
Abstract
l-arginine (l-Arg) is metabolized by nitric oxide synthase and arginase enzymes. The gastric pathogen Helicobacter pylori causes peptic ulcer disease and gastric cancer. We have shown that alterations in l-Arg availability and metabolism into polyamines contribute significantly to the dysregulation of the host immune response to this infection. Nitric oxide (NO) derived from inducible NO synthase (iNOS) can kill H. pylori. There are multiple mechanisms leading to failure of this process, including competition for l-Arg substrate by H. pylori arginase, and induction of host macrophage arginase II (Arg2) and ornithine decarboxylase (ODC). Generation of spermine by ODC inhibits iNOS translation and NO-mediated H. pylori killing. Expression of ODC is dependent on formation of a unique AP-1 complex, leading to upregulation of c-Myc as a transcriptional enhancer. Macrophage apoptosis is mediated by oxidation of spermine via the enzyme spermine oxidase (SMO) that generates hydrogen peroxide (H2O2), and thus oxidative stress-induced mitochondrial membrane polarization. Our studies have demonstrated that apoptosis occurs through a pERK → pc-Fos/c-Jun → c-Myc → ODC → SMO pathway. In gastric epithelial cells, activation of oxidative stress by H. pylori is dependent on SMO induction and results in both apoptosis and DNA damage, such that inhibition or knockdown of SMO markedly attenuates these events. In summary, l-Arg metabolism by the arginase–ODC pathway and the activation of SMO leads to H. pylori-induced DNA damage and immune dysregulation through polyamine-mediated oxidative stress and impairment of antimicrobial NO synthesis. Our studies indicate novel targets for therapeutic intervention in H. pylori-associated diseases, including gastritis, ulcer disease, and gastric cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Helicobacter pylori is a microaerophilic, Gram-negative bacterium that selectively colonizes the human stomach and causes chronic gastritis, peptic ulcers, and gastric cancer (Marshall and Warren 1984; Correa 1992; Uemura et al. 2001). Gastric adenocarcinoma is the second leading cause of cancer-related death worldwide, and chronic gastritis induced by H. pylori is the strongest known risk factor for this malignancy (Nomura et al. 1991; Parsonnet et al. 1991; Peek and Blaser 2002). Of those infected, approximately 10% develop peptic ulcers and 1% develop carcinoma (Nomura et al. 1991; Parsonnet et al. 1991; Peek and Blaser 2002). Factors shown to contribute to the risk for development of gastric cancer include host genetic susceptibility (El-Omar et al. 2000), phylogenetic origin (de Sablet et al. 2011) and virulence factors (Basso et al. 2008; Blaser et al. 1995) of H. pylori strains, and diet (Dorant et al. 1996; Piazuelo et al. 2008). Moreover, the persistence of H. pylori in the gastric mucosa despite eliciting a vigorous innate and adaptive immune response is a hallmark of the infection and is considered to be a major cause for malignant transformation (Wilson and Crabtree 2007; Peek et al. 2010; Wroblewski et al. 2010). Thus, various mechanisms have been proposed to explain how H. pylori evades host immune responses, such as induction of apoptosis in T cells (Wang et al. 2001a) and macrophages (Gobert et al. 2002; Chaturvedi et al. 2004; Cheng et al. 2005; Asim et al. 2010; Menaker et al. 2004). In addition, increased regulatory T cells have been implicated (Rad et al. 2006).
Polyamines have been shown to attenuate immune responses by inhibiting cytokine production in inflammatory diseases. Moreover, polyamine catabolism by the enzyme spermine oxidase (SMO; PAOh1) generates reactive oxygen species (ROS), which may cause DNA damage and cell apoptosis (Wang et al. 2001b; Vujcic et al. 2002; Pledgie et al. 2005; Chaturvedi et al. 2004; Xu et al. 2004). In this review, we will discuss the mechanisms by which polyamines dysregulate the host immune response, modulate apoptosis, and induce oxidative damage in gastric epithelial cells during H. pylori infection.
Biosynthesis of polyamines in cells infected with H. pylori
Polyamines are polycationic amino acids that are synthesized by the arginase–ornithine decarboxylase (ODC) pathway. Arginase metabolizes l-arginine (l-Arg) into l-ornithine plus urea; in the cytosol, l-ornithine is converted to putrescine by ODC, and in the mitochondria into l-proline by ornithine aminotransferase (Li et al. 2002; Pegg and McCann 1982). ODC, a rate-limiting enzyme, converts l-ornithine into putrescine, which can then be converted to spermidine and spermine by spermidine synthase and spermine synthase, respectively (Pegg and McCann 1982). We have shown that H. pylori induces arginase II (Arg2) (Gobert et al. 2002; Lewis et al. 2010, 2011) and ODC (Gobert et al. 2002; Chaturvedi et al. 2004; Bussiere et al. 2005; Cheng et al. 2005; Asim et al. 2010; Chaturvedi et al. 2010) in macrophages in vitro and in vivo and causes an increase in polyamine levels (Chaturvedi et al. 2004, 2010) (Fig. 1).

Schematic of the inhibition of iNOS-dependent innate immune response. H. pylori upregulates Arg2 which reduces l-Arg in the cytosol that is needed for iNOS translation; ODC converts l-ornithine into the polyamines putrescine, spermidine, and spermine; and spermine inhibits l-Arg uptake and thus iNOS protein translation and NO production. Inhibition of NO synthesis leads to decreased killing of H. pylori and thus its survival in the gastric niche
Induction of arginase
Arginase enzymes are the endogenous antagonists to inducible nitric oxide (NO) synthase (iNOS) because they compete for the same l-Arg substrate by metabolizing it to l-ornithine and urea (Wu and Morris 1998). The latter is used by ODC to produce the polyamine putrescine, which is further metabolized to form spermidine and spermine. There are two isoforms of arginase: arginase I (Arg1) is abundant in liver and is important for the urea cycle, and arginase II (Arg2) is abundant in kidney and localizes to mitochondria (Nissim et al. 2005; Li et al. 2001; Wu and Morris 1998). H. pylori infection causes an increase in Arg2 expression in the RAW 264.7 murine macrophage cell line and in primary peritoneal macrophages (Gobert et al. 2002; Lewis et al. 2010). A time course study showed that Arg2 mRNA expression was upregulated after 2 h of activation with H. pylori, which continued out to 24 h; in contrast, Arg1 was not induced, and a decrease was observed at 6 h and later time points (Gobert et al. 2002). This increase in Arg2 expression was confirmed with Northern blot analysis (Gobert et al. 2002).
Arg2 protein expression is also significantly enhanced in response to H. pylori in macrophages (Fig. 1), and Arg1 protein is not induced (Gobert et al. 2002; Lewis et al. 2010). Immunofluorescence detection with double staining for Arg2 and MitoTracker dye showed that Arg2 localizes to mitochondria (Lewis et al. 2010). A dramatic increase in arginase activity was observed in H. pylori-stimulated macrophages after 12 h (Lewis et al. 2010). When arginase activity was assessed in cytosolic and mitochondrial subcellular fractions, it was only increased in the latter (Lewis et al. 2010), which further confirms that H. pylori-induced Arg2 is a mitochondrial protein.
Upregulation of Arg2 in RAW 264.7 cells was also observed when live H. pylori were separated from macrophages by a Transwell filter support (Gobert et al. 2002). These data suggest that H. pylori-derived factors are potent activators of Arg2. We have also demonstrated that Arg2 levels are increased in mouse and human H. pylori gastritis tissues (Gobert et al. 2002; Lewis et al. 2011). There is a marked and consistent increase in Arg2 mRNA levels compared with control tissues, and the basal expression of Arg1 in uninfected control tissues is actually decreased in H. pylori-infected tissues (Gobert et al. 2002; Lewis et al. 2011). Immunofluorescence staining of formalin-fixed, paraffin-embedded gastric tissue sections from wild-type (WT) and Arg2−/− mice on the C57BL/6 background demonstrated increased Arg2 levels in H. pylori-infected tissues from WT mice that localized to F4/80+ macrophages (Lewis et al. 2011). Taken together, these studies provide evidence that H. pylori induces Arg2 in macrophages in vitro, and in gastric mucosa of mice and humans.
Biological consequences of upregulation of Arg2
Simultaneous induction of both iNOS and arginase in macrophages is uncommon, as studies have demonstrated that induction of one usually leads to the inhibition of the other. However, several pathogens have devised strategies to upregulate arginase to suppress iNOS-dependent host defense. For example, downregulation of NO production by macrophages has been attributed to induction of Arg1 by the intracellular parasites Leishmania major (Iniesta et al. 2001) and Toxoplasma gondii (El Kasmi et al. 2008), and the intracellular bacterium Mycobacterium tuberculosis (El Kasmi et al. 2008); and to induction of Arg2 by the extracellular parasite Trypanosoma brucei brucei (Duleu et al. 2004) and the intracellular bacteria Chlamydia psittaci and C. pneumoniae (Huang et al. 2002).
Decrease in NO production in these studies has been attributed to substrate competition. In contrast, in H. pylori infection, increased Arg2 activity inhibits iNOS translation, and this is the mechanism of inhibition of NO production (Lewis et al. 2010) (Fig. 1). We reported that inhibition of arginase with S-(2-boronoethyl)-l-cysteine (BEC) or with small interfering RNA (siRNA) significantly enhanced NO generation in H. pylori-stimulated RAW 264.7 macrophages by enhancing iNOS protein translation (Lewis et al. 2010). The resulting increase in NO resulted in increased killing of H. pylori, which was verified by attenuated antimicrobial action of macrophages with an NO scavenger (Lewis et al. 2010). Similarly, H. pylori-stimulated iNOS protein expression and NO production were increased in peritoneal macrophages isolated from Arg2−/− mice (Lewis et al. 2010). In H. pylori-infected mice, treatment with BEC or deletion of Arg2 increased iNOS protein levels and NO generation in gastric macrophages (Lewis et al. 2010). This study indicated that Arg2 activation decreased the availability of intracellular l-Arg for iNOS translation and NO production. Consistent with our data, decreased l-Arg levels have been shown to inhibit iNOS translation in astrocytes (Lee et al. 2003).
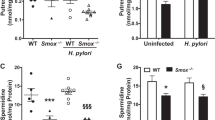
Using a chronic H. pylori infection model, we determined that Arg2 impairs host defense in vivo (Lewis et al. 2011). Arg2−/− mice exhibited increased histologic gastritis and decreased bacterial colonization compared with WT mice. Importantly, when individual mice were considered, in Arg2−/− mice gastritis scores were inversely correlated with H. pylori colonization; thus more inflammatory response was associated with less colonization, but in WT mice this benefit of the inflammatory response did not occur (Lewis et al. 2011). Consistent with more effective host defense to H. pylori, when compared with WT mice, Arg2−/− mice had more gastric macrophages, more of these cells were iNOS+, and these cells expressed higher levels of iNOS protein (Lewis et al. 2011). To assess NO levels in situ, we used nitrotyrosine staining as a marker, and found that it was increased in the inflammatory cells in infected Arg2−/−, but not WT mice (Lewis et al. 2011). Because Arg2−/− mice had more gastric macrophages, we assessed apoptosis and found that it was markedly decreased during acute infection when measured by flow cytometry in isolated cells and in situ by staining for cleaved caspase-3 (Lewis et al. 2011). We also demonstrated enhanced mRNA expression levels of IFN-γ, IL-17a, and IL-12p40, and reduced IL-10 levels by real-time PCR, consistent with a more vigorous Th1/Th17 response (Lewis et al. 2011). These studies demonstrate that Arg2 contributes to the immune evasion of H. pylori by limiting macrophage iNOS protein expression and NO production (Fig. 1), causing macrophage apoptosis, and inhibiting proinflammatory cytokine responses. A remaining question is the role of polyamines as downstream mediators of the effects of Arg2 in macrophages in vivo. To address this, we intend to measure polyamine levels in Arg2−/− mice at several time points post-inoculation, and to cross Arg2−/− mice with mice that have heterozygous deletion of ODC and reduced polyamine synthesis.
Induction of ODC in macrophages
H. pylori induces ODC, the rate-limiting enzyme for polyamine synthesis (Fig. 1). We initially described an increase in ODC mRNA expression and activity in H. pylori-stimulated macrophages (Gobert et al. 2002). A detailed study of ODC activity (Chaturvedi et al. 2004) showed a biphasic increase that peaked at 6 h, followed by a decline at 12 h, and a second peak at 18 h (Fig. 2a). We demonstrated that H. pylori induced ODC promoter activity that correlated with mRNA and protein expression (Fig. 3) (Cheng et al. 2005). As shown in Fig. 3a, H. pylori addition to RAW 264.7 macrophages resulted in a time-dependent, 12-fold increase in ODC promoter activity that peaked at 6 h after bacterial stimulation. There was a nearly identical degree and time course of induction of ODC mRNA expression, determined by real-time PCR (Fig. 3b), and protein expression as assessed by Western blot analysis (Fig. 3c). These data indicated that in H. pylori-stimulated macrophages, a time-dependent increase in ODC promoter activation leads directly to an increase in ODC activity, and thus modulation of polyamine content (Cheng et al. 2005).

Time course of induction of enzyme activity of ODC and alterations in polyamine levels in response to H. pylori. RAW 264.7 macrophages were stimulated with H. pylori lysate at a multiplicity of infection of 100 a ODC activity was measured by conversion of L-[14C]ornithine to 14CO2. b Polyamine levels in cellular lysates, c enzyme activity levels *p < 0.05; **p < 0.01 versus time 0; § p < 0.05; §§ p < 0.01 versus peak level (data are from Chaturvedi et al. 2004)

Time course of induction of ODC in H. pylori-stimulated macrophages. RAW 264.7 cells were exposed to H. pylori lysate for the times indicated; a promoter activity determined by luciferase reporter assay using a −264-bp functional ODC promoter; b mRNA levels determined by real-time PCR; c Western blot for ODC (53 kDa) and β-actin (42 kDa); * p < 0.05; ** p < 0.01 versus time 0 (data are from Cheng et al. 2005)
H. pylori increases levels of polyamines in macrophages
Measurements of the levels of putrescine, spermidine, and spermine provide biological significance for increased ODC in macrophages stimulated with H. pylori. As shown in Fig. 2b, after exposure to lysates of H. pylori, there was no detectable level of putrescine observed up to 12 h, but a marked increase was observed at 18 h, and this was significantly reduced at 24 h (Chaturvedi et al. 2004). In contrast, there was an increase in spermidine at 6 and 12 h and an increase in spermine at 12 h, followed by a decline in these two polyamines that was inversely proportional to the increase in putrescine from 12 to 24 h (Fig. 2b). These polyamine data are consistent with the back-conversion of spermine and spermidine mediated by the induction of catabolic enzymes from 6 to 18 h, as shown in Fig. 2c (Chaturvedi et al. 2004). Induction of catabolic enzymes and effects on polyamine levels will be discussed in detail in this review.
Lack of induction of ODC in gastric epithelial cells
In our studies, we have tested several gastric epithelial cell lines (AGS and MKN28 human carcinoma cells, and conditionally immortalized mouse stomach cells) and found no increase in the levels of ODC mRNA in H. pylori-infected cells (Asim et al. 2010). In fact, it has been reported that H. pylori decreases ODC activity in gastric epithelial cells (Takashima et al. 2002). As discussed below, our tissue studies have demonstrated that ODC is not upregulated in gastric epithelial cells in infected tissues.
Induction of ODC in gastric tissues
Epidemiological studies have demonstrated that ODC mRNA expression is associated with an increased risk of gastric cancer. It has been reported that H. pylori eradication decreases ODC mRNA expression and activity (Konturek et al. 2003), but the source had not been known. It had also been reported that there are increased polyamine levels in gastric tissues from H. pylori-infected human subjects (Linsalata et al. 1998) and that spermine levels were increased in intestinal-type gastric tumors, typical of H. pylori-induced cancer, compared with diffuse gastric tumors (Russo et al. 1997). We have recently reported (Chaturvedi et al. 2010) that ODC mRNA is increased in human H. pylori infection compared with H. pylori-negative gastritis or normal tissues, and that experimental infection in mice also induces ODC mRNA expression. We also demonstrated by immunohistochemistry that ODC localizes to the lamina propria mononuclear cells in human H. pylori gastritis (Chaturvedi et al. 2010) and that ODC protein expression is increased in isolated gastric macrophages from infected mice when assessed by flow cytometry (Asim et al. 2010).
Molecular mechanism of ODC induction
Ornithine decarboxylase is known to be a transcriptional target of c-Myc, and the ODC promoter has a putative c-Myc binding sequence in the untranslated exon1 region (Bello-Fernandez et al. 1993). We therefore tested whether c-Myc binding was involved in the transcriptional activation of ODC in response to H. pylori. c-Myc mRNA and protein expression were induced in murine macrophages (Cheng et al. 2005). In response to H. pylori, newly synthesized c-Myc translocates to the nucleus and binds to the ODC promoter, which we demonstrated by electromobility shift assay (Cheng et al. 2005). Inhibition of c-Myc binding to the promoter region by a dominant-negative c-Myc construct or by an antennapedia-linked peptide binding inhibitor resulted in inhibition of ODC promoter activity, mRNA expression, and protein expression (Cheng et al. 2005). However, using deletion constructs, we found that even in the presence of c-Myc induction and an intact c-Myc binding site, there was no upregulation of ODC promoter activity unless the minimal upstream −264 bp ODC promoter was left intact, indicating that there is an H. pylori response element (HPRE) is in this region (Figs. 4, 5). These findings suggested that c-Myc served as a transcriptional enhancer in conjunction with the HPRE to activate ODC expression in response to H. pylori.

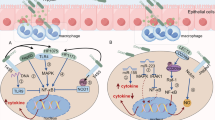
Schematic of the molecular mechanism of c-Myc and ODC induction in H. pylori-stimulated macrophages. H. pylori causes nuclear translocation of phosphorylated ERK leading to generation of a phospho-c-Fos/c-Jun AP-1 complex that binds to the c-Myc promoter to increase c-Myc expression that enhances ODC expression in conjunction with activation of an H. pylori response element

Schematic presentation of the mechanism for apoptosis in H. pylori-stimulated macrophages. H. pylori induced synthesis of polyamines by the Arg2-ODC pathway is followed by release of H2O2 by SMO that causes apoptosis
When we assessed the activation of these events further, we found that H. pylori induced phosphorylation of ERK1/2 and that phosphorylated ERK translocated to the nucleus and activated an AP-1 transcription complex (Asim et al. 2010) (Fig. 4). Inhibition of ERK phosphorylation by chemical inhibitors or inhibition of AP-1 complex binding by ectopic overexpression of a dominant-negative form of c-Fos inhibited c-Myc and ODC expression in parallel (Asim et al. 2010). Immunoprecipitation, fluorescence resonance energy transfer, and oligonucleotide pull-down assays showed that the H. pylori-induced AP-1 complex was uniquely composed of a phospho-c-Fos and c-Jun heterodimer (Asim et al. 2010) (Fig. 4). The formation of this unique AP-1 complex also depends on phosphorylation of ERK and its translocation to the nucleus. Chromatin immunoprecipitation and fluorescence polarization assays using antibodies against phospho-c-Fos and c-Jun confirmed the binding of these AP-1 components to the c-Myc promoter (Asim et al. 2010). Our studies were consistent with previous reports that H. pylori induced AP-1 activity (Kudo et al. 2007; Yamauchi et al. 2008), but the formation of a specific phospho-c-Fos and c-Jun heterodimer appeared to be unique to macrophages, and the involvement of this AP-1 complex in c-Myc expression had not been previously identified.
The ERK-pc-Fos/cJun-c-Myc pathway provides mechanistic insight into how ODC is induced in macrophages. In addition, it should be noted that this pathway was not activated in macrophages stimulated by other bacteria, such as Camplyobacter jejuni or Citrobacter rodentium, which were tested because they are enteric pathogens that cause colitis in mice (Asim et al. 2010). Also, H. pylori does not induce this specific AP-1 complex in gastric epithelial cells, and the induction of ERK phosphorylation is much more transient in macrophages than epithelial cells in response to H. pylori (Asim et al. 2010). These findings may help explain why ODC is differentially regulated in macrophages versus gastric epithelial cells. The importance of our findings is further highlighted by the fact that gene polymorphisms of ODC were not identified in H. pylori infection (You et al. 2005). In addition, ODC has not emerged as a gene undergoing epigenetic regulation during H. pylori infection (Schneider et al. 2010).
Biological consequences of ODC induction
We have reported that in macrophages, H. pylori upregulates iNOS and antimicrobial NO production, but in parallel there is induction of Arg2, generating ornithine, and of ODC, generating polyamines (Fig. 1). Spermine, in particular, has been shown to restrain immune response in activated macrophages by inhibiting proinflammatory gene expression (Zhang et al. 1997). We have reported that spermine inhibits NO production in a dose-dependent manner in a murine macrophage cell line and in peritoneal macrophages with an IC50 of 9.2 and 9.0 μM, respectively (Bussiere et al. 2005) (Fig. 1). Spermine did not alter iNOS mRNA expression levels, but inhibited iNOS protein expression levels (Bussiere et al. 2005). Using [35S] methionine for radiolabeling of proteins, we showed that spermine inhibits de novo synthesis of iNOS protein expression (Bussiere et al. 2005). Pulse chase experiments confirmed that spermine inhibits iNOS protein synthesis, but not the stability of iNOS protein (Bussiere et al. 2005). Further studies involving knockdown of ODC expression by siRNA demonstrated that depletion of endogenous polyamines increases de novo synthesis of iNOS protein and results in an increase in NO production, which leads to more effective killing of H. pylori (Bussiere et al. 2005) (Fig. 1). Treatment of macrophages with spermidine also inhibited H. pylori-stimulated NO production and iNOS protein levels, but to a lesser degree than that which occurred with spermine, and putrescine had no inhibitory effect (Bussiere et al. 2005). These data suggest a specific mechanism by which H. pylori suppresses iNOS expression at the translational level, which allows this pathogen to avoid the innate antimicrobial response of the host.
In addition, we have reported that NO generation in H. pylori-stimulated macrophages is strongly dependent on l-Arg availability in the culture medium and requires concentrations well above the circulating physiologic level of l-Arg in mice and human beings of 0.1 mM to maximize response (Chaturvedi et al. 2007). This effect of l-Arg was shown to be concentration dependent and was mediated primarily by an enhancement of iNOS translation with increasing l-Arg levels (Chaturvedi et al. 2007). In cells starved of l-Arg, there was nearly complete loss of global protein translation, but this was fully restored at 0.1 mM l-Arg and did not increase further at higher levels of l-Arg in culture medium. Similarly, other H. pylori-inducible enzymes, ODC and cyclooxygenase-2, were absent without l-Arg, but fully restored at 0.1 mM l-Arg. Thus the concentration-dependent effect of l-Arg was specific to iNOS (Chaturvedi et al. 2007).
l-Arg uptake in macrophages has been attributed to the y+ cationic amino acid (CAT) transport system (Kakuda et al. 1999). CAT2, a member of the CAT family of proteins, transports l-Arg into macrophages (Nicholson et al. 2001). We have reported that H. pylori induces CAT2 mRNA expression in macrophages in vitro and in infected gastric tissues from mice and humans, with localization of CAT2 protein to lamina propria macrophages (Chaturvedi et al. 2010). H. pylori increases l-Arg uptake in a biphasic, time-dependent manner. Spermine did not decrease the levels of CAT2 mRNA or protein expression, but inhibited l-Arg uptake in macrophages when added exogenously to the cell culture medium (Chaturvedi et al. 2010) (Fig. 1). Specifically, we detected decreases in l-Arg uptake of 85.3 ± 8.2 and 99.8 ± 2.4% at 6 and 24 h after activation with H. pylori (Chaturvedi et al. 2010). Similar to the effect of spermine, knockdown of CAT2 with siRNA decreased iNOS protein expression and production of NO, but not expression of iNOS mRNA (Chaturvedi et al. 2010). Furthermore, knockdown of ODC with siRNA reduces intracellular levels of spermine (Bussiere et al. 2005) and increases l-Arg uptake in macrophages in a sustained manner, and led to earlier, more sustained, and higher levels of iNOS expression and NO production (Chaturvedi et al. 2010). Taken together, these results indicate a novel pathway for inhibition of host defense by H. pylori, such that increased endogenous levels of spermine in macrophages due to induction of ODC causes an inhibition of l-Arg uptake and thus suboptimal levels of iNOS and NO that are needed to kill the bacterium (Fig. 1).
To directly determine the effect of spermine accumulation on the NO-dependent innate immune response to H. pylori, we administered the ODC inhibitor α-difluoromethylornithine (DFMO) to mice (1% wt/vol in the drinking water) for the 4-month course of infection. With H. pylori infection, there was a twofold increase in polyamine levels in gastric tissues that were reduced to basal levels with DFMO treatment (Chaturvedi et al. 2010). In freshly isolated gastric macrophages from H. pylori-infected mice, the levels of l-Arg uptake, iNOS protein, and NO were enhanced significantly in cells from mice treated with DFMO when compared with cells from infected mice receiving water alone (Chaturvedi et al. 2010). Importantly, direct assessment of gastric colonization with H. pylori revealed that DFMO treatment of mice resulted in a significant, 2-log-order reduction in bacterial levels when assessed by culture or quantitative PCR (Chaturvedi et al. 2010). In parallel, there was a significant reduction in gastric inflammation in DFMO-treated mice.
In addition to affecting iNOS translation, polyamines also modulate H. pylori-induced apoptosis. H. pylori causes apoptosis in macrophages in a time-dependent manner that parallels the Arg2 and ODC activity kinetics in H. pylori-stimulated macrophages (Gobert et al. 2002). Two different arginase inhibitors, N (omega) hydroxy-l-arginine and N(omega)-hydroxy-nor-l-arginine, each attenuated H. pylori-stimulated macrophage apoptosis (Gobert et al. 2002). Inhibition of ODC, which utilizes l-ornithine as substrate generated by arginase activity, also blocked apoptosis in an H. pylori-stimulated murine macrophage cell line (Gobert et al. 2002) (Fig. 5). We observed inhibition of H. pylori-induced apoptosis when ODC expression was blocked by either inhibiting c-Myc binding to its partner protein by a dominant-negative form of c-Myc protein or treating cells with a cell-permeable peptide that inhibits binding of the c-Myc/Max complex to its putative DNA-binding sequence in the ODC promoter (Cheng et al. 2005). Furthermore, inhibition of phospho-c-Fos/cJun AP-1 complex formation in H. pylori-activated macrophages by the ERK inhibitor, PD 98059, or by overexpression of a vector expressing a dominant-negative form of c-Fos resulted in inhibition of c-Myc/ODC-mediated apoptosis in macrophages (Asim et al. 2010) (Fig. 4).
In a mouse model of H. pylori infection, using flow cytometry and staining of macrophages with the cell surface marker F4/80, a rapid, 12-fold increase in the number of macrophages was observed in mouse gastric tissues, peaking at 2 days post-inoculation, but the number of these cells rapidly decreased to levels of uninfected mice by 3 days post-inoculation (Asim et al. 2010). This decrease in cell counts was accompanied by a large increase in macrophage apoptosis that peaked at 2 days post-inoculation (Asim et al. 2010). Based on our in vitro findings, we tested the ERK inhibitor, PD 98059, in vivo, and found that pretreatment with this agent before infection with H. pylori markedly decreased levels of phospho-ERK, phospho-c-Fos, the phospho-c-Fos/c-Jun AP1 complex, ODC, and apoptosis in gastric macrophages (Asim et al. 2010). Similarly, apoptosis has been linked to polyamines in intestinal epithelial cells (Yuan et al. 2002).
Catabolism of polyamines in cells
Back-conversion of spermidine and spermine by catabolic enzymes has been proposed to induce apoptosis in lung and breast cancer cell lines (Ha et al. 1997; Pledgie et al. 2005). We therefore postulated that H. pylori might induce polyamine catabolic enzymes that produce H2O2. In one pathway, spermine or spermidine is metabolized by the enzyme spermidine/spermine N 1-acetyltransferase (SSAT) to acetylspermine and acetylspermidine, respectively, which are then acted upon by peroxisomal acetyl polyamine oxidase (APAO) to form spermidine and putrescine, respectively (Pegg and McCann 1982). Spermine can be oxidized directly and specifically to spermidine without the intermediate acetylation step by spermine oxidase. Initially, this enzyme was called PAOh1, but was later renamed as spermine oxidase (SMO) (Wang et al. 2001b; Vujcic et al. 2002; Pledgie et al. 2005; Chaturvedi et al. 2004; Xu et al. 2004).
Induction of catabolic enzymes by H. pylori
In macrophages, H. pylori increases levels of ODC, SSAT, and SMO mRNA expression, but not that of APAO, in macrophages (Chaturvedi et al. 2004). Ornithine decarboxylase enzyme activity shows a biphasic increase that initially peaks at 6 h and then again at 18 h after stimulation (Fig. 2a). Spermine oxidase activity peaks at 18 h, but is significantly increased as early as 6 h after stimulation, whereas SSAT activity does not increase until 18 h after stimulation and APAO activity is not increased as shown in Fig. 2c (Chaturvedi et al. 2004). Polyamine measurements showed a parallel between catabolic enzyme activity and levels of putrescine, spermidine, and spermine; there was an increase in levels of spermidine and spermine from 6 to 12 h after stimulation by H. pylori, followed by a clear decline in these two polyamines that was inversely proportional to the increase in putrescine from 12 to 18 h (Fig. 2b). These polyamine data are consistent with the back-conversion of spermine to spermidine that is mediated by the induction of SMO from 6 to 18 h, and the subsequent increase in putrescine is likely due to the late increase in SSAT activity and subsequent back-conversion of acetylated polyamines by APAO (Fig. 1).
We have also reported that H. pylori induces a significant increase in SMO mRNA expression in the gastric epithelial AGS cell line (Xu et al. 2004). Stimulation with H. pylori resulted in a significant increase in SMO promoter activity with the −1,117-bp minimal promoter construct (Xu et al. 2004), suggesting that the observed increase in SMO mRNA is regulated at the transcriptional level and is associated with H. pylori infection. We have observed SMO induction in other gastric cancer cell lines as well as in non-malignant immortalized gastric epithelial cells. We have observed induction of SMO with several H. pylori strains that we have tested.
In addition, we have found a significant increase in SMO mRNA expression in mouse and human H. pylori gastritis tissues (Xu et al. 2004). Furthermore, we observed that levels of SMO in human gastritis tissues from H. pylori-negative patients with gastritis were only modestly increased, whereas tissues from H. pylori-infected patients exhibited consistently higher levels of expression (Xu et al. 2004). We also showed that SMO was increased in gastric epithelium by assessing SMO mRNA expression levels in epithelial cells harvested by laser capture micro-dissection from gastric tissues (Xu et al. 2004).
Biological consequences of induction of SMO
MDL 72527, an inhibitor of SMO activity, attenuated H. pylori-induced apoptosis in macrophages and this observation was confirmed with knockdown studies using siRNA to SMO (Chaturvedi et al. 2004). Further, MDL 72527 decreased levels of ROS measured by flow cytometry, using CM-H2DCFDA, and levels of H2O2 measured by Amplex red (Chaturvedi et al. 2004). Inhibition of SMO by MDL 72527 also blocked mitochondrial membrane depolarization induced by H. pylori and cytochrome c release and activation of caspase-3 in macrophages (Fig. 5). Inhibition of these events by MDL 72527 or siRNA knockdown of SMO indicates that apoptosis in macrophages during H. pylori stimulation is due to oxidation of polyamines, specifically spermine (Chaturvedi et al. 2004). Exogenous overexpression of SMO also resulted in an induction of apoptosis (Chaturvedi et al. 2004). Because APAO was not induced at the mRNA level and the activity was also not increased by H. pylori, we focused on SMO; when we performed siRNA knockdown, which resulted in complete attenuation of the H. pylori-induced apoptosis in macrophages (Chaturvedi et al. 2004). In addition, in the mouse model of H. pylori infection, gastric macrophage SMO mRNA levels were increased by 10.3 ± 3.4-fold (p < 0.001, data not shown).
In summary, in response to H. pylori, l-Arg is transported into macrophages and metabolized by Arg2 into l-ornithine, which ODC then converts into polyamines. H. pylori-induced SMO can then back-convert spermine into spermidine and release H2O2 that depolarizes mitochondrial membrane potential leading to release of cytochrome c into the cytosol, activation of caspase-3, and apoptosis in macrophages (Fig. 5).
In epithelial cells, H. pylori-induced apoptosis is also mediated by H2O2 generated by SMO. We implicated polyamine oxidation as the source of oxidative stress by using MDL 72527 (Xu et al. 2004). We reported that knockdown of SMO with siRNA resulted in marked attenuation of both apoptosis and DNA damage, directly demonstrating the role of SMO in these events in H. pylori-stimulated cells (Xu et al. 2004). Importantly, we found that H. pylori did not induce any increase in APAO activity in gastric epithelial cells (Xu et al. 2004). As in macrophages, inhibition of SMO in epithelial cells attenuated mitochondrial membrane depolarization, cytochrome c release, and activation of caspase-3 (Xu et al. 2004). Oxidative stress that occurs during interactions between H. pylori and gastric epithelial cells can lead to DNA damage in vivo in humans and mice by causing nucleotide oxidation (Bagchi et al. 2002; Davies et al. 1994; Farinati et al. 1998). Chronic H. pylori infection increases mutation frequency leading to base substitutions and frame shift mutations (Touati et al. 2003), and accumulation of cells with damaged DNA may occur by downregulation of pro-apoptotic proteins (Wei et al. 2010). These findings highlight the importance of oxidative stress during H. pylori infection, as the fate of cells with damaged DNA is critical in the development of H. pylori-induced cancer.
In our epithelial cell model of H. pylori infection, DNA damage was shown to be increased by infection, when assessed by comet assay (Fig. 6a–d) or by levels of 8-oxoguanosine (Fig. 6e) (Xu et al. 2004). The latter was measured using an FITC-labeled binding peptide, specifically indicating that there was oxidative DNA damage. The H. pylori-induced DNA damage was attenuated by the H2O2 detoxifying agent, catalase, or by MDL 72527 to a similar degree (Fig. 6a–d), and in addition, knockdown of SMO with siRNA also effectively abrogated the oxidative DNA damage (Fig. 6e) (Xu et al. 2004). Figure 7 demonstrates the proposed pathway for generation of oxidative stress in gastric epithelial cells exposed to H. pylori. We have emphasized the back-conversion of spermine to spermidine by SMO, but the SSAT/APAO pathway is also likely to participate in the metabolism of polyamines in this model and would allow for conversion of spermidine to putrescine. It is important to note that some ROS-induced mutations may not be lethal and may lead to growth and/or survival advantages during gastrointestinal inflammation (Meira et al. 2008). 8-oxoguanosine, as a DNA adduct that results from H2O2 production, can produce G to T transversion mutations that are commonly found in tumor suppressor genes and oncogenes (Hussain and Harris 1998; Cheng et al. 1992). Additionally, we have reported that H. pylori-induced phosphorylation of the epidermal growth factor receptor (EGFR), which results in its transactivation, leads to attenuation of H. pylori-induced apoptosis in gastric epithelial cells (Yan et al. 2009). Cells with DNA damage that avoid apoptotic death could in time accumulate sufficient numbers of mutations to become neoplastically transformed (Fig. 7). To this point, we have observed that in conditionally immortalized gastric epithelial cells, and in isolated cells from infected stomachs of gerbils and hypergastrinemic mice, there is a population of cells that are resistant to apoptosis despite exhibiting DNA damage.

H. pylori-induced DNA damage in gastric epithelial cells. Comet assay after alkaline electrophoresis and propidium iodide staining after 6 h of stimulation with: a control, b H. pylori, c H. pylori + MDL 72527, d H. pylori + catalase; e flow cytometry of 8-oxoguanosine levels showing a shift to the right with H. pylori in cells transfected with scrambled siRNA that was abrogated in cells transfected with SMO (PAOh1) siRNA (data are from Xu et al. 2004)

Schematic of the role of SMO in gastric epithelial cells exposed to H. pylori. Induction of SMO generates oxidative stress that can result in both apoptosis and DNA damage, with increased survival of cells with DNA damage when apoptosis is attenuated by EGFR transactivation resulting in heightened risk for acquisition of molecular alterations that can lead to neoplastic transformation
Clinical implications
In general, rates of H. pylori infection correlate with gastric cancer and are greatest in underdeveloped countries (Peek and Blaser 2002). The fact that infection rates are high in underdeveloped countries raises practical issues related to the management of H. pylori-associated disease, including: cost of antibiotic treatment, high rates of treatment failure in these regions (Camargo et al. 2007; Mera et al. 2005), and frequent reinfection after antibiotic treatment (Mera et al. 2005). H. pylori may often behave as a commensal organism (Blaser 1997), making it more difficult to determine which individuals should be treated, and in addition the infection is inversely correlated with asthma (Chen and Blaser 2008) and esophageal reflux disease/cancer (Varanasi et al. 1998; Islami and Kamangar 2008), a counter-argument to universal treatment. Therefore, enhanced understanding of the immunopathogenesis of the infection and the stratification of cancer risk is needed. We contend that the Arg2-ODC pathway in macrophages is a major mediator of the failed immune response to H. pylori. Further, the generation of DNA damage from the oxidation of spermine by SMO induction may be a central mechanism in the development of neoplastic transformation.
Taken together, these concepts suggest that an effective target for therapeutic intervention in H. pylori infection could be interference with synthesis and/or oxidation of polyamines. A similar strategy has been used for the treatment of tropical diseases such as trypanosomiasis, Chagas’ disease, and leishmaniasis with inhibitors of polyamine synthesis (Heby et al. 2007). In our chemoprevention study in mice, inhibition of ODC with DFMO and subsequent reduction in polyamines leads to enhanced host defense against this infection (Chaturvedi et al. 2010). We speculate that inhibition of polyamine synthesis could have an additional benefit of reducing oxidative stress (and associated DNA damage) by depleting polyamines, including spermine, which was reduced in our chronic treatment model (Chaturvedi et al. 2010). To this end, studies are underway in laboratory on mouse and gerbil models of gastric cancer using DFMO and MDL 72527.
Another intriguing point is that we have recently reported that treatment of H. pylori with DFMO in vitro reduces its growth and leads to bacteria of a smaller size that are similar to the less virulent coccoid form of H. pylori (Barry et al. 2011). In addition, DFMO reduces protein levels of CagA, an H. pylori-derived virulence factor that is translocated into gastric epithelial cells by a type IV secretion system. This was associated with decreased translocation, as evidenced by less phosphorylated CagA in gastric epithelial cells in a co-culture model (Barry et al. 2011). However, the direct effects of DFMO on H. pylori were reversible, indicating that it was unlikely to be the main mechanism of inhibition of H. pylori colonization in vivo; in fact, we found that pretreatment of H. pylori had no effect on colonization when mice were not treated with DFMO (Barry et al. 2011). Further, we have recent evidence that when ODC+/− mice were infected with H. pylori, there was decreased colonization compared with WT mice, and this was associated with increased macrophage iNOS levels. Notably, DFMO has been shown to be effective in the chemoprevention of intestinal neoplastic lesions in the ApcMin/+ mouse model (Yerushalmi et al. 2006) and of recurrent adenomatous colon polyps when used in combination with the non-steroidal anti-inflammatory drug sulindac in human studies (Meyskens et al. 2008). Thus, inhibition of the polyamine metabolic pathway could have several important benefits in H. pylori infection by improving host defense and reducing oxidative stress and neoplastic risk. It could prove to be a useful adjunctive treatment for H. pylori infection.
Abbreviations
- H. pylori :
-
Helicobacter pylori
- NO:
-
Nitric oxide
- iNOS:
-
Inducible NO synthase
- l-Arg:
-
l-Arginine
- Arg1:
-
Arginase I
- Arg2:
-
Arginase II
- ODC:
-
Ornithine decarboxylase
- PAO1:
-
Polyamine oxidase 1
- SMO:
-
Spermine oxidase
- H2O2 :
-
Hydrogen peroxide
- ROS:
-
Reactive oxygen species
- BEC:
-
S-(2-Boronoethyl)-l-cysteine
- siRNA:
-
Small interfering RNA
- DFMO:
-
α-Difluoromethylornithine
- CAT:
-
Cationic amino acid transporter
- SSAT:
-
Spermidine/spermine N 1-acetyltransferase
- APAO:
-
Acetyl polyamine oxidase
- EGFR:
-
Epidermal growth factor
- WT:
-
Wild type
References
Asim M, Chaturvedi R, Hoge S, Lewis ND, Singh K, Barry DP, Algood HS, de Sablet T, Gobert AP, Wilson KT (2010) Helicobacter pylori induces ERK-dependent formation of a phospho-c-Fos c-Jun activator protein-1 complex that causes apoptosis in macrophages. J Biol Chem 285:20343–20357
Bagchi D, McGinn TR, Ye X, Bagchi M, Krohn RL, Chatterjee A, Stohs SJ (2002) Helicobacter pylori-induced oxidative stress and DNA damage in a primary culture of human gastric mucosal cells. Dig Dis Sci 47:1405–1412
Barry DP, Asim M, Leiman DA, de Sablet T, Singh K, Casero RA, Chaturvedi R, Wilson KT (2011) Difluoromethylornithine is a novel inhibitor of Helicobacter pylori growth, CagA translocation, and interleukin-8 Induction. PLoS One 6:e17510
Basso D, Zambon CF, Letley DP, Stranges A, Marchet A, Rhead JL, Schiavon S, Guariso G, Ceroti M, Nitti D, Rugge M, Plebani M, Atherton JC (2008) Clinical relevance of Helicobacter pylori cagA and vacA gene polymorphisms. Gastroenterology 135:91–99
Bello-Fernandez C, Packham G, Cleveland JL (1993) The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc Natl Acad Sci USA 90:7804–7808
Blaser MJ (1997) Ecology of Helicobacter pylori in the human stomach. J Clin Invest 100:759–762
Blaser MJ, Perez–Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, Stemmermann GN, Nomura A (1995) Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res 55:2111–2115
Bussiere FI, Chaturvedi R, Cheng Y, Gobert AP, Asim M, Blumberg DR, Xu H, Kim PY, Hacker A, Casero RA Jr, Wilson KT (2005) Spermine causes loss of innate immune response to Helicobacter pylori by inhibition of inducible nitric-oxide synthase translation. J Biol Chem 280:2409–2412
Camargo MC, Piazuelo MB, Mera RM, Fontham ET, Delgado AG, Yepez MC, Ceron C, Bravo LE, Bravo JC, Correa P (2007) Effect of smoking on failure of H. pylori therapy and gastric histology in a high gastric cancer risk area of Colombia. Acta Gastroenterol Latinoam 37:238–245
Chaturvedi R, Cheng Y, Asim M, Bussiere FI, Xu H, Gobert AP, Hacker A, Casero RA Jr, Wilson KT (2004) Induction of polyamine oxidase 1 by Helicobacter pylori causes macrophage apoptosis by hydrogen peroxide release and mitochondrial membrane depolarization. J Biol Chem 279:40161–40173
Chaturvedi R, Asim M, Lewis ND, Algood HM, Cover TL, Kim PY, Wilson KT (2007) l-Arginine availability regulates inducible nitric oxide synthase-dependent host defense against Helicobacter pylori. Infect Immun 75:4305–4315
Chaturvedi R, Asim M, Hoge S, Lewis ND, Singh K, Barry DP, de Sablet T, Piazuelo MB, Sarvaria AR, Cheng Y, Closs EI, Casero RA, Jr., Gobert AP, Wilson KT (2010) Polyamines impair immunity to Helicobacter pylori by inhibiting l-arginine uptake required for nitric oxide production. Gastroenterology 139:1686–1698, 1698 e1681–1686
Chen Y, Blaser MJ (2008) Helicobacter pylori colonization is inversely associated with childhood asthma. J Infect Dis 198:553–560
Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA (1992) 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G–T and A–C substitutions. J Biol Chem 267:166–172
Cheng Y, Chaturvedi R, Asim M, Bussiere FI, Xu H, Casero RA Jr, Wilson KT (2005) Helicobacter pylori-induced macrophage apoptosis requires activation of ornithine decarboxylase by c-Myc. J Biol Chem 280:22492–22496
Correa P (1992) Human gastric carcinogenesis: a multistep and multifactorial process–First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res 52:6735–6740
Davies GR, Banatvala N, Collins CE, Sheaff MT, Abdi Y, Clements L, Rampton DS (1994) Relationship between infective load of Helicobacter pylori and reactive oxygen metabolite production in antral mucosa. Scand J Gastroenterol 29:419–424
de Sablet T, Piazuelo MB, Shaffer CL, Schneider BG, Asim M, Chaturvedi R, Bravo LE, Sicinschi LA, Delgado AG, Mera RM, Israel DA, Romero-Gallo J, Peek RM, Jr, Cover TL, Correa P, Wilson KT (2011) Phylogeographic origin of Helicobacter pylori is a determinant of gastric cancer risk. Gut (in press)
Dorant E, van den Brandt PA, Goldbohm RA, Sturmans F (1996) Consumption of onions and a reduced risk of stomach carcinoma. Gastroenterology 110:12–20
Duleu S, Vincendeau P, Courtois P, Semballa S, Lagroye I, Daulouede S, Boucher JL, Wilson KT, Veyret B, Gobert AP (2004) Mouse strain susceptibility to trypanosome infection: an arginase-dependent effect. J Immunol 172:6298–6303
El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, Basaraba RJ, Konig T, Schleicher U, Koo MS, Kaplan G, Fitzgerald KA, Tuomanen EI, Orme IM, Kanneganti TD, Bogdan C, Wynn TA, Murray PJ (2008) Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol 9:1399–1406
El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, Lanyon G, Martin M, Fraumeni JF Jr, Rabkin CS (2000) Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 404:398–402
Farinati F, Cardin R, Degan P, Rugge M, Mario FD, Bonvicini P, Naccarato R (1998) Oxidative DNA damage accumulation in gastric carcinogenesis. Gut 42:351–356
Gobert AP, Cheng Y, Wang JY, Boucher JL, Iyer RK, Cederbaum SD, Casero RA Jr, Newton JC, Wilson KT (2002) Helicobacter pylori induces macrophage apoptosis by activation of arginase II. J Immunol 168:4692–4700
Ha HC, Woster PM, Yager JD, Casero RA Jr (1997) The role of polyamine catabolism in polyamine analogue-induced programmed cell death. Proc Natl Acad Sci USA 94:11557–11562
Heby O, Persson L, Rentala M (2007) Targeting the polyamine biosynthetic enzymes: a promising approach to therapy of African sleeping sickness, Chagas’ disease, and leishmaniasis. Amino Acids 33:359–366
Huang J, DeGraves FJ, Lenz SD, Gao D, Feng P, Li D, Schlapp T, Kaltenboeck B (2002) The quantity of nitric oxide released by macrophages regulates Chlamydia-induced disease. Proc Natl Acad Sci USA 99:3914–3919
Hussain SP, Harris CC (1998) Molecular epidemiology of human cancer: contribution of mutation spectra studies of tumor suppressor genes. Cancer Res 58:4023–4037
Iniesta V, Gomez-Nieto LC, Corraliza I (2001) The inhibition of arginase by N(omega)-hydroxy-l-arginine controls the growth of Leishmania inside macrophages. J Exp Med 193:777–784
Islami F, Kamangar F (2008) Helicobacter pylori and esophageal cancer risk: a meta-analysis. Cancer Prev Res (Phila Pa) 1:329–338
Kakuda DK, Sweet MJ, Mac Leod CL, Hume DA, Markovich D (1999) CAT2-mediated l-arginine transport and nitric oxide production in activated macrophages. Biochem J 340(Pt 2):549–553
Konturek PC, Rembiasz K, Konturek SJ, Stachura J, Bielanski W, Galuschka K, Karcz D, Hahn EG (2003) Gene expression of ornithine decarboxylase, cyclooxygenase-2, and gastrin in atrophic gastric mucosa infected with Helicobacter pylori before and after eradication therapy. Dig Dis Sci 48:36–46
Kudo T, Lu H, Wu JY, Ohno T, Wu MJ, Genta RM, Graham DY, Yamaoka Y (2007) Pattern of transcription factor activation in Helicobacter pylori-infected Mongolian gerbils. Gastroenterology 132:1024–1038
Lee J, Ryu H, Ferrante RJ, Morris SM Jr, Ratan RR (2003) Translational control of inducible nitric oxide synthase expression by arginine can explain the arginine paradox. Proc Natl Acad Sci USA 100:4843–4848
Lewis ND, Asim M, Barry DP, Singh K, de Sablet T, Boucher JL, Gobert AP, Chaturvedi R, Wilson KT (2010) Arginase II restricts host defense to Helicobacter pylori by attenuating inducible nitric oxide synthase translation in macrophages. J Immunol 184:2572–2582
Lewis ND, Asim M, Barry DP, de Sablet T, Singh K, Piazuelo MB, Gobert AP, Chaturvedi R, Wilson KT (2011) Immune evasion by Helicobacter pylori is mediated by induction of macrophage arginase II. J Immunol 186:3632–3641
Li H, Meininger CJ, Hawker JR Jr, Haynes TE, Kepka-Lenhart D, Mistry SK, Morris SM Jr, Wu G (2001) Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am J Physiol Endocrinol Metab 280:E75–E82
Li H, Meininger CJ, Kelly KA, Hawker JR Jr, Morris SM Jr, Wu G (2002) Activities of arginase I and II are limiting for endothelial cell proliferation. Am J Physiol Regul Integr Comp Physiol 282:R64–R69
Linsalata M, Russo F, Notarnicola M, Berloco P, Di Leo A (1998) Polyamine profile in human gastric mucosa infected by Helicobacter pylori. Ital J Gastroenterol Hepatol 30:484–489
Marshall BJ, Warren JR (1984) Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1:1311–1315
Meira LB, Bugni JM, Green SL, Lee CW, Pang B, Borenshtein D, Rickman BH, Rogers AB, Moroski-Erkul CA, McFaline JL, Schauer DB, Dedon PC, Fox JG, Samson LD (2008) DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest 118:2516–2525
Menaker RJ, Ceponis PJ, Jones NL (2004) Helicobacter pylori induces apoptosis of macrophages in association with alterations in the mitochondrial pathway. Infect Immun 72:2889–2898
Mera R, Fontham ET, Bravo LE, Bravo JC, Piazuelo MB, Camargo MC, Correa P (2005) Long term follow up of patients treated for Helicobacter pylori infection. Gut 54:1536–1540
Meyskens FL, McLaren CE, Pelot D, Fujikawa-Brooks S, Carpenter PM, Hawk E, Kelloff G, Lawson MJ, Kidao J, McCracken J, Albers CG, Ahnen DJ, Turgeon DK, Goldschmid S, Lance P, Hagedorn CH, Gillen DL, Gerner EW (2008) Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prev Res Phila Pa 1:9–11
Nicholson B, Manner CK, Kleeman J, MacLeod CL (2001) Sustained nitric oxide production in macrophages requires the arginine transporter CAT2. J Biol Chem 276:15881–15885
Nissim I, Luhovyy B, Horyn O, Daikhin Y, Yudkoff M (2005) The role of mitochondrially bound arginase in the regulation of urea synthesis: studies with [U-15N4]arginine, isolated mitochondria, and perfused rat liver. J Biol Chem 280:17715–17724
Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez–Perez GI, Blaser MJ (1991) Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med 325:1132–1136
Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK (1991) Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 325:1127–1131
Peek RM Jr, Blaser MJ (2002) Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer 2:28–37
Peek RM Jr, Fiske C, Wilson KT (2010) Role of innate immunity in Helicobacter pylori-induced gastric malignancy. Physiol Rev 90:831–858
Pegg AE, McCann PP (1982) Polyamine metabolism and function. Am J Physiol 243:C212–C221
Piazuelo MB, Camargo MC, Mera RM, Delgado AG, Peek RM Jr, Correa H, Schneider BG, Sicinschi LA, Mora Y, Bravo LE, Correa P (2008) Eosinophils and mast cells in chronic gastritis: possible implications in carcinogenesis. Hum Pathol 39:1360–1369
Pledgie A, Huang Y, Hacker A, Zhang Z, Woster PM, Davidson NE, Casero RA Jr (2005) Spermine oxidase SMO(PAOh1), Not N1-acetylpolyamine oxidase PAO, is the primary source of cytotoxic H2O2 in polyamine analogue-treated human breast cancer cell lines. J Biol Chem 280:39843–39851
Rad R, Brenner L, Bauer S, Schwendy S, Layland L, da Costa CP, Reindl W, Dossumbekova A, Friedrich M, Saur D, Wagner H, Schmid RM, Prinz C (2006) CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology 131:525–537
Russo F, Linsalata M, Giorgio I, Caruso ML, Armentano R, Di Leo A (1997) Polyamine levels and ODC activity in intestinal-type and diffuse-type gastric carcinoma. Dig Dis Sci 42:576–579
Schneider BG, Peng DF, Camargo MC, Piazuelo MB, Sicinschi LA, Mera R, Romero-Gallo J, Delgado AG, Bravo LE, Wilson KT, Peek RM Jr, Correa P, El-Rifai W (2010) Promoter DNA hypermethylation in gastric biopsies from subjects at high and low risk for gastric cancer. Int J Cancer 127:2588–2597
Takashima T, Fujiwara Y, Watanabe T, Tominaga K, Oshitani N, Higuchi K, Matsumoto T, Arakawa T, Hasuma T, Yano Y, Otani S (2002) High molecular protein of Helicobacter pylori responsible for inhibition of ornithine decarboxylase activity of human gastric cultured cells. Aliment Pharmacol Ther 16(Suppl 2):167–173
Touati E, Michel V, Thiberge JM, Wuscher N, Huerre M, Labigne A (2003) Chronic Helicobacter pylori infections induce gastric mutations in mice. Gastroenterology 124:1408–1419
Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ (2001) Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 345:784–789
Varanasi RV, Fantry GT, Wilson KT (1998) Decreased prevalence of Helicobacter pylori infection in gastroesophageal reflux disease. Helicobacter 3:188–194
Vujcic S, Diegelman P, Bacchi CJ, Kramer DL, Porter CW (2002) Identification and characterization of a novel flavin-containing spermine oxidase of mammalian cell origin. Biochem J 367:665–675
Wang J, Brooks EG, Bamford KB, Denning TL, Pappo J, Ernst PB (2001a) Negative selection of T cells by Helicobacter pylori as a model for bacterial strain selection by immune evasion. J Immunol 167:926–934
Wang Y, Devereux W, Woster PM, Stewart TM, Hacker A, Casero RA Jr (2001b) Cloning and characterization of a human polyamine oxidase that is inducible by polyamine analogue exposure. Cancer Res 61:5370–5373
Wei J, Nagy TA, Vilgelm A, Zaika E, Ogden SR, Romero-Gallo J, Piazuelo MB, Correa P, Washington MK, El-Rifai W, Peek RM, Zaika A (2010) Regulation of p53 tumor suppressor by Helicobacter pylori in gastric epithelial cells. Gastroenterology 139:1333–1343
Wilson KT, Crabtree JE (2007) Immunology of Helicobacter pylori: insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology 133:288–308
Wroblewski LE, Peek RM Jr, Wilson KT (2010) Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev 23:713–739
Wu G, Morris SM Jr (1998) Arginine metabolism: nitric oxide and beyond. Biochem J 336:1–17
Xu H, Chaturvedi R, Cheng Y, Bussiere FI, Asim M, Yao MD, Potosky D, Meltzer SJ, Rhee JG, Kim SS, Moss SF, Hacker A, Wang Y, Casero RA Jr, Wilson KT (2004) Spermine oxidation induced by Helicobacter pylori results in apoptosis and DNA damage: implications for gastric carcinogenesis. Cancer Res 64:8521–8525
Yamauchi K, Choi IJ, Lu H, Ogiwara H, Graham DY, Yamaoka Y (2008) Regulation of IL-18 in Helicobacter pylori infection. J Immunol 180:1207–1216
Yan F, Cao H, Chaturvedi R, Krishna U, Hobbs SS, Dempsey PJ, Peek RM Jr, Cover TL, Washington MK, Wilson KT, Polk DB (2009) Epidermal growth factor receptor activation protects gastric epithelial cells from Helicobacter pylori-induced apoptosis. Gastroenterology 136(1297–1307):e1291–e1293
Yerushalmi HF, Besselsen DG, Ignatenko NA, Blohm-Mangone KA, Padilla-Torres JL, Stringer DE, Guillen JM, Holubec H, Payne CM, Gerner EW (2006) Role of polyamines in arginine-dependent colon carcinogenesis in Apc(Min) (/+) mice. Mol Carcinog 45:764–773
You WC, Hong JY, Zhang L, Pan KF, Pee D, Li JY, Ma JL, Rothman N, Caporaso N, Fraumeni JF Jr, Xu GW, Gail MH (2005) Genetic polymorphisms of CYP2E1, GSTT1, GSTP1, GSTM1, ALDH2, and ODC and the risk of advanced precancerous gastric lesions in a Chinese population. Cancer Epidemiol Biomarkers Prev 14:451–458
Yuan Q, Ray RM, Johnson LR (2002) Polyamine depletion prevents camptothecin-induced apoptosis by inhibiting the release of cytochrome c. Am J Physiol Cell Physiol 282:C1290–C1297
Zhang M, Caragine T, Wang H, Cohen PS, Botchkina G, Soda K, Bianchi M, Ulrich P, Cerami A, Sherry B, Tracey KJ (1997) Spermine inhibits proinflammatory cytokine synthesis in human mononuclear cells: a counterregulatory mechanism that restrains the immune response. J Exp Med 185:1759–1768
Acknowledgments
This work was supported by the National Institutes of Health grants R01DK053620, R01AT004821, 3R01AT004821-02S1, P01CA028842, and P01CA116087 (to K.T.W.), the Flow Cytometry Core of the Vanderbilt University Digestive Disease Research Center grant (P30DK058404), a Merit Review Grant from the Office of Medical Research, Department of Veterans Affairs (to K.T.W.), and the Philippe Foundation (T.S. and A.P.G.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chaturvedi, R., de Sablet, T., Coburn, L.A. et al. Arginine and polyamines in Helicobacter pylori-induced immune dysregulation and gastric carcinogenesis. Amino Acids 42, 627–640 (2012). https://doi.org/10.1007/s00726-011-1038-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-011-1038-4