
Abstract
The analysis by electrospray-ionization tandem mass spectrometry of amino acids with butyl esterification and isotopically labeled internal standard is routine in newborn screening laboratories worldwide. In the present study, we established a direct analysis method of higher accuracy that uses a non-deuterated internal standard. The automatic sampler and the pump of an LC apparatus were used to inject sample and mobile phase to MS, but no LC column was needed. The dry blood spot (DBS) material was prepared at levels of low, medium and high concentration; the running time was 1 min. In parallel to the new procedure, we applied the established method to analyze nine amino acids on DBS of healthy newborns and phenylketonuria newborns. The newly proposed method of product ion confirmation scan along with multiple reaction monitoring resulted in a very accurate identification of each amino acid. Our innovative protocol had high sensitivity and specificity in the analysis of cases of suspected metabolic diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Disorders of amino acid metabolism are a group of rare inherited diseases. Newborn infants with amino acid disorders cannot process certain amino acids (AA) due to the shortage or reduced activity of specific enzymes. The unprocessed AA, along with other toxic substances, then build up in the body and threaten health. For example, elevated phenylalanine results in phenylketonuria (PKU) (Guthrie and Suzi 1963), elevated branched-chain amino acids valine and leucine give rise to maple syrup urine disease (MSUD) (Naylor and Guthrie 1978). So, it is important to simultaneously determine the content in free AA of newborn body fluids as important biomarkers for disorders of amino acid metabolism.
Since most of the AA do not have ultraviolet absorption (UV) or fluorescent emission, the methods that have been used for AA analysis involved a chromatographic separation always with pre-column or post-column derivatization of the AA, with UV absorptive or fluorescent functional group detection (Sarwar and Botting 1993; Fekkes 1996). The most frequently applied method for the quantitative analysis of physiological AA in body fluids is ion-exchange chromatography (IEX-LC) with post-column ninhydrin derivatization (Spackman et al. 1958; Deyl et al. 1986; Qu et al. 2001). However, this technique requires dedicated equipment as classic amino acid analysis and is time consuming, often requiring 2–3 h per sample. The use of HPLC is another conventional technique utilized for AA quantifications with pre-column derivatization (Dale et al. 2003). It has the advantage of producing reliable, cost-effective results and implies no instrument specificity; however, the disadvantage is that samples must be derivatized before processing, and excess reagents impact on the analytical performance. In comparison with HPLC, due to its cost and the requirement of skillful staff, MS instrumentation is less widely diffused in research and routine laboratories but it offers the advantage of rapid and accurate analysis, which makes it more suitable than HPLC for high sample throughput in clinics.
Tandem mass spectrometry (MS/MS) has rapidly gained world-wide support as the method of choice for the screening of newborn metabolic disorders, including the analysis of AA. The analytical process requires that AA are first extracted from DBS, and then chemically modified by butyl esterification. The subsequent analysis by MS/MS focuses on a neutral loss (NL) of 102 Da from neutral and acidic amino acids, of 119 Da from basic amino acids, to improve the chances of detecting the target AA in complex biological matrices (Chace et al. 1993, 1995, 1999; Zytkovicz et al. 2001; Chace and Kalas 2005; Dietzen and Weindel 2010). Although the method provides good sensitivity, the manual derivatization step decreases the reproducibility of the results. LC–MS/MS improves the separation among AA and reduces the noise due to matrix components. The use of the ion-pair reagent in LC separation allows omitting the derivatization step but the ion-pair reagent may eventually produce ion suppression in the mass spectrometry step, and the high proportion of aqueous phase in LC separation shortens the lifetime of the column (Qu et al. 2002; Piraud et al. 2003, 2005a, b; Zoppa et al. 2006; Waterval et al. 2009).
A recent report shows that only minor differences are observed between quantitative values obtained with butylesters and underivatized samples in the analysis of four amino acids (De Jesus et al. 2010). In general, however, we lack knowledge about the quantification by MS/MS of underivatized AA with non-deuterated internal standard.
On this background, this paper presents a method to quantify nine amino acids using MS/MS without processing by butyl esterification; the analysis time with this protocol is only 1 min. To accurately quantify the underivatized AA in DBS, the method of the product ion confirmation scan (PICS) is introduced. The protocol is simple, rapid, sensitive and inexpensive, and can be effectively applied to the screening of a large number of samples in clinics thanks to its short run time and the simple processing of DBS samples.
Experimental
Instrumentation and chemicals
A Perkin Elmer blood spot puncher (1296-071 Delfia) (Milan, Italy) was used for punching discs from blood spots on filter paper. Waters Xevo TQ MS spectrometer with Ionspray source (Micromass UK, Manchester, UK) and a two-way valve connecting the system were utilized for amino acid analysis. Data were recorded on an IBM personal computer using the MassLynx4.0 acquisition and calculation software (Micromass UK).
The amino acids such as l-Phenylalanine (Phe), l-Tyrosine (Tyr), l-Proline (Pro), l-Valine (Val), l-Threonine (Thr), l-Leucine (Leu), l-Lysine (Lys), l-Methionine (Met), l-Glutamic acid (Glu) and the N,N-Dimethyl-l-phenylalanine (N,N-Phe) as internal standard were obtained from Sigma–Aldrich (St. Louis, MO, USA). HPLC grade formic acid was obtained from Tedia (USA). HPLC grade methanol was purchased from Fisher Scientific (Fairlawn, NJ). All the other chemicals were of analytical grade. The standard stock solutions of each unlabeled amino acid were 500 μmol/L and the internal standard stock solution was 400 μmol/L; they were stored at −20°C until used.
Blood spot collection and sample preparation
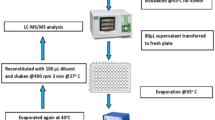
Blood spot samples were from newborns tested by the local Newborn Screening Program. Samples from a group of healthy newborns (n = 100) and a group of PKU newborns (n = 12) were collected on to S&S903 specimen collection paper and allowed to dry for at least 24 h before measurement of amino acid concentrations. Blood spots were immediately used or stored at 4°C. Samples were prepared for MS/MS analysis as follows. About 60 μL of the prepared spiked blood standards (60 μmol/L, 300 μmol/L, and 1.8 mmol/L, of each amino acid) and blood from newborns were spotted directly on to S&S903 filter paper. From every filter paper three blood spot discs (each with diameter = 3.2 mm and ≈3.4 μL whole blood) (Chace et al. 2003) were punched out and transferred to 0.5 ml Eppendorf tubes for three separated analytical runs (n = 9). Then 100 μL of methanol containing 0.1% formic acid with 5 μmol/L internal standard was added to Eppendorf tubes for the extraction of the amino acids. The samples were extracted under sonication for 30 min at room temperature, and no derivatization was carried out. The extract solution was centrifuged at 10,000g and 20°C for 5 min, and the resulting supernatant was used for analysis by MS/MS.
Analysis method
The automatic sampler and pump of an LC apparatus were used to inject the sample and send the flow phase to MS. The mobile phase solvent was 100% methanol. The flow rate was kept constant at 0.4 mL/min, and 2 μL of sample was injected. The running time was 1 min. MS/MS analysis was done in positive ion mode and in MRM mode following the ion transitions with appropriate instrumental parameters as described in Table 1. Source voltage was 3 kV, source temperature 150°C, desolvation temperature 350°C, gas flow desolvation 800 L/h., and the collision gas argon was kept at a pressure of 1.4 × 10−3 mbar.
Results and discussion
MS/MS parameters
At first, we used the Intellistart function to optimize the cone voltages and the collision energies to find the most specific and sensitive detection parameters for each amino acid in MRM mode. Every amino acid solution (2 μmol/L) was directly injected into the mass spectrometer with the aid of a syringe pump, at the rate of 20 μL/min. The appropriate instrumental parameters are reported in Table 1. MS/MS analysis was done in positive ion mode and in MRM mode following the ion transitions with the parameters listed in Table 1. The MRM scan for underivatized amino acids is different from that of the butylesters, in that a neutral loss of 46 Da is often observed.
Preparation of DBS samples
We have tried various organic solvents to increase the effectiveness of AA extraction from DBS. The blood spots were soaked with different extraction media, such as methanol, acetonitrile, methanol containing 0.1, 0.5 and 1% formic acid, and acetonitrile containing 0.1, 0.5 and 1% formic acid. The best recovery of all AA from DBS was obtained using methanol with 0.1% formic acid. Furthermore, the addition of formic acid also improved the response signal of the analytes in comparison with that of pure methanol. Accordingly, we used a volume of 100 μL of methanol, containing 0.1% formic acid and 5 μmol/L N,N-Phe internal standard, as the extraction solvent in all the tests.
Calibration, linearity, and limit of detection (LOD)
Calibration has been performed using scalar concentrations of a mixture of nine amino acids with identical concentration of internal standard. Two sets of calibration curves were evaluated: one using methanol (without matrix) (Fig. 1) and one spiked with dried blood amino acids (with matrix) for the assay of blood spots (data not shown). Results for Phe in methanol solution and in DBS samples were in close agreement in the MS/MS assay (Fig. 2). Linearity of the MS/MS assay without and with matrix was found between 0.1 and 200 μmol/L and between 0.5 and 20 μmol/L of each AA, with a LOD of 0.1 and 0.5 μmol/L, respectively (Figs. 1, 2).

Typical calibration (dose–response) curves for nine amino acids/internal standard (without matrix). Calibrations are corrected with the weight coefficient 1/X 2 (the range of concentration of each amino acid was 0–6 mmol/L corresponding to the concentration in whole blood)

Calibration curve for Phe (mean, SD) from measurements by MS/MS with the new assay on methanol and blood spot calibrators (data for other amino acids not shown). The equation and correlation coefficient were y = 0.0892265x−0.0015847 (r 2 = 0.9964) and y = 0.0891267x + 0.088384 (r 2 = 0.9920), respectively
Precision and recovery
Precision and recovery results are shown in Table 2. On the whole, an analytical recovery of 93.0–110.5% meets the requirements for a bioanalytical assay. Precision and recovery studies (within-day, n = 10, between-day, n = 10) were carried out using DBS from healthy neonates, stored at 4°C.
Application to DBS samples
The above-characterized new procedure was applied to the analysis of nine amino acids in DBS from healthy newborns (n = 100) and from PKU newborn patients (n = 12) (Table 3). The levels of each AA in the control group were similar to Qu’s study except for the concentration of Glu (Qu et al. 2002).
In our study, we reassessed PKU patients first diagnosed through newborn screening, and quantified Phe and other eight amino acids in their DBS with our method. We found significant differences in the concentration of a number of amino acids between PKU and healthy newborns. In the PKU group (n = 12), the level of Tyr, XIe, Lys-Gln and Met did not change (P > 0.05), the level of Phe and Val significantly increased (P < 0.001 and P < 0.01) and the level of Pro, Thr and Glu slightly decreased (P < 0.05).
The phenomenon of ion suppression is to be taken into account in MS/MS. We thus verified whether the samples containing high concentration of Phe would inhibit the detection of other amino acids. We used 10 DBS from healthy neonates that were divided equally into two groups. The first group of samples was prepared with 100 μL methanol containing 0.1% formic acid with 5 μmol/L internal standard; the second group contained in addition 40 μmol/L Phe. The amount of amino acids (except Phe) detected by MS/MS was not significantly different between the two groups: so, the effect of ion suppression was excluded. The changes of AA other than Phe in DBS of PKU newborns need further research to be confirmed and to be clarified in their biological reasons.
Samples numbered 1–12 were from newborns diagnosed as affected by PKU using commercial BIA diagnostic kits and retested with the new MS/MS method. The results for Phe and Tyr concentrations and the molar ratios of Phe to Tyr in those samples are summarized in Table 4. For the control group, the mean ± SD Phe/Tyr ratio was 0.67 ± 0.26 (range 0.20–1.33, n = 100). For all cases of PKU, the mean ratio was 12.87 ± 9.75 (range 2.40–24.78, n = 12). We found that both the concentrations of Phe and the molar ratios of Phe/Tyr were remarkably higher in PKU-positive than in control newborns. Therefore, false positives connected with using only Phe concentration as the marker for PKU diagnosis, which is the basis for the BIA method, can be avoided with the proposed protocol. Our new procedure is also superior to the n-butanol derivatization MS/MS method in the elimination of the laborious derivatization procedure.
Application of PICS to enhance the reliabilty of AA determination in DBS of PKU patientss
The PICS method was used to enhance the reliabilty of our protocol. When the response intensity of the target compound exceeds a set threshold of background noise level in the collected MRM, the PICS could be collected during a single run, by acquiring at once the daughter ion spectrum. To prevent the occurrence of false positives, we also compared the daughter ion spectrum of AA in blood samples with that of the target analyte standard. It was possible to quantitatively evaluate the chromatographic peak and at the same time to improve the qualitative results. The limit was that in a single run only one or two important amino acids could be set on the channel with PICS; with more PICS channels a little sensitivity would be lost. But the use of PICS proved essential to avoid false positives when some amino acids interfered with a complex matrix. An example of PICS application to a PKU newborn is shown in Fig. 3.

Phe 166 > 120 ion transition in the PICS spectrum of a PKU newborn compared to the MS/MS spectrum of the corresponding Phe standard. The forward fit and the reverse fit was as a reference to check for false positives
Conclusion
The above results demonstrate that the method we have established is reliable for the quantification of underivatized AA in blood.
For example, the amino acids related with phenylketonuria (PKU) and tyrosinemia (TYR I and II) could be adequately detected with a 1 min run. The use of the PICS function greatly reduces false positive results. With our method sample preparation is simple and fast, the derivatization step is omitted, and the expensive isotopically labeled internal standard is replaced by N,N-dimethyl-l-phenylalanine (N,N-Phe). The protocol could be effectively applied on a routine basis to analyze a large number of samples in newborn screening and patient monitoring.
References
Chace DH, Kalas TA (2005) A biochemical perspective on the use of tandem mass spectrometry for newborn screening and clinical testing. Clin Biochem 38:296–309
Chace DH, Milhington DS, Terada N, Kahler SG, Roe CR, Hofman LF (1993) Rapid diagnosis of phenylketonuria by quantitative analysis for phenylalanine and tyrosine in neonatal blood spots by tandem mass spectrometry. Clin Chem 39:66–71
Chace DH, Hillman SL, Milhngton DS, Kahier SG, Roe CR, Naylor EW (1995) Rapid diagnosis of maple-syrup-urine-disease in blood spots from newborns by tandem mass-spectrometry. Clin Chem 41:62–68
Chace DH, Adam BW, Jay Smith S, Richard Alexander J, Hillman SL, Harry Hannon W (1999) Validation of accuracy-based amino acid reference materials in dried-blood spots by tandem mass spectrometry for newborn screening assays. Clin Chem 45:1269–1277
Chace DH, Kalas TA, Naylor EW (2003) Use of tandem mass spectrometry for multianalyte screening of dried blood specimens from newborns. Clin Chem 49:1797–1817
Dale Y, Mackey V, Mushi R, Nyanda A, Maleque M, Ike J (2003) Simultaneous measurement of phenylalanine and tyrosine in phenylketonuric plasma and dried blood by high-performance liquid chromatography. J Chromatogr B 788:1–8
De Jesus VR, Chace DH, Lim TH, Mei JV, Harry Hannon W (2010) Comparison of amino acids and acylcarnitines assay methods used in newborn screening assays by tandem mass spectrometry. Clin Chim Acta 411:684–689
Deyl Z, Hyanek J, Horakova M (1986) Profiling of amino acids in body fluids and tissues by means of liquid chromatography. J Chromatogr 379:177–250
Dietzen DJ, Weindel AL (2010) Comprehensive determination of amino acids for diagnosis of inborn errors of metabolism. Methods Mol Biol 603:27–36
Fekkes D (1996) State-of-the-art of high-performance liquid chromatographic analysis of amino acids in physiological samples. J Chromatogr Biomed Appl 682:3–22
Guthrie R, Suzi A (1963) A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 32:338–343
Naylor EW, Guthrie R (1978) Newborn screening for maple syrup urine disease (branched-chain ketoaciduria). Pediatrics 61:262–266
Piraud M, Vianey-Saban C, Petritis K (2003) ESI-MS/MS analysis of underivatised amino acids: a new tool for the diagnosis of inherited disorders of amino acid metabolism. Fragmentation study of 79 molecules of biological interest in positive and negative ionisation mode. Rapid Commun Mass Spectrom 17:1297–1311
Piraud M, Vianey-Saban C, Bourdin C (2005a) A new reversed-phase liquid chromatographic/tandem mass spectrometric method for analysis of underivatised amino acids: evaluation for the diagnosis and the management of inherited disorders of amino acid metabolism. Rapid Commun Mass Spectrom 19:3287–3297
Piraud M, Vianey-Saban C, Petritis K (2005b) Ion-pairing reversed-phase liquid chromatography/electrospray ionization mass spectrometric analysis of 76 underivatized amino acids of biological interest: a new tool for the diagnosis of inherited disorders of amino acid metabolism. Rapid Commun Mass Spectrom 19:1587–1602
Qu Y, Slocum RH, Fu J, Rasmussen WE, Rector HD, Miller JB, Coldwell JG (2001) Quantitative amino acid analysis using a Beckman system gold HPLC 126AA analyzer. Clin Chim Acta 312:153–162
Qu J, Wang YM, Luo GA, Wu ZP, Yang CD (2002) Validated quantitation of underivatized amino acids in human blood samples by volatile ion-pair reversed-phase liquid chromatography coupled to isotope dilution tandem mass spectrometry. Anal Chem 74:2034–2040
Sarwar G, Botting HG (1993) Evaluation of liquid-chromatographic analysis of nutritionally important amino-acids in food and physiological samples. J Chromatogr Biomed Appl 615:1–22
Spackman DH, Stein WH, Moore S (1958) Automatic recording apparatus for use in chromatography of amino acids. Anal Chem 30:1190–1206
Waterval WAH, Scheijen JLJM, Ortmans-Ploemen MMJC (2009) Quantitative UPLC-MS/MS analysis of underivatised amino acids in body fluids is a reliable tool for the diagnosis and follow-up of patients with inborn errors of metabolism. Clin Chim Acta 407:36–42
Zoppa M, Gallo L, Zacchello F, Giordano G (2006) Method for the quantification of underivatized amino acids on dry blood spots from newborn screening by HPLC-ESI-MS/MS. J Chromatogr B 831:267–273
Zytkovicz TH, Fitzgerald EF, Deborah M (2001) Tandem mass spectrometric analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: A two-year summary from the New England newborn screening program. Clin Chem 47:1945–1955
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No.81073040). The authors thank Prof. Zhenlai Li for providing blood spot samples of Changchun Maternal and Child Health Care Center.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Wang, C., Zhang, W., Song, F. et al. A simple method for the analysis by MS/MS of underivatized amino acids on dry blood spots from newborn screening. Amino Acids 42, 1889–1895 (2012). https://doi.org/10.1007/s00726-011-0910-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-011-0910-6