conditions by heating in methanol/3 N HCl. The target amino acid 117 was isolated by ion-exchange chromatography in greater than 95 % purity. It should be mentioned that the reaction between complex 12 and o-dibromoxylylene conducted under standard conditions for dialkylation with NaO-t-Bu in DMF was complicated by the formation of undesired byproducts and afforded Ni(II) complex 115 in 70 % yield.

Synthesis of 2-aminoindane-2-carboxylic acid 117
Asymmetric alkylation of Ni(II) complex 12 in CH2Cl2 catalyzed by (S) or (R) 2-amino-2′-hydroxy-1,1′-binaphthyl (NOBIN) 119 with activated alkyl halides, conducted at room temperature, gave monoalkylated Ni(II) complexes 118 in good chemical yields and up to 93–98 % enantiomerical purity (Belokon et al. 2001, 2003a) (Scheme 35). Relatively low yields of the alkylated products were obtained with unactivated alkyl halides, although the enantiomeric purity of the resulting amino acids was reasonably high. Increasing the polarity of the solvent resulted in lower enantioselectivities, down from 96 % in CH2Cl2 to 17 % in MeCN. Attempts of using BINOL or 2,2′-diamino-1,1′-binaphtyl as catalysts resulted in both low enantioselectivity and chemical yields of the alkylation products. The enantiomeric purity of isolated amino acids could be further improved by crystallization of the intermediate alkylated Ni(II) complexes. The target amino acids could be easily obtained by hydrolysis of the alkylation products with aqueous HCl in MeOH followed by ion-exchange chromatography.

Catalytic asymmetric alkylation of Ni(II) complex 12 under phase-transfer conditions
On the other hand, catalysis by (R)-NOBIN of the benzylation of Ni(II) complex 30 derived from alanine proved to be very slow and gave resulting amino acid 121 in only 44 % ee (Scheme 36).

Catalytic asymmetric benzylation of Ni(II) complex 30 under phase-transfer conditions
Alkylation of new generation of Ni(II)-complexes
Achiral Ni(II) complex 14c incorporating a dibutylamine moiety (Soloshonok et al. 2009b) was found to be efficient in dialkylation reactions which were conducted under homogeneous conditions in DMF at ambient temperature with only 2.5 equivalents of NaO-t-Bu and benzyl, allyl or cinnamyl bromide (Ellis et al. 2006) (Scheme 37). Most of the reactions were completed in about 15 min giving rise to α,α-dialkylation products 122 in high chemical yields (Soloshonok et al. 2008b). No alkylation products on the N,N-di-n-butyl glycine methylene moiety were observed in the crude reaction mixtures. Introduction of two unactivated alkyl groups could also be accomplished using the more reactive methyl iodide. Isolation of the free α,α-disubstituted α-amino acids was conducted by standard procedure along with recovery of ligand 35c. Operationally convenient experimental procedures, mild reaction conditions, as well as high chemical yields render this method practical for preparing α,α-disubstituted α-amino acids and their analogs.

Dialkylation of achiral Ni(II) complex 14c under homogeneous conditions
Unusual chemoselectivity was demonstrated during the alkylation reactions of Ni(II) complex 15c containing a secondary amino group. For example, the alkylation of t-butylamine-derived NH–Ni(II) complex with one equivalent of benzyl, cinnamyl or propargyl bromides under phase-transfer conditions produced the corresponding homologated products 123 in 90–94 % yield and in diastereomerically pure form (Ellis and Soloshonok 2006) (Scheme 38). The complete C–H chemoselectivity observed in these reactions suggested that coordination of nitrogen to a metal has a significant synthetic potential as protecting a group without the need of introducing a transient N–C substituent.

Alkylation of Ni(II) complex 15c under phase-transfer conditions
Conclusions
The data discussed in this Review, clearly suggest that alkylation of the Ni(II) complexes (S)-9, 12–15 is a mature methodology for preparation of natural as well as various unnatural amino acids. In particular, asymmetric synthesis via homologation of glycine and alanine-derived complexes (S)-9 and (S,S)-18a, respectively, provides a reliable and synthetically attractive approach for preparation of the target amino acids in enantiomerically pure form. It would be beneficial for the readers, to emphasize in this section the major features of this methodology which make it markedly different from other numerous literature approaches. First of all, these Ni(II) complexes, including the chiral (S)-9, are very inexpensive and can be easily prepared on large scale. Second, the alkylation reactions of glycine and alanine complexes can be conducted under operationally convenient conditions without recourse to specially dry solvents, moisture/air-sensitive bases, like n-BuLi, or extreme low temperatures. Third, the corresponding alkylation products are highly crystalline compounds and can be further purified to diastereomerically pure form simply by crystallization. Fourth, glycine (S)-9 and alanine (S,S)-18a-derived complexes can be alkylated by chiral (racemic) secondary alkyl halides with appreciable kinetic resolution providing the most direct approach to extremely valuable sterically constrained β-substituted amino acids. Fifth, in most of cases, the alkylation reactions occur at high reaction rates rendering this approach as a method of choice for time-sensitive projects such as preparation of radioactive amino acid derivatives for positron-emitting tomography (PET). Finally, the alkylation reactions can be conducted under variety of conditions, such as phase-transfer and homogeneous, to accommodate various functional groups on the alkylation reagents. Furthermore, recently developed bis-alkylation procedure allows for straightforward preparation of bis-amino acids, relatively unexplored class of compounds. Here, we also should mention some drawbacks of this approach. Probably the most important one is incomplete diastereoselectivity, in particular under the kinetically controlled condition, necessitating the additional purification step. Also, relatively large molecular weight of these Ni(II) complexes provides for some inconvenience. Thus, only about 20 % of the total weight of an alkylated product carry the target amino acid and the rest is the ligand and Ni(II), requiring to use large quantities of solvents per gram of the target amino acid. Nevertheless, all these drawbacks play rather minor role as compared to the synthetic advantages provided by this methodology. It should also be stated that this approach attracts an increasing attention of chemists as the most simple and reliable method for preparation of various amino acids.
References
Aceña JL, Sorochinsky AE, Soloshonok VA (2012) Recent advances in asymmetric synthesis of α-(trifluoromethyl)-containing α-amino acids. Synthesis 44:1591–1602
Alves I, Cowell S, Lee YS, Tang X, Davis P, Porreca F, Hruby VJ (2004) A novel 3-step enantioselective synthesis of pyrenylalanine with subsequent incorporation into opioid, CCK, and melanotropin ligands. Biochem Biophys Res Commun 318:335–340
Belokon YN (1992) Chiral complexes of Ni(II), Cu(II), and Cu(I) as reagents, catalysts and receptors for asymmetric synthesis and chiral recognition of amino acids. Pure Appl Chem 64:1917–1924
Belokon YN, Zel’tzer E, Bakhmutov VI, Saporovskaya MB, Ryzhov MG, Yanovsky AI, Struchkov YT, Belikov VM (1983) Asymmeric synthesis of threonine and partial resolution and retroracemization of α-amino acids via copper(II) complexes of their Schiff bases with (S)-2-N-(N′-benzylprolyl)aminobenzaldehyde and (S)-2-N-(N′-benzylprolyl)aminoacetophenone. Crystal and molecular structure of a copper(II) complex of glycine Schiff base with (S)-2-N-(N′benzylprolyl)aminoacetophenone. J Am Chem Soc 105:2010–2017
Belokon YN, Bulychev AG, Vitt SV, Struchkov YT, Batsanov AS, Timofeeva TV, Tsyryapkin VA, Ryzhov MC, Lysova LA, Bakhmutov VI, Belikov VM (1985a) General method of diastereo- and enantioselective synthesis of β-hydroxy-α-amino acids by condensation of aldehydes and ketones with glycine. J Am Chem Soc 107:4252–4259
Belokon YN, Chernoglazova NI, Kochetkov CA, Garbalinskaya NS, Belikov VM (1985b) Preparation of optically pure α-methyl-α-amino acids via alkyñation of the nickel(II) Schiff base of (R,S)-alanine with (S)-2-N-(N′-benzylprolyl)aminobenzledehyde. J Chem Soc Chem Commun 171–172
Belokon YN, Chernoglazova NI, Botsanov AS, Garbalinskaya NS, Bakhmutov VI, Struchkov YT, Belikov VM (1987) Asymmetric synthesis of (2S,4S)-2,4-diaminoglutaric and (2S,3S)-2,3-diamino-2,3-dimethylsuccinic acids using chiral Ni(II) complexes. Russ Chem Bull 36:779–784
Belokon YN, Bakhmutov VI, Chernoglazova NI, Kochetkov KA, Vitt SV, Garbalinskaya NS, Belikov VM (1988) General method for the asymmetric synthesis of α-amino acids via alkylation of the chiral nickel(II) Schiff base complexes of glycine and alanine. J Chem Soc Perkin Trans 1:305–311
Belokon YN, Kochetkov KA, Churkina TD, Ikonnikov NS, Orlova SA, Smirnov VV, Chesnokov AA (1997) Asymmetric Michael addition of a glycine synthon to methyl methacrylate, mediated by disodium TADDOLate. Mendeleev Commun 7:137–138
Belokon YN, Tararov VI, Maleev VI, Savel’eva TF, Ryzhov MG (1998) Improved procedures for the synthesis of (S)-2-[N-(N′-benzylprolyl)amino]-benzophenone (BPB) and Ni(II) complexes of Schiff’s bases derived from BPB and amino acids. Tetrahedron Asymmetry 9:4249–4252
Belokon YN, Kochetkov KA, Churkina TD, Ikonnikov NS, Larionov OV, Harutyunyan S, North M, Vyskošil Š, Kagan HB (2001) Highly efficient catalytic synthesis of α-amino acids under phase-transfer conditions with a novel catalyst/substrate pair. Angew Chem Int Ed 40:1948–1951
Belokon YN, Maleev VI, Petrrosyan AA, Savel’eva TF, Ikonnikov NS, Peregudov AS, Khrustalev VN, Saghiyan AS (2002) Halo-substituted (S)-N-(2-benzoylphenyl)-1-benzylpyrolidine-2-carboxamides as new chiral auxiliaries for the asymmetric synthesis of (S)-α-amino acids. Russ Chem Bull 51:1593–1599
Belokon YN, Bespalova NB, Churkina TD, Cisarova I, Ezernitskaya MG, Harutyunyan SR, Hrdina R, Kagan HB, Kocovsky P, Kochetkov KA, Larionov OV, Lyssenko KA, North M, Polasek M, Peregudov AS, Prisyazhnyuk VV, Vyskocil S (2003a) Synthesis of α-amino acids via asymmetric phase transfer-catalyzed alkylation of achiral nickel(II) complexes of glycine-derived Schiff bases. J Am Chem Soc 125:12860–12871
Belokon YN, Kochetkov KA, Borkin DA (2003b) Asymmetric synthesis of unusual α-amino acids. Mendeleev Commun 13:132–134
Boyall D, Frantz DE, Carreira EM (2002) Efficient enantioselective additions of terminal alkynes and aldehydes under operationally convenient conditions. Org Lett 4:2605–2606
Burck S, van Assema SGA, Lastdrager B, Slootweg JC, Ehlers AW, Otero JM, Dacunha-Marinho B, Llamas-Saiz AL, Overhand M, van Raaij MJ, Lammertsma K (2009) Bisphosphine-functionalized cyclic decapeptides based on the natural product gramicidin S: a potential scaffold for transition-metal coordination. Chem Eur J 15:8134–8145
Cai C, Yamada T, Tiwari R, Hruby VJ, Soloshonok VA (2004) Application of (S)- and (R)-methyl pyroglutamates as inexpensive, yet highly efficient chiral auxiliaries in the asymmetric Michael addition reactions. Tetrahedron Lett 45:6855–6858
Chaykovski MM, Bae LC, Cheng M-C, Murray JH, Tortolani KE, Zhang R, Seshadri K, Findlay JHBC, Hsieh S-Y, Kalverda AP, Homans SW, Miles Brown JM (2003) Methyl side-chain dynamics in proteins using selective enrichment with a single isotopomer. J Am Chem Soc 125:15767–15771
Chen S, Wang L, Fahmi NE, Benkovic SJ, Hecht SM (2012) Two pyrenylalanines in dihydrofolate reductase form an excimer enabling the study of protein dynamics. J Am Chem Soc 134:18883–18885
Chinchilla R, Mazón P, Nájera C (2000) Asymmetric synthesis of α-amino acids using polymer-supported Cinchona alkaloid-derived ammonium salts as chiral phase-transfer catalysts. Tetrahedron Asymmetry 11:3277–3281
Collet S, Bauchat P, Danion-Bougot R, Danion D (1998) Stereoselective, nonracemic synthesis of ω-borono-α-amino acids. Tetrahedron Asymmetry 9:2121–2131
Collet S, Carreaux F, Boucher J-L, Pethe S, Lepoivre M, Danion-Bougot R, Danion D (2000) Synthesis and evaluation of ω-borono-α-amino acids as active site probes of arginase and nitric oxide synthases. J Chem Soc Perkin Trans 1, pp 177–182
Coppola GM, Schuster HF (1987) Asymmetric synthesis: construction of chiral molecules using amino acids. Wiley-Interscience, New York
Corey EJ, Xu F, Noe MC (1997) A rational approach to catalytic enantioselective enolate alkylation using a structurally rigidified and defined chiral quaternary ammonium salt under phase transfer conditions. J Am Chem Soc 119:12414–12415
De BB, Thomas NR (1997) Optimization of the retroracemisation procedure for α-amino acids using (S)-2-[(N-alkylprolyl)amino]benzophenones, recyclable chiral auxiliaries. Tetrahedron Asymmetry 8:2687–2691
Deng G, Wang J, Zhou Y, Jiang H, Liu H (2007) One-pot, large-scale synthesis of nickel(II) complexes derived from 2-[N-(α-picolyl)amino]benzophenone (PABP) and α- or β-amino acids. J Org Chem 72:8932–8934
Deng G, Ye D, Li Y, He L, Zhou Y, Wang J, Li J, Jiang H, Liu H (2008) Synthesis of (S)-, (R)-, and (rac)-2-amino-3,3-bis(4-fluorophenyl)propanoic acids and an evaluation of the DPP IV inhibitory activity of Denagliptin diastereomers. Tetrahedron 64:10512–10516
Duthaler RO (1994) Recent developments in the stereoselective synthesis of α-amino acids. Tetrahedron 50:1539–1560
Ellis TK, Soloshonok VA (2006) Design and synthesis of a new generation of ‘NH’–Ni(II) complexes of glycine Schiff bases and their unprecedented C–H vs. N–H chemoselectivity in alkyl halide alkylations and Michael addition reactions. Synlett 17(4):533–538
Ellis TK, Martin CH, Ueki H, Soloshonok VA (2003a) Efficient, practical synthesis of symmetrically α, α-disubstituted α-amino acids. Tetrahedron Lett 44:1063–1066
Ellis TK, Martin CH, Tsai GM, Ueki H, Soloshonok VA (2003b) Efficient synthesis of sterically constrained symmetrically α, α-disubstituted α-amino acids under operationally convenient conditions. J Org Chem 68:6208–6214
Ellis TK, Hochla VM, Soloshonok VA (2003c) Efficient synthesis of 2-aminoindane-2-carboxylic acid via dialkylation of nucleophilic glycine equivalent. J Org Chem 68:4973–4976
Ellis TK, Ueki H, Yamada T, Ohfune Y, Soloshonok VA (2006) Design, synthesis, and evaluation of a new generation of modular nucleophilic glycine equivalents for the efficient synthesis of sterically constrained α-amino acids. J Org Chem 71:8572–8578
Ellis TK, Ueki H, Tiwari R, Soloshonok VA (2009) Michael addition reactions between various nucleophilic glycine equivalents and (S, E)-1-enoyl-5-oxo-N-phenylpyrrolidine-2-carboxamide, and optimal type of chiral Michael acceptor in the asymmetric synthesis of β-pheyl pyroglutamic acid and related compounds. Tetrahedron Asymmetry 20:2629–2634
Etayo P, Vidal-Ferrán A (2013) Rhodium-catalyzed asymmetric hydrogenation as a valuable synthetic tool for the preparation of chiral drugs. Chem Soc Rev 42:728–754
Evans DA, Weber AE (1986) Asymmetric glycine enolate aldol reaction: synthesis of cyclosporin’s unusual amino acid, MeBmt. J Am Chem Soc 108:6757–6761
Fishwick CWG, Sanderson JM, Findlay JBC (1994) An efficient route to S-N-(9-fluorenymethoxycarbony)-4′-(l-azi-2,2,2-trifluoroethyl)phenylalanine. Tetrahedron Lett 35:4611–4614
Fitzi R, Seebach D (1988) Resolution and use in α-amino acid synthesis of imidazolidinone glycine derivatives. Tetrahedron 44:5277–5292
Gu X, Tang X, Cowell S, Ying J, Hruby VJ (2002) A novel strategy toward [6,5]-bicyclic β-turn dipeptide. Tetrahedron Lett 43:6669–6672
Gu X, Cowell S, Ying J, Tang X, Hruby VJ (2003) Synthesis of β-phenyl-ω, ε-unsaturated amino acids and stereoselective introduction of side chain groups into [4,3,0]-bicyclic β-turn dipeptides. Tetrahedron Lett 44:5863–5866
Gu X, Ndungu JM, Qiu W, Ying J, Carducci MD, Wooden H, Hruby VJ (2004) Large scale enantiomeric synthesis, purification, and characterization of ω-unsaturated amino acids via a Gly-Ni(II)-BPB-complex. Tetrahedron 60:8233–8243
Hashimoto T, Maruoka K (2007) Recent development and application of chiral phase-transfer catalysts. Chem Rev 107:5656–5682
Hashimoto M, Hatanaka Y, Sadakane Y, Nabeta K (2002) Synthesis of tag introducible (3-trifluoromethyl)phenyldiazirine based photoreactive phenylalanine. Bioorg Med Chem Lett 12:2507–2510
Houck D, Aceña JL, Soloshonok VA (2012) Alkylations of chiral Ni(II)-complexes of glycine under phase-transfer conditions. Helv Chim Acta 95:2672–2679
Hung K-y, Harris PWR, Brimble MA (2010) Synthesis of methyl N-Boc-(2S,4R)-4-methylpipecolate. J Org Chem 75:8728–8731
Izumi Y, Chibata I, Itoh T (1978) Production and utilization of amino acids. Angew Chem Int Ed Engl 17:176–183
Kawamoto SA, Coleska A, Ran X, Yi H, Yang C-Y, Wang S (2012) Design of triazole-stapled BCL9 α-helical peptides to target the β-catenin/B cell CLL/lymphoma 9 (BCL9) protein–protein interaction. J Med Chem 55:1137–1146
Kim JK, Sieburth SM (2012) Synthesis and properties of a sterically unencumbered δ-silanediol amino acid. J Org Chem 77:2901–2906
Kožíšek J, Fronc M, Skubák P, Popkov A, Breza M, Fuess H, Paulmann C (2004) Electronic structure of the nickel(II) complex of the Schiff base of (S)-N-(2-benzoylphenyl)-1-benzylprolinamide and glycine. Acta Cryst A60:510–516
Kukhar VP, Belokon YN, Svistunova NY, Soloshonok VA, Rozhenko AB, Kuzmina NA (1993) Asymmetric synthesis of organoelement analogues of natural products; part 12: general method for the asymmetric synthesis of fluorine-containing phenylalanines and α-methyl(phenyl)alanines via alkylation of chiral nickel(II) Schiff’s base complexes of glycine and alanine. Synthesis 117–121
Kukhar VP, Soloshonok VA, Solodenko VA (1994) Asymmetric synthesis of phosphorus analogs of amino acids. Phosphorus Sulfur Silicon Relat Elem 92:239–264
Larionov OV, Savel’eva TF, Kochetkov KA, Ikonnokov NS, Kozhushkov SI, Yufit DS, Howard JAK, Khrustalev VN, Belokon YN, de Meijere A (2003) Productive asymmetric synthesis of all four diastereomers of 3-(trans-2-nitrocyclopropyl)alanine from glycine with (S)- or (R)-2-[(N-benzylprolyl)amino]benzophenone as a reusable chiral auxiliary. Eur J Org Chem 2003(5):869–877
Le Chevalier Isaad A, Barbetti F, Rovero P, D’Ursi AM, Chelli M, Chorev M, Papini AM (2008) N α-Fmoc-protected ω-azido- and ω-alkynyl-l-amino acids as building blocks for the synthesis of “clickable” peptides. Eur J Org Chem 2008(31):5308–5314
Lygo B, Andrews BI (2004) Asymmetric phase-transfer catalysis utilizing chiral quaternary ammonium salts: asymmetric alkylation of glycine imines. Acc Chem Res 37:518–525
Lygo B, Wainwright PG (1997) A new class of asymmetric phase-transfer catalysts derived from chincona alkaloids—application in the enantioselective synthesis of α-amino acids. Tetrahedron Lett 38:8595–8598
Lygo B, Crosby J, Lowdon TR, Peterson JA, Wainwright PG (2001) Studies on the enantioselective synthesis of α-amino acids via asymmetric phase-transfer catalysis. Tetrahedron 57:2403–2409
Lygo B, Andrews BI, Crosby J, Peterson JA (2002) Asymmetric alkylation of glycine imines using in situ generated phase-transfer catalysts. Tetrahedron Lett 43:8015–8018
Ma J-A (2003) Recent developments in the catalytic asymmetric synthesis of α- and β-amino acids. Angew Chem Int Ed 42:4290–4299
Maruoka K, Ooi T (2003) Enantioselective amino acid synthesis by chiral phase-transfer catalysis. Chem Rev 103:3013–3028
Moore JL, Taylor SM, Soloshonok VA (2005) An efficient and operationally convenient general synthesis of tertiary amines by direct alkylation of secondary amines with alkyl halides in the presence of Hunig’s base. ARKIVOC 2005(6):287–292
Myers AG, Gleason JL, Yoon T, Kung DW (1997) Highly practical methodology for the synthesis of d- and l-α-amino acids, N-protected α-amino acids, and N-methyl-α-amino acids. J Am Chem Soc 119:656–673
Nádvorník M, Popkov A (2002) Improved synthesis of the Ni(II) complex of the Schiff base of (S)-2-[N-(NA-benzylprolyl)amino]benzophenone and glycine. Green Chem 4:71–72
Nádvorník M, Langer V, Jirásko R, Holčapek M, Weidlich T, Lyčka A, Popkov A (2008) Syntheses, X-ray, MSn, NMR and CD structure determination of nickel(II) complexes of Schiff bases of (S)-N-(2-benzoylphenyl)-1-benzylpyrrolidine-2-carboxamide and aromatic α-amino acids. Polyhedron 27:3477–3483
Nájera C, Sansano JM (2007) Catalytic asymmetric synthesis of α-amino acids. Chem Rev 107:4584–4671
O’Donnell MJ (2001) The preparation of optically active α-amino acids from the benzophenone imines of glycine derivatives. Aldrichimica Acta 34:3–15
O’Donnell MJ (2004) The enantioselective synthesis of α-amino acids by phase transfer catalysis with achiral Schiff base esters. Acc Chem Res 37:506–517
O’Donnell MJ, Bennet WD, Wu S (1989) The stereoselective synthesis of α-amino acids by phase transfer catalysis. J Am Chem Soc 111:2353–2355
O’Donnell MJ, Eckrich TM (1978) The synthesis of amino acid derivatives by catalytic phase-transfer alkylations. Tetrahedron Lett 19:4625–4628
O’Donnell MJ, Bennett WD, Bruder WA, Jacobsen WN, Knuth K, LeClef B, Polt RL, Bordwell FG, Mrozack SR, Cripe TA (1988) Acidities of glycine Schiff bases and alkylation of their conjugate bases. J Am Chem Soc 110:8520–8525
Ooi T, Maruoka K (2007) Recent advances in asymmetric phase-transfer catalysis. Angew Chem Int Ed 46:4222–4266
Ooi T, Kameda M, Maruoka K (1999) Molecular design of a C2-symmetric chiral phase-transfer catalyst for practical asymmetric synthesis of α-amino acids. J Am Chem Soc 121:6519–6520
Ooi T, Kameda M, Maruoka K (2003) Design of N-spiro C2-symmetric chiral quaternary ammonium bromides as novel chiral phase-transfer catalysts: synthesis and application to practical asymmetric synthesis of α-amino acids. J Am Chem Soc 125:5139–5151
Park H-g, Jeong B-S, Yoo M-S, Lee J-H, M-k Park, Lee Y-J, Kim M-J, Jew S-s (2002) Highly enantioselective and practical chincona-derived phase-transfer catalysts for the synthesis of α-amino acids. Angew Chem Int Ed 41:3036–3038
Popkov A, De Spiegeleer BD (2012) Chiral nickel(II) complexes in the preparation of 11C- and 18F-labelled enantiomerically pure α-amino acids. Dalton Trans 41:1430–1440
Popkov A, Gree A, Nádvorník M, Lyčka A (2002) Chiral nucleophilic glycine and alanine synthons: nickel(II) complexes of Schiff bases of (S)-N-(2,4,6-trimethylbenzyl)proline (2-benzoylphenyl)amide and glycine or alanine. Trans Met Chem 27:884–887
Popkov A, Langer V, Manorik PA, Weidlich T (2003) Long-range spin–spin interactions in the 13C-n.m.r. spectra of the nickel(II) complex of the Schiff base of (S)-N-benzylproline (2-benzoylphenyl)amide and glycine. Quantum-chemical calculations and possible donation of electron density from the π-system of the benzyl group to nickel. Trans Met Chem 28:475–481
Qiu W, Soloshonok VA, Cai C, Tang X, Hruby VJ (2000) Convenient, large-scale asymmetric synthesis of enantiomerically pure trans-cinnamylglycine and -α-alanine. Tetrahedron 56:2577–2582
Qiu W, Gu X, Soloshonok VA, Carducci MD, Hruby VJ (2001) Stereoselective synthesis of conformationally constrained reverse turn dipeptide mimetics. Tetrahedron Lett 42:145–148
Saghiyan AS, Dadayan SA, Petrosyan SG, Manasyan LL, Geolchanyan AV, Djamgaryan SM, Andreasyan SA, Maleev VI, Khrustalev VN (2006) New chiral NiII complexes of Schiff’s bases of glycine and alanine for efficient asymmetric synthesis of α-amino acids. Tetrahedron Asymmetry 17:455–467
Saghiyan AS, Dadayan AS, Dadayan SA, Mkrtchyan AF, Geolchanyan AV, Manasyan LL, Ajvazyan HR, Khrustalev VN, Hambardzumyan HH, Maleev VI (2010) Rapid asymmetric synthesis of amino acids via NiII complexes based on new fluorine containing chiral auxiliaries. Tetrahedron Asymmetry 21:2956–2965
Schöllkopf U (1983a) Enantioselective synthesis of non-proteinogenic amino acids via metalated bis-lactim ethers of 2,5-diketopiperazines. Tetrahedron 39:2085–2091
Schöllkopf U (1983b) Asymmetric syntheses of amino acids via metalated bis-lactim ethers of 2,5-diketopiperazines. Pure Appl Chem 55:1799–1806
Schöllkopf U, Groth U, Deng C (1981) Enantioselective syntheses of (R)-amino acids using l-valine as chiral agent. Angew Chem Int Ed Engl 20:798–799
Sebahar PR, Williams RM (2000) The asymmetric total synthesis of (+)- and (−)-spirotryprostatin B. J Am Chem Soc 122:5666–5667
Seebach D, Sting AR, Hoffmann M (1996) Self-regeneration of stereocenters (SRS)—applications, limitations, and abandonment of a synthetic principle. Angew Chem Int Ed Engl 35:2708–2748
Shibuguchi T, Fukuta Y, Akachi Y, Sekine A, Ohshima T, Shibasaki M (2002) Development of new asymmetric two-center catalysts in phase-transfer reactions. Tetrahedron Lett 43:9539–9543
Shirakawa S, Maruoka K (2013) Recent developments in asymmetric phase-transfer reactions. Angew Chem Int Ed 52:4312–4348
Sinclair PJ, Zhai D, Reibenspies J, Williams RM (1986) Electrophilic glycinates: new and versatile templates for asymmetric amino acid synthesis. J Am Chem Soc 108:1103–1104
Soloshonok VA (2002) Highly diastereoselective Michael addition reactions between nucleophilic glycine equivalents and β-substituted-α, β-unsaturated carboxylic acid derivatives; a general approach to the stereochemically defined and sterically χ constrained α-amino acids. Curr Org Chem 6:341–364
Soloshonok VA, Berbasov DO (2004) Synthesis of fluorine-containing compounds under operationally convenient conditions. J Fluorine Chem 125:1757–1763
Soloshonok VA, Ono T (2009) Operationally convenient asymmetric synthesis of (S)-2-amino-3,3-bis-(4-fluorophenyl)propanoic acid. J Fluorine Chem 130:547–549
Soloshonok VA, Sorochinsky AE (2010) Practical methods for the synthesis of symmetrically α,α-disubstituted α-amino acids. Synthesis 2319–2344
Soloshonok VA, Ueki H (2007) Design, synthesis, and characterization of binuclear Ni(II) complexes with inherent helical chirality. J Am Chem Soc 129:2426–2427
Soloshonok VA, Belokon YN, Kukhar VP, Chernoglazova NI, Saporovskaya MB, Bakhmutov VI, Kolycheva MT, Belikov VM (1990) Asymmetric synthesis of organoelement analogs of natural products. 2. Convenient synthetic approach to enantiomerically pure (S)-(−)-o, m, p-fluorophenylalanines and their 2-methyl derivatives. Izv Akad Nauk SSSR Ser Khim 1630–1636
Soloshonok VA, Svistunova NY, Kukhar VP, Solodenko VA, Kuzmina NA, Rozhenko AB, Galushko SV, Shishkina IP, Gudima AO, Belokon YN (1992a) Asymmetric synthesis of organoelement analogs of natural products. 6. (S)-α-Amino-ω-phosphonocarboxylic acids. Izv Akad Nauk SSSR Ser Khim 397–402
Soloshonok VA, Belokon YN, Kuzmina NA, Maleev VI, Svistunova NY, Solodenko VA, Kukhar VP (1992b) Asymmetric synthesis of phosphorus analogues of dicarboxylic α-amino acids. J Chem Soc Perkin Trans 1, pp 1525–1529
Soloshonok VA, Kukhar VP, Galushko SV, Svistunova NY, Avilov DV, Kuzmina NA, Raevski NI, Struchkov YT, Pisarevsky AP, Belokon YN (1993) General method for the synthesis of enantiomerically pure β-hydroxy-α-amino acids, containing fluorine atoms in the side chains. Case of stereochemical distinction between methyl and trifluoromethyl groups. X-ray crystal and molecular structure of the nickel(II) complex of (2S,3S)-2-(trifluoromethyl)threonine. J Chem Soc Perkin Trans 1, pp 3143–3155
Soloshonok VA, Kirilenko NA, Fokina NA, Shishkina IP, Galushko SV, Kukhar VP, Svedas VK, Kozlova EV (1994a) Biocatalytic resolution of β-fluoroalkyl-β-amino acids. Tetrahedron Asymmetry 5:1119–1126
Soloshonok VA, Kirilenko AG, Galushko SV, Kukhar VP (1994b) Catalytic asymmetric synthesis of β-fluoroalkyl β-amino acids via biomimetic [1,3]-proton shift reaction. Tetrahedron Lett 35:5063–5064
Soloshonok VA, Avilov DV, Kukhar VP, Tararov VI, Saveleva TF, Churkina TD, Ikonnikov NS, Kochetkov KA, Orlova SA, Pysarevsky AP, Struchkov YT, Raevsky NI, Belokon YN (1995) Asymmetric aldol reactions of chiral Ni(II)-complex of glycine with aldehydes. Stereodivergent synthesis of syn-(2S)- and syn-(2R)-β-alkylserines. Tetrahedron Asymmetry 6:1741–1756
Soloshonok VA, Avilov DV, Kukhar’ VP (1996a) Highly diastereoselective asymmetric aldol reactions of chiral Ni(II)-complex of glycine with trifluoromethyl ketones. Tetrahedron Asymmetry 7:1547–1550
Soloshonok VA, Avilov DV, Kukhar VP (1996b) Asymmetric aldol reactions of trifluoromethyl ketones with a chiral Ni(II) complex of glycine: stereocontrolling effect of the trifluoromethyl group. Tetrahedron 52:12433–12442
Soloshonok VA, Avilov DV, Kukhar VP, Van Meervelt L, Mischenko N (1997) Highly diastereoselective aza-aldol reactions of a chiral Ni(II) complex of glycine with imines. An efficient asymmetric approach to 3-perfluoroalkyl-2,3-diamino acids. Tetrahedron Lett 38:4671–4674
Soloshonok VA, Cai C, Hruby VJ (1999) Asymmetric Michael addition reactions of chiral Ni(II)-complex of glycine with (N-trans-enoyl)oxazolidines: improved reactivity and stereochemical outcome. Tetrahedron Asymmetry 10:4265–4269
Soloshonok VA, Cai C, Hruby VJ, Van Meervelt L, Yamazaki T (2000a) Rational design of highly diastereoselective, organic base-catalyzed, room-temperature Michael addition reactions. J Org Chem 65:6688–6696
Soloshonok VA, Cai C, Hruby VJ (2000b) A unique case of face diastereoselectivity in the Michael addition reactions between Ni(II)-complexes of glycine and chiral 3-(E-enoyl)-1,3-oxazolidin-2-ones. Tetrahedron Lett 41:9645–9649
Soloshonok VA, Cai C, Hruby VJ (2000c) Toward design of a practical methodology for stereocontrolled synthesis of χ-constrained pyroglutamic acids and related compounds. Virtually complete control of simple diastereoselectivity in the Michael addition reactions of glycine Ni(II) complexes with N-(enoyl)oxazolidinones. Tetrahedron Lett 41:135–139
Soloshonok VA, Cai C, Hruby VJ (2000d) (S)- or (R)-3-(E-enoyl)-4-phenyl-1,3-oxazolidin-2-ones: ideal Michael acceptors to afford a virtually complete control of simple and face diastereoselectivity in addition reactions with glycine derivatives. Org Lett 2:747–750
Soloshonok VA, Tang X, Hruby VJ (2001a) Large-scale asymmetric synthesis of novel sterically constrained 2′,6′-dimethyl- and α,2′,6′-trimethyltyrosine and -phenylalanine derivatives via alkylation of chiral equivalents of nucleophilic glycine and alanine. Tetrahedron 57:6375–6382
Soloshonok VA, Tang X, Hruby VJ, Van Meervelt L (2001b) Asymmetric synthesis of α, β-dialkyl-α-phenylalanines via direct alkylation of a chiral alanine derivative with racemic α-alkylbenzyl bromides. A case of high enantiomer differentiation at room temperature. Org Lett 3:341–343
Soloshonok VA, Ueki H, Tiwari R, Cai C, Hruby VJ (2004) Virtually complete control of simple and face diastereoselectivity in the Michael addition reactions between achiral equivalents of a nucleophilic glycine and (S)- or (R)-3-(E-enoyl)-4-phenyl-1,3-oxazolidin-2-ones: practical method for preparation of β-substituted pyroglutamic acids and prolines. J Org Chem 69:4984–4990
Soloshonok VA, Cai C, Yamada T, Ueki H, Ohfune Y, Hruby VJ (2005a) Michael addition reactions between chiral equivalents of a nucleophilic glycine and (S)- or (R)-3-(E-enoyl)-4-phenyl-1,3-oxazolidin-2-ones as a general method for efficient preparation of β-substituted pyroglutamic acids. Case of topographically controlled stereoselectivity. J Am Chem Soc 127:15296–15303
Soloshonok VA, Ueki H, Ellis TK (2005b) New generation of nucleophilic glycine equivalents. Tetrahedron Lett 46:941–944
Soloshonok VA, Ueki H, Ellis TK, Yamada T, Ohfune Y (2005c) Application of modular nucleophilic glycine equivalents for truly practical asymmetric synthesis of β-substituted pyroglutamic acids. Tetrahedron Lett 46:1107–1110
Soloshonok VA, Ohkura H, Yasumoto M (2006a) Operationally convenient asymmetric synthesis of (S)- and (R)-3-amino-4,4,4-trifluorobutanoic acid. Part I: enantioselective biomimetic transamination of isopropyl 4,4,4-trifluoro-3-oxobutanoate. J Fluorine Chem 127:924–929
Soloshonok VA, Ohkura H, Yasumoto M (2006b) Operationally convenient asymmetric synthesis of (S)- and (R)-3-amino-4,4,4-trifluorobutanoic acid. Part II: enantioselective biomimetic transamination of 4,4,4-trifluoro-3-oxo-N-[(R)-1-phenylethyl)butanamide. J Fluorine Chem 127:930–935
Soloshonok VA, Yamada T, Ueki H, Moore AM, Cook TK, Arbogast KL, Soloshonok AV, Martin CH, Ohfune Y (2006c) Operationally convenient, efficient asymmetric synthesis of enantiomerically pure 4-aminoglutamic acids via methylene dimerization of chiral glycine equivalents with dichloromethane. Tetrahedron 62:6412–6419
Soloshonok VA, Boettiger TU, Bolene SB (2008a) Asymmetric synthesis of (2S,3S)- and (2R,3R)-α,β-dialkyl-α-amino acids via alkylation of chiral nickel(II) complexes of aliphatic α-amino acids with racemic α-alkylbenzyl bromides. Synthesis 2008(16):2594–2602
Soloshonok VA, Ueki H, Ellis TK (2008b) Modular approach to the design of nucleophilic glycine equivalents with tailorable physico-chemical properties. Chimica Oggi/Chem Today 26:51–54
Soloshonok VA, Yamada T, Sakaguchi K, Ohfune Y (2009a) Concise asymmetric synthesis of configurationally stable 4-trifluoromethyl thalidomide. Future Med Chem 1:897–908
Soloshonok VA, Ueki H, Ellis TK (2009b) New generation of modular nucleophilic glycine equivalents for the general synthesis of α-amino acids. Synlett 2009(5):704–715
Soloshonok VA, Ellis TK, Ueki H, Ono T (2009c) Resolution/deracemization of chiral α-amino acids using resolving reagents with flexible stereogenic centers. J Am Chem Soc 131:7208–7209
Soloshonok VA, Ono T, Ueki H, Vanthuyne N, Balaban TS, Bürck J, Fliegl H, Klopper W, Naubron J-V, Bui TTT, Drake AF, Roussel C (2010) Ridge-tile-like chiral topology: synthesis, resolution, and complete chiroptical characterization of enantiomers of edge-sharing binuclear square planar complexes of Ni(II) bearing achiral ligands. J Am Chem Soc 132:10477–10483
Sorochinsky AE, Hisanori U, Aceña JL, Ellis TK, Moriwaki H, Sato T, Soloshonok VA (2013) Chemical deracemization and (S) to (R) interconversion of some fluorine-containing α-amino acids. J Fluor Chem. doi:10.1016/j.jfluchem.2013.02.022
Stork G, Leong AYW, Touzin AM (1976) Alkylation and Michael additions of glycine ethyl ester. Use in α-amino acid synthesis and as acyl carbanion equivalent. J Org Chem 41:3491–3493
Tang X, Soloshonok VA, Hruby VJ (2000) Convenient, asymmetric synthesis of enantiomerically pure 2′,6′-dimethyltyrosine (DMT) via alkylation of chiral equivalent of nucleophilic glycine. Tetrahedron Asymmetry 11:2917–2925
Tararov VI, Saveleva TF, Kuznetsov NY, Ikonnikov NS, Orlova SA, Belokon YN, North M (1997) Synthesis of both enantiomers of β, β-diphenyl-α-alanine (Dip) from glycine using (S)- or (R)-2-[(N-benzylpropyl)amino]benzophenone as reusable chiral auxiliary. Tetrahedron Asymmetry 8:79–83
Taylor SM, Yamada T, Ueki H, Soloshonok VA (2004) Asymmetric synthesis of enantiomerically pure 4-aminoglutamic acids via methylenedimerization of chiral glycine equivalents with dichloromethane under operationally convenient conditions. Tetrahedron Lett 45:9159–9162
Ueki H, Ellis TK, Martin CH, Boettiger TU, Bolene SB, Soloshonok VA (2003a) Improved synthesis of proline-derived Ni(II) complexes of glycine: versatile chiral equivalents of nucleophilic glycine for general asymmetric synthesis of α-amino acids. J Org Chem 68:7104–7107
Ueki H, Ellis TK, Martin CH, Soloshonok VA (2003b) Efficient large-scale synthesis of picolinic acid-derived nickel(II) complexes of glycine. Eur J Org Chem 2003(10):1954–1957
Undheim K (2008) The Schöllkopf chiron and transition metal mediated reactions, a powerful combination for stereoselective construction of cyclic α-quaternary-α-amino acid derivatives. Amino Acids 34:357–402
Vadon-Legoff S, Dijols S, Mansuy D, Boucher J-L (2005) Improved and high yield synthesis of the potent arginase inhibitor: 2(S)-amino-6-boronohexanoic acid. Org Process Res Dev 9:677–679
Wang J, Liu X, Feng X (2011a) Asymmetric Strecker reactions. Chem Rev 111:6947–6983
Wang J, Lin D, Zhou S, Ding X, Soloshonok VA, Liu H (2011b) Asymmetric synthesis of sterically and electronically demanding linear ω-trifluoromethyl containing amino acids via alkylation of chiral equivalents of nucleophilic glycine and alanine. J Org Chem 76:684–687
Wang J, Liu H, Aceña JL, Houck D, Takeda R, Moriwaki H, Sato T, Soloshonok VA (2013) Synthesis of bis-α, α′-amino acids through diastereoselective bis-alkylations of chiral Ni(II)-complexes of glycine. Org Biomol Chem 11:4508–4515
Williams RM, Im M-N (1991) Asymmetric synthesis of monosubstituted and α, α-disubstituted α-amino acids via diastereoselective glycine enolate alkylations. J Am Chem Soc 113:9276–9286
Williams RM, Sinclair PJ, Zhai W (1988) Asymmetric synthesis of β-carboxyaspartic acid. J Am Chem Soc 110:482–483
Yamada T, Okada T, Sakaguchi K, Ohfune Y, Ueki H, Soloshonok VA (2006) Efficient asymmetric synthesis of novel 4-substituted and configurationally stable analogues of thalidomide. Org Lett 8:5625–5628
Yamada T, Sakaguchi K, Shinada T, Ohfune Y, Soloshonok VA (2008) Efficient asymmetric synthesis of the functionalized pyroglutamate core unit common to oxazolomycin and neooxazolomycin using Michael reaction of nucleophilic glycine Schiff base with α, β-disubstituted acrylate. Tetrahedron Asymmetry 19:2789–2795
Yasumoto M, Ueki H, Soloshonok VA (2007) Base-free, thermal 1,3-proton shift reaction and its application for operationally convenient and improved synthesis of α-(trifluoromethyl)benzylamine. J Fluorine Chem 128:736–739
Zlatopolskiy BD, Loscha K, Alvermann P, Kozhushkov SI, Nikolaev SV, Zeeck A, de Meijere A (2004) Final elucidation of the absolute configuration of the signal metabolite hormaomycin. Chem Eur J 10:4708–4717
Acknowledgments
We thank IKERBASQUE, Basque Foundation for Science; Basque Government (SAIOTEK S-PE12UN044) and Hamari Chemicals (Osaka, Japan) for generous financial support.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sorochinsky, A.E., Aceña, J.L., Moriwaki, H. et al. Asymmetric synthesis of α-amino acids via homologation of Ni(II) complexes of glycine Schiff bases; Part 1: alkyl halide alkylations. Amino Acids 45, 691–718 (2013). https://doi.org/10.1007/s00726-013-1539-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-013-1539-4