
Abstract
The thermostability of microbial transglutaminase (MTG) of Streptomyces mobaraensis was further improved by saturation mutagenesis and DNA-shuffling. High-throughput screening was used to identify clones with increased thermostability at 55°C. Saturation mutagenesis was performed at seven “hot spots”, previously evolved by random mutagenesis. Mutations at four positions (2, 23, 269, and 294) led to higher thermostability. The variants with single amino acid exchanges comprising the highest thermostabilities were combined by DNA-shuffling. A library of 1,500 clones was screened and variants showing the highest ratio of activities after incubation for 30 min at 55°C relative to a control at 37°C were selected. 116 mutants of this library showed an increased thermostability and 2 clones per deep well plate were sequenced (35 clones). 13 clones showed only the desired sites without additional point mutations and eight variants were purified and characterized. The most thermostable mutant (triple mutant S23V-Y24N-K294L) exhibited a 12-fold higher half-life at 60°C and a 10-fold higher half-life at 50°C compared to the unmodified recombinant wild-type enzyme. From the characterization of different triple mutants differing only in one amino acid residue, it can be concluded that position 294 is especially important for thermostabilization. The simultaneous exchange of amino acids at sites 23, 24, 269 and 289 resulted in a MTG-variant with nearly twofold higher specific activity and a temperature optimum of 55°C. A triple mutant with amino acid substitutions at sites 2, 289 and 294 exhibits a temperature optimum of 60°C, which is 10°C higher than that of the wild-type enzyme.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transglutaminases (protein-glutamine γ-glutamyltransferase, EC 2.3.2.13, TG) are enzymes catalyzing an acyl-transfer reaction between the γ-carboxamide group of a peptide-bound glutamine and the ε-amino group of a lysine. TGs were found in eukaryotes and prokaryotes (Beninati and Piacentini 2004; Ando et al. 1989).
Microbial TG (MTG) can be used in a variety of applications. The MTG is widely used in the food industry for modification of proteins in meat and fish and to support gelling of yogurt and cheese (de Jong and Koppelman 2002). Other applications of the MTG are the site-directed covalent modification of therapeutic proteins, by e.g., the PEGylation of recombinant human interleukin 2 (Sato 2002) or recombinant granulocyte colony-stimulating factor (Maullu et al. 2009). Another field of application is the production of protein-based materials, e.g., films from casein (Patzsch et al. 2010). The properties of the proteins, like water solubility or elasticity, can be influenced by the MTG-catalyzed protein cross-linking (Chambi and Grosso 2006; Oh et al. 2004). For thermoplastic processing by extrusion it would be advantageous to carry out the cross-linking reaction at higher temperatures. For this approach, a thermostable transglutaminase is required.
In vivo toxic enzymes, e.g., the protein cross-linking TG are usually produced as inactive pro-enzymes. For any application of the respective enzyme, activation by proteolytic cleavage of the pro-sequence is required. Recently a MTG from Streptomyces mobaraensis (DSM 40847) carrying a histidine tag (pro-MTG-His6) was overproduced in soluble form in E. coli (Marx et al. 2007) and a suitable activating protease was identified (Marx et al. 2008a; Sommer et al. 2011a).
Directed evolution is a strategy to improve the properties of proteins, e.g., thermostability and activity (Yuan et al. 2005). This technology has been continuously expanded by the availability of molecular biology tools and upcoming of high-throughput screening technologies. One possible approach of directed evolution is the random mutagenesis where mutations are introduced in the gene of an enzyme by, e.g., error-prone PCR (Cadwell and Joyce 1992, 1994). Previously, random mutagenesis based on error-prone PCR was used to improve the thermostability of the MTG from S. mobaraensis. The screening of 5,500 clones for increased thermostability revealed six variants with single and one variant with two amino acid exchanges. The exchanges occurred at certain “hot spots” in the N-terminal domain and resulted in a higher thermostability at 60°C compared to the recombinant wild-type enzyme (Marx et al. 2008b). These mutants were isolated and characterized in regard to their thermostability and their specific activity at different temperatures. The most thermostable variant (S2P) exhibited a 270% increased half-life at 60°C and a twofold higher specific activity than the recombinant wild-type enzyme (FRAP-MTG-His6) (Marx et al. 2008b). With a single base mutation, only 5.7 amino acid substitutions are accessible on an average for a given amino acid residue. Marx et al. (2008b) utilized a mutation rate between one and two base pairs for the random mutagenesis by error-prone PCR.
Saturation mutagenesis was already successfully used to enhance the thermal stability of other enzymes (McLachlan et al. 2008; Miyazaki and Arnold 1999). For example, the optimization of the phosphite dehydrogenase from Pseudomonas stutzeri showed 12 important positions. Saturation mutagenesis of each of these 12 positions resulted in 4 positions, where an amino acid exchange led to an increased thermostability (McLachlan et al. 2008).
Early investigations in the field of directed evolution showed that different mutations in one protein, screened with the same selection pressure, could be additively cooperate (Lowman and Wells 1993; Stemmer 1994). DNA-shuffling developed by Stemmer in 1994 is one possible approach for an in vitro recombination, e.g., the thermostability of an esterase from Bacillus subtilis was increased by several rounds of random mutagenesis and DNA-shuffling (Giver et al. 1998). The combination of two thermostable cytidine deaminases of two Bacillus strains by DNA-shuffling led to a 150% increase in the half-life at 70°C (Park et al. 2009).
In the present work, the results of the saturation mutagenesis of the microbial transglutaminase at previously identified thermostabilizing sites (2, 23, 24, 257, 269, 289, and 294) are presented. For this approach an improved method for the primer design was used which enhances the PCR amplification efficiency (Zheng et al. 2004). Due to the poor annealing of the primers for site 2, site-directed mutagenesis was preformed in order to introduce all possible amino acid residues. In a second step, the seven thermostabilizing sites revealed by site-directed, saturation or random mutagenesis were combined by DNA-shuffling. In vitro recombination was based on the method described by Stemmer (1994), with modifications in the fragmentation reaction as described by Lorimer and Pastan (1995).
Materials and methods
General
Unless otherwise stated, all chemicals were of analytical grade and were purchased from Sigma-Aldrich (Taufkirchen, Germany). The protein marker (Protein Molecular Weight Marker) used for SDS-PAGE was purchased from Fermentas (St. Leon-Rot, Germany). The DNA marker (Massruler DNA Ladder Mix and 50 bp DNA Ladder) used for agarose gel electrophoresis was purchased from Fermentas (St. Leon-Rot, Germany). Material and equipment (Äkta Explorer) for chromatography was purchased from GE Healthcare (Freiburg, Germany). The following commercial enzymes were used: Lysozyme (order No 8259.2, Carl Roth GmbH & Co. KG, Karlsruhe, Germany), Benzonase (order No 1.01695.0001, Merck KGaA, Darmstadt, Germany), Dispase I (order No 354235, BD Biosciences, Heidelberg) and Proteinase K (order No #EO0491, Fermentas GmbH, St. Leon-Rot). Deionized water was used throughout the experiments.
Bacterial strain
Escherichia coli BL21Gold(DE3) was purchased from Stratagene (Amsterdam, The Netherlands).
Saturation mutagenesis
Saturation mutagenesis was used to generate libraries of MTG genes encoding all possible amino acids at sites 2, 23, 24, 257, 269, 289 and 294. The mutagenesis of the MTG gene was carried out using the QuikChange site-directed Mutagenesis Kit (Stratagene, Amsterdam, The Netherlands) according to the manufacturers manual (Stratagene 2007). The primer pairs for each site were designed by the method of Zheng et al. (2004), using 5′-NNG/T for the sense and 5′-A/CNN for the anti-sense strand. The reaction was performed in a total volume of 50 μl, and the reaction mixture was prepared according to the QuikChange™ protocol, using 50 ng of template DNA (MTG gene in plasmid pDJ1-3 (Marx et al. 2007), GenBank™ accession number: EU301664). The PCR conditions were: Initial denaturation 3 s at 95°C followed by 16 cycles of 30 s 95°C, 1 min annealing at 55°C, 5 min elongation at 68°C (performed in a Tpersonal 48, Whatman Biometra, Göttingen, Germany). The parent plasmid was digested with 10 U DpnI (Fermentas, St. Leon-Rot, Germany) for 1 h at 37°C, after which another aliquot of DpnI (10 U) was added and again incubated for 1 h. The reaction mixture was purified with MSB® Spin PCRapace Kit (Invitek, Berlin Germany). 2 μl of the purified plasmids containing the mutated MTG gene was transformed into 40 μl competent cells of E. coli BL21Gold(DE3) (Stratagene, Amsterdam, The Netherlands) using electroporation.
Site-directed mutagenesis
Site-directed mutagenesis was used to introduce amino acids at site 2 which could not be obtained by the saturation mutagenesis and for the generation of the double mutant S23V-Y24N-MTG. The site-directed mutagenesis was performed by using a Pfu DNA polymerase (order No EP0571, Fermentas, St. Leon-Rot, Germany). The reaction mixture contained 1× Pfu Buffer with 2 mM MgSO4, 0.2 mM of each dNTP, 1 μM fw- and rev-Primer, 1.25 U Pfu DNA polymerase and 50 ng template DNA (MTG gene in plasmid pDJ1-3 (Marx et al. 2007), GenBank™ accession number: EU301664). The extension reaction was initiated by pre-heating the reaction mixture to 94°C for 10 min. After adding the Pfu DNA Polymerase, 16 cycles of heating at 95°C for 30 s, annealing at 55–65°C for 30 s according to the melting temperature of the primer pair, followed by elongation at 68°C for 13 min were carried out. The template DNA was digested with 10 U DpnI (Fermentas, St. Leon-Rot, Germany) for 1 h at 37°C, after which another aliquot of DpnI (10 U) was added and incubated again for 1 h. The reaction mixture was purified with MSB® Spin PCRapace Kit (Invitek, Berlin Germany). 2 μl of the purified plasmids containing the mutated MTG gene was transformed into 40 μl competent cells of E. coli BL21Gold(DE3) (Stratagene, Amsterdam, The Netherlands) using electroporation.
DNA-shuffling
The six ~1.1 kbp DNA fragments containing the pro-MTG genes from plasmids pKB2Y, pUHS23V-Y24N, pCM211, pKB269S, pCM224 and pKB294L were amplified by PCR using the primers fw-MTG-NdeI 5′ G GAA TTC CAT ATG GAC AAT GGC GCG GGG GAA G 3′ and rev-MTG-XhoI 5′ CCG CTC GAG CGG CCA GCC CTG CTT TAC CTT GTC 3′ (Marx et al. 2007). Equal amounts of the six gene preparations were mixed and digested with 0.3 U DNase I (order No EN0525 Fermentas, St. Leon-Rot, Germany) in the presence of 2 mM Mn2+ for 2 min at 15°C as described previously (Lorimer and Pastan 1995). After inactivation of DNase I, the DNA fragments in the range of 50 bp were purified using a centrisep column (Princeton Separation Inc., Adelphia, USA). Afterwards, the purified DNA fragments were subjected to 40 cycles of the reassembling-PCR without primers [94°C, 3 min (94°C, 1 min; 55°C 1 min; 72°C, 1 min + 5 s cycle−1, 40 cycles), 72°C, 7 min]. To generate the MTG libraries, a second PCR was performed with the primers fw-MTG-NdeI and rev-MTG-XhoI as described above. The resulting libraries were cloned into the NdeI/XhoI site of pET20b and transformed into E. coli BL21Gold(DE3) (Stratagene, Amsterdam, The Netherlands) using electroporation.
Cultivation and expression of libraries
The cultivation of the mutant libraries and the expression of the enzyme variants were carried out as described for the libraries of random mutagenesis (Marx et al. 2008b). The cells were harvested by centrifugation at 3,222g for 20 min (Centrifuge 5810 R, Eppendorf, Hamburg, Germany). The microtiter plates (MTPs) containing the cell pellet were stored at −80°C until cell lysis.
Cell lysis and activation of pro-MTG-His6 and its mutants
Cell lysis and activation of the pro-MTG-His6 and its mutants were carried out as described previously (Marx et al. 2008b).
Activity test of FRAP-MTG-His6 and its mutants
The activity of MTG-variants was assayed by the colorimetric hydroxamate procedure (Folk and Cole 1966) adapted to MTP format. To 50 μl of supernatant containing the respective enzyme, 90 μl substrate solution (final concentrations: 200 mM Tris/HCl-buffer, 100 mM hydroxylamine, 10 mM reduced glutathione, 30 mM Z-Gln-Gly, pH 6.0) were added. After incubation at 37°C for 10 min in a thermomixer, the reaction was stopped with 160 μl reagent A (1 vol. 3 M HCl, 1 vol. 12% trichloroacetic acid, 1 vol. 5% FeCl3·6H2O (in 0.1 M HCl)). 200 μl of the reaction mixture was transferred to a transparent MTP (No 701304, Brand GmbH & Co. KG, Wertheim, Germany) and the extinction was measured at 525 nm using a microtiter plate reader (FluoStar, BMG Labtech GmbH, Offenburg, Germany). One unit of TG activity is defined as the formation of 1 μmol l-glutamic acid γ-monohydroxamate per min at 37°C.
Screening for thermostable MTG-variants
The screening for thermostable variants of FRAP-MTG-His6 was performed as described previously (Marx et al. 2008b). Briefly, a 96-well MTP with 50 μl of the supernatant containing the activated MTG (variant) was incubated at 55°C using a water bath. After 30 min, the MTP was cooled down in another water bath to 20°C. Afterwards the enzyme activity was determined. Mutants with higher residual activity than the recombinant wild-type enzyme and the respective mutant at the same position achieved by random mutagenesis (Marx et al. 2008b) were selected for a detailed characterization.
Expression and purification of selected variants
Expression and purification of selected variants were carried out as described previously (Marx et al. 2007, 2008a). The pro-MTG-variants were activated with 5 U Proteinase K (Fermentas GmbH, St. Leon-Rot, Germany) per gram cell wet mass for 1 h at 37°C. The incubation was followed by centrifugation for 10 min at 3,222g. After affinity purification, MTG-His6 containing fractions were pooled and the buffer was exchanged to 50 mM Tris/HCl pH 8.0 by size exclusion chromatography (PD-10 column, Sephadex G-25 M, GE Healthcare).
Characterization of selected variants
To determine the rate of inactivation at 60°C, the solution containing the purified enzyme (ten parallel samples, 35 μl each, 15 U ml−1) were incubated in PCR tubes in a PCR thermocycler (Tpersonal 48, Whatman Biometra, Göttingen, Germany) at 60°C for 1–10 min. After every minute, the samples were cooled on ice. The activity was measured at 37°C using the standard assay with 140 μl pre-incubated substrate solution, starting with 10 μl enzyme solution, and stopping with 150 μl reagent A after 20 min.
To determine the rate of inactivation at 50°C, the solution containing the purified enzyme (parallel samples, 35 μl each, 15 U ml−1) was incubated in a PCR tube in a PCR thermocycler (Tpersonal 48, Whatman Biometra, Göttingen, Germany) at 50°C for 0–360 min. After every 30 min, a sample was taken and cooled on ice. The activity was measured as described above, stopping the reaction after 10 min.
The half-life at 50 and 60°C was calculated using exponential fitting of the data points according to the literature (Longo and Combes 1999; O’Fagain 2003).
To determine the specific activity at different temperatures, enzyme samples were diluted to 5 U ml−1 and assayed by the activity test at different temperatures (10–80°C). The protein concentration was determined by measuring the UV-absorption at 280 nm (Pace and Schmid 1997) (FRAP-MTG-His6 ε 1%280 nm = 18.36). In the MTG literature, usually the Bradford assay is used and bovine serum albumin (BSA) for calibration. In order to reduce the time required for the measurements, the factor between the values obtained by direct UV measurement at 280 nm and by the Bradford assay was determined. Protein concentrations determined by direct UV measurement at 280 nm were multiplied by this factor of 1.4 to obtain comparable protein concentrations determined by Bradford assays in the literature.
Results and discussion
Construction of the mutant libraries and estimation of the mutation rate
In order to increase the thermostability of the microbial transglutaminase, saturation mutagenesis was performed separately on each of the following sites of the parent MTG template: 2, 23, 24, 257, 269, 289 and 294. The mutant libraries for each position were constructed using a commercial mutagenesis kit as described in “Materials and methods”. The mutation rate was estimated by sequencing randomly five clones of every library. The mutation efficiency for all thermostabilizing positions of the MTG was determined to be 60 to 100%. With this mutation efficiency, the mutant libraries were constructed in up to four 96-deep-well-plates with 84 clones per plate.
Site-directed mutagenesis of position 2
As described above, the saturation mutagenesis was also performed for position 2 of the MTG. For this site, the mutation efficiency was very poor, probably because of the GC-rich gene sequence. Sequencing revealed only nine genes with the desired exchanges. Most probably, some primers of the degenerated mixture annealed preferentially with the template. To avoid this effect, site-directed mutagenesis for the nine missing amino acids of position 2 was carried out. The mutant library for site 2 was constructed in one deep-well-plate with four clones for every amino acid. The screening for thermostable variants was carried out as described below.
Screening for thermostable variants of FRAP-MTG-His6
The mutant libraries of positions 2, 23, 24, 257, 269, 289, and 294 obtained by saturation and site-directed mutagenesis were screened for increased thermostability at 55°C using a microtiter plate based assay. As described in “Materials and methods”, more thermostable variants were detected in comparison to the respective mutant at the same position previously obtained by random mutagenesis (Marx et al. 2008b). The libraries obtained by saturation mutagenesis of the sites 24, 257, and 289 showed no MTG-variant with a higher residual activity after incubation at 55°C. For the sites 2, 23, 269, and 294 several clones exhibited an increased residual activity. Sequencing showed that three amino acid substitutions at the sites 23 and 294 and two at sites 2 and 269 resulted in a higher residual activity. The sequencing results are summarized in Table 1.
Recombination of thermostabilizing amino acids and screening for improved thermostability
In order to further increase the thermostability, the single amino acid exchanges resulting in the highest MTG thermostabilities were combined by DNA-shuffling. Recombination was performed as explained in “Materials and methods”. Since it is unlikely, to combine the neighbored positions 23 and 24 by DNA-shuffling, a double mutant (S23V-Y24N) was produced by site-directed mutagenesis and used for the shuffling experiments. The recombination event was verified by sequencing some randomly picked clones. 1,500 clones were screened for improved thermostability of MTG at 55 and 37°C and yielded 116 mutants showing a higher residual activity than the wild-type enzyme. The positive clones with the highest ratio of residual activity at 55 and 37°C were selected and sequenced. Of the 35 sequenced genes, 13 mutants showed only the desired sites without additional point mutations. Among the 13 mutants, 6 different clones were detected. Two more mutants showed an additional point mutation at position 23 (S23Y) which also led to an increased thermostability. The sequencing results of the eight clones are summarized in Table 2.
Partial characterization of selected thermostable mutants obtained by saturation mutagenesis and DNA-shuffling
The mutants obtained by saturation mutagenesis and DNA-shuffling and showing an increased thermostability were expressed in shaking flasks, purified by affinity chromatography and characterized as described in “Materials and methods”. The results are summarized in Table 3.
Mutants obtained by saturation mutagenesis
Compared to the wild-type enzyme, all mutants obtained by saturation mutagenesis showed an increased residual activity after pre-incubation at 60°C for 10 min and 50°C for 360 min. The substitution of serine at site 2 shows remarkable effects with respect to the thermostability and the specific activity. Replacing S2 by a tyrosine increased the half-life t 1/2 (60°C) by a factor of 2. As reported before (Marx et al. 2008b), a serine to proline mutation at position 2 obtained by random mutagenesis resulted in only a 1.4-fold increase in thermostability at 60°C. Shimba et al. (2002) introduced three substitutions of S2 (S2R, S2D and S2Y) by site-directed mutagenesis. The specific activity was slightly increased but the thermostability was not investigated. The slight increase reported for the S2Y variant was not confirmed by our own experiments for the same variant. An S2P variant showed a doubled specific activity (Sommer et al. 2011b). According to Kashiwagi et al. (2002), the structure of MTG is stabilized by polar and hydrophobic interactions between the N-terminal loop (Asp1–Ala10) and a loop between Asn276 and Met288. Position 2 is located in the N-terminal loop and obviously affecting the interactions. The beneficial mutation S2Y obviously increases interactions between these two loops resulting in a further stabilization of MTG.
For residue 23, valine, isoleucine, and tyrosine substitutions of serine increased the thermostability by 1.4- to 1.6-fold. Site 23 is closely located to the N-terminal region of the first β-strand ranging from position 26 to 30 (Kashiwagi et al. 2002). It was shown previously that the mutation G25S led to increased heat-sensitivity (Marx et al. 2008b). Obviously, hydrophobic interactions contribute to the stabilization of the MTG in this region.
A lysine to serine substitution at residue 269 resulted in an increased thermostability by a factor of 1.4.
The most thermostable variant (K294L) created by saturating mutagenesis, exhibited a 4.5-fold higher t 1/2 (50°C) and a 2.3-fold higher t 1/2 (60°C). The previously evolved thermostable MTG-mutant (N184S-K294M) exhibited two amino acid substitutions (Marx et al. 2008b). At this time, it was unclear, which site was responsible for the increased thermostability. However, only the position 294 is located in the N-terminal domain where all the other thermostabilizing exchanges are located (Marx et al. 2008b). Therefore, in the present paper, saturation mutagenesis was performed only at site 294 leaving the N184 unchanged. It was confirmed, that the single amino acid exchange K294L was enough to increase the thermostability of the MTG-variant.
Compared to the wild-type enzyme the specific activities of several mutants are increased. The mutants K269D and K294I exhibit a specific activity of 44 and 42 U mg−1, respectively. These values are nearly as high as determined for the S2P mutant obtained by random mutagenesis (46.1 U mg−1) (Sommer et al. 2011b).
Mutants obtained by DNA-shuffling
All mutants obtained by DNA-shuffling showed an increased residual activity after pre-incubation at 50 and 60°C and increased half-life. The inactivation curves at 50 and 60°C of selected variants are shown in Figs. 1 and 2, respectively. Of the eight analyzed mutants, at 60°C UH306, UH308-B, and UH309 showed the highest residual activities compared to the non-mutated FRAP-MTG-His6. After pre-incubation at 50°C over 6 h, the mutants showed a remarkable residual activity of 40–60%, whereas the wild-type enzyme is almost inactive after 2 h. For the mutant UH307 with four combined thermostabilizing sites (23, 24, 269 and 289) a half-life (t 1/2 (50°C)) of 391 min was determined, reflecting a 9.3-fold increase. Compared to the unmodified wild-type enzyme, the most thermostable mutant UH308-B with amino acid substitutions at sites 23, 24, and 294 exhibits a 10- and 12-fold higher half-life at 50 and 60°C, respectively. This mutant is an example for a significantly increased thermostability of an enzyme by combination of the most stabilizing sites without decreasing the activity. As shown previously by directed evolution of a thermostable esterase, enhanced thermostability does not necessarily come at the cost of activity (Giver et al. 1998). As shown by McLachlan et al. (2008), the combination of the most thermostabilizing mutations of a phosphite dehydrogenase resulted in a variant exhibiting a 100-fold increased half-life at 62°C. The mutation K294L was obtained by saturation mutagenesis and resulted in a 2.3-fold increase of the half-life at 60°C. By site-directed mutagenesis a double mutant at sites 23 and 24 was created, showing a 4.4 times higher half-life at 60°C. For the triple mutant UH308-B, the combination of these mutations by DNA-shuffling resulted in a 12-fold higher half-life at 60°C. Compared to the wild-type enzyme, the specific activity of the triple mutant was also increased.

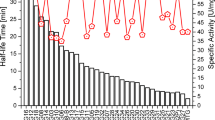
Thermostability of selected thermostable variants at 50°C. The mutants were incubated at 50°C for the respective time, followed by the standard hydroxamate procedure over 10 min (Folk and Cole 1966) (pDJ1-3: recombinant wild-type MTG (Marx et al. 2007), UH306: S2Y-S23Y-H289Y; UH307: S23V-Y24N-G257S-H289Y; UH308-B: S23V-Y24N-K294L)

Thermostability of selected thermostable variants at 60°C. The mutants were incubated at 60°C for the respective time, followed by the standard hydroxamate procedure over 20 min (Folk and Cole 1966) (pDJ1-3: recombinant wild-type MTG (Marx et al. 2007), UH306: S2Y-S23Y-H289Y; UH308-B: S23V-Y24N-K294L; UH309: S2Y-S23Y-K294L)
In Fig. 3, the activity profiles of four thermostable mutants obtained by DNA-shuffling are compared with the wild-type FRAP-MTG-His6. The optimal temperature of the wild-type enzyme is around 50°C.

Specific activity at different temperatures of selected thermostable variants. The substrate solution was pre-incubated for 2 min at the respective temperature, followed by the hydroxamate assay for 10 min (Folk and Cole 1966) (pDJ1-3: recombinant wild-type MTG (Marx et al. 2007), UH302: S23V-Y24N-K269S-H289Y; UH303: S2Y-H289Y-K294L; UH306: S2Y-S23Y-H289Y; UH308-B: S23V-Y24N-K294L)
In Fig. 4, the 3D structure of the microbial transglutaminase is shown. Amino acids which have been introduced during the mutagenesis are shown as sticks and have been labeled in order to visualize the important sites.

Schematic illustration of MTG-Variants evolved by DNA-shuffling. Crystal structure of MTG from Streptomyces mobaraensis (PDB 1iu4) (Kashiwagi et al. 2002). Amino acids were exchanged with the program swiss pdb viewer (http://spdbv.vital-it.ch/) (Guex and Peitsch 1997). The mutated amino acids are colored in red and the Cys64 in the active site cleft is also depicted (yellow color, not mutated). The original figure given by Kashiwagi et al. was turned by 180°, to show the amino acid substitutions, which otherwise would be located on the back side. The figures were produced using PyMOL (Schrodinger 2010)
The combination of the thermostabilizing mutations S2Y, H289Y and K294L resulted in a MTG-variant (UH303) with a temperature optimum at 60°C, a specific activity of 37 U mg−1, and a 9.1-fold increased half-life at 50°C. This is a significant increase in comparison to the unmodified wild-type enzyme. The mutations H289Y and K294L are located in the β7-strand of the MTG structure as defined by Kashiwagi et al. (2002). This sheet, together with a loop (Asn276–Met288) and the β6-strand constitutes the left site wall of the active cleft. Many aromatic residues, like Trp59, Tyr62, Trp69, Tyr75, Tyr278, Tyr291, and Tyr302 are placed on the surface of the active cleft. The mutations H289Y and K294L obviously increase the hydrophobic interactions in this region and stabilize the 3D-structure at higher temperatures without reducing the specific activity. Recently, Yokoyama et al. (2010) also published a slightly increased specific activity for MTG-variants with amino acid substitutions at site 289 to tyrosine or phenylalanine. As already described for the single amino acid exchange, an additional stabilization effect can be assigned to the S2Y mutation.
The specific activity of the quadruple MTG-variant UH302 (S23V-Y24N-K269S-H289Y) is nearly twofold higher than the one of the unmodified wild-type enzyme and as high as the single mutant S2P (Table 3). Interestingly, the position 2 also leading to a doubled specific activity (e.g., S2P) was not detected in this mutant, indicating some potential for further optimization. The mutation K269S is located in vicinity of the β6-strand which is part of the left site wall of the active cleft. Sites 23 and 24 are located closely to the first β-strand. As reported previously, some exchanges located in the first 32 amino acids of the MTG resulted in increased specific activity (Yokoyama et al. 2010). The possible effect of H289Y was discussed above.
The MTG-variants UHS23V-Y24N, UH301 (S23V-Y24N-H289Y), and UH307 (S23V-Y24N-G257S-H289Y) show an increased thermostability at 50 and 60°C. In contrast to UHS23V-Y24N, for the latter two mutants a reduced specific activity was detected (Table 3). The combination of the substitutions H289Y and S23V-Y24N obviously influence the activity negatively. Interestingly, this negative effect is reversed if a K269S mutation is added, as was found for the highly active quadruple mutant UH302 (S23V-Y24N-K269S-H289Y). Among the eight thermostable MTG mutants which were characterized, only one was not carrying a hydrophobic amino acid substitution at site 23 (UH303).
The relatively low activity of UH307 is probably caused by the G257S substitution. Gly257 is located in line with Trp258 and Thr273 which form a hydrogen bond between the main chains of the β5- and β6-strands (Kashiwagi et al. 2002). It is possible that the introduction of Ser at site 257 stabilizes this hydrogen bond network, resulting in increased thermostability. On the other hand, position 257 is located close to the active site Asp255 which could be an explanation for the influence on the specific activity. It would be very interesting to investigate the influence of the respective amino acid substitutions on the substrate specificity and to compare the results to the standard substrate CBZ-Gln-Gly.
Conclusions
In the present paper, the further improvement of thermostable microbial transglutaminase is described. Thermostable MTGs are desired to improve, e.g., the production of biodegradable polymers by extrusion. In this case it is advantageous to carry out the cross-linking reaction with increased activity and at higher temperature.
The first optimization of the microbial transglutaminase from Streptomyces mobaraensis was carried out by random mutagenesis (Marx et al. 2008b). Seven thermostabilizing sites located in the N-terminal domain have been identified. The present paper describes the saturation mutagenesis performed at these “hot spots”. For each site, a library of clones was screened for increased thermostability. Of the seven previously identified sites, only four resulted in amino acid substitutions which led to further increased stability.
The mutations obtained by random mutagenesis and saturation mutagenesis were then combined by DNA-shuffling. Six different MTG genes with altogether seven amino acid substitutions were used for recombination. A mutant library of 1,500 clones was screened for improved thermostability. Improved MTG-variants were sequenced and found to carry different combinations of the mutations as well as new point mutations. Eight variants were characterized. Characterization of the investigated thermostable MTG-variants revealed different sites with different importance for the thermostability. In the most thermostable mutant with a single amino acid exchange, site K294 is replaced by leucine. Also, in different thermostable MTG-variants obtained by DNA-shuffling, the K294L mutation was detected (UH306, UH308-B, and UH309) showing the importance of especially this amino acid exchange.
The most thermostable mutant (triple mutant S23V-Y24N-K294L) exhibited a 12-fold higher half-life at 60°C and a 10-fold higher half-life at 50°C compared to the unmodified recombinant wild-type enzyme.
Abbreviations
- FRAP-MTG-His6 :
-
Recombinant wild-type MTG
- MTG:
-
Microbial transglutaminase
- TG:
-
Transglutaminase
- t 1/2 :
-
Half-life
References
Ando H, Adachi M, Umeda K, Matsuura A, Nonaka M, Uchio R, Tanaka H, Motoki M (1989) Purification and characteristics of a novel transglutaminase derived from microorganisms. Agric Biol Chem 53(10):2613–2617
Beninati S, Piacentini M (2004) The transglutaminase family: an overview: minireview article. Amino Acids 26(4):367–372
Cadwell RC, Joyce GF (1992) Randomization of genes by PCR mutagenesis. PCR Methods Appl 2(1):28–33
Cadwell RC, Joyce GF (1994) Mutagenic PCR. PCR Methods Appl 3(6):S136–S140
Chambi H, Grosso C (2006) Edible films produced with gelatin and casein cross-linked with transglutaminase. Food Res Int 39:458–466
de Jong GA, Koppelman SJ (2002) Transglutaminase catalyzed reactions: impact on food applications. J Food Sci 67(8):2798–2806
Folk JE, Cole PW (1966) Transglutaminase: mechanistic features of the active site as determined by kinetic and inhibitor studies. Biochim Biophys Acta 122(2):244–264
Giver L, Gershenson A, Freskgard P-O, Arnold FH (1998) Directed evolution of a thermostable esterase. Proc Natl Acad Sci USA 95(22):12809–12813
Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-PdbViewer. An environment for comparative protein modeling. Electrophoresis 18(15):2714–2723
Kashiwagi T, Yokoyama K, Ishikawa K, Ono K, Ejima D, Matsui H, Suzuki E (2002) Crystal structure of microbial transglutaminase from Streptoverticillium mobaraense. J Biol Chem 277(46):44252–44260
Longo MA, Combes D (1999) Thermostability of modified enzymes: a detailed study. J Chem Technol Biotechnol 74(1):25–32
Lorimer IAJ, Pastan I (1995) Random recombination of antibody single chain Fv sequences after fragmentation with DNasel in the presence of Mn2+. Nucleic Acids Res 23(15):3067–3068
Lowman HB, Wells JA (1993) Affinity maturation of human growth hormone by monovalent phage display. J Mol Biol 234(3):564–578
Marx C, Hertel T, Pietzsch M (2007) Soluble expression of a pro-transglutaminase from Streptomyces mobaraensis in Escherichia coli. Enzyme Microb Technol 40:1543–1550
Marx CK, Hertel TC, Pietzsch M (2008a) Purification and activation of a recombinant histidine-tagged pro-transglutaminase after soluble expression in E. coli and characterization of the active enzyme. Enzyme Microb Technol 42:568–575
Marx CK, Hertel TC, Pietzsch M (2008b) Random mutagenesis of a recombinant microbial transglutaminase for the generation of thermostable and heat sensitive variants. J Biotechnol 136:156–162
Maullu C, Raimondo D, Caboi F, Giorgetti A, Sergi M, Valentini M, Tonon G, Tramontano A (2009) Site-directed enzymatic PEGylation of the human granulocyte colony-stimulating factor. FEBS J 276(22):6741–6750
McLachlan MJ, Johannes TW, Zhao H (2008) Further improvement of phosphite dehydrogenase thermostability by saturation mutagenesis. Biotechnol Bioeng 99(2):268–274
Miyazaki K, Arnold FH (1999) Exploring nonnatural evolutionary pathways by saturation mutagenesis: rapid improvement of protein function. J Mol Evol 49(6):716–720
O’Fagain C (2003) Enzyme stabilization—recent experimental progress. Enzyme Microb Technol 33(2–3):137–149
Oh J-H, Wang B, Field PD, Aglan HA (2004) Characteristics of edible films made from dairy proteins and zein hydrolysate cross-linked with transglutaminase. Int J Food Sci Technol 39(3):287–294
Pace CN, Schmid FX (1997) How to determine the molar absorption coefficient of a protein. Protein structure: a practical approach. IRL Press, Oxford
Park Y-M, Phi QT, Song B-H, Ghim S-Y (2009) Thermostability of chimeric cytidine deaminase variants produced by DNA shuffling. J Microbiol Biotechnol 19(12):1536–1541
Patzsch K, Riedel K, Pietzsch M (2010) Parameter optimization for protein film production using microbial transglutaminase. Biomacromolecules 11:896–903
Sato H (2002) Enzymatic procedure for site-specific pegylation of proteins. Adv Drug Deliv Rev 54(4):487–504
Schrodinger LLC (2010) The PyMOL Molecular Graphics System, Version 1.3r1
Shimba N, Shinohara M, Yokoyama K, Kashiwagi T, Ishikawa K, Ejima D, Suzuki E (2002) Enhancement of transglutaminase activity by NMR identification of its flexible residues affecting the active site. FEBS Lett 517(1–3):175–179
Sommer C, Hertel TC, Schmelzer CEH, Pietzsch M (2011a) Investigations on the activation of recombinant microbial pro-transglutaminase: in contrast to Proteinase K, Dispase removes the Histidine-tag. Amino Acids. doi:10.1007/s00726-011-1016-x (this issue)
Sommer C, Volk N, Pietzsch M (2011b) Model based optimization of the fed-batch production of a highly active transglutaminase variant in E. coli. Protein Expr Purif 77:9–19
Stemmer WPC (1994) Rapid evolution of a protein in vitro by DNA shuffling. Nature (London) 370(6488):389–391
Stratagene (2007) QuikChange site-directed Mutagenesis Kit. Manufacturer’s Manual
Yokoyama K, Utsumi H, Nakamura T, Ogaya D, Shimba N, Suzuki E, Taguchi S (2010) Screening for improved activity of a transglutaminase from Streptomyces mobaraensis created by a novel rational mutagenesis and random mutagenesis. Appl Microbiol Biotechnol 87(6):2087–2096
Yuan L, Kurek I, English J, Keenan R (2005) Laboratory-directed protein evolution. Microbiol Mol Biol Rev 69(3):373–392
Zheng L, Baumann U, Reymond JL (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res 32(14):e115/111–e115/115
Acknowledgments
This research was supported by the “Fachagentur für Nachwachsende Rohstoffe” (FNR, BMELV, Germany, Project Code 22021807).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Buettner, K., Hertel, T.C. & Pietzsch, M. Increased thermostability of microbial transglutaminase by combination of several hot spots evolved by random and saturation mutagenesis. Amino Acids 42, 987–996 (2012). https://doi.org/10.1007/s00726-011-1015-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-011-1015-y