
Abstract
Data concerning the pathophysiological role of extracellular S100A4, a member of the multigenic family of Ca2+-modulated S100 proteins, and its interaction with the receptor for advanced glycation endproducts (RAGE) or other putative receptors in tumorigenesis, metastasis, and inflammatory processes in vivo are scarce. One reason is the shortage of suitable radiotracer methods. We report a novel methodology using recombinant human S100A4 as potential probe for molecular imaging and functional characterization of this interaction. Therefore, human S100A4 was cloned as GST fusion protein in the bacterial expression vector pGEX-6P-1 and expressed in E. coli strain BL21. Purified recombinant human S100A4 was radiolabeled with the positron emitter fluorine-18 (18F) by conjugation with N-succinimidyl-4-[18F]fluorobenzoate ([18F]SFB). The radioligand [18F]fluorobenzoyl-S100A4 (18F-S100A4) was used in cell binding experiments in RAGE-bearing human melanoma cells and endothelial cells in vitro, and in both biodistribution experiments and small animal positron emission tomography (PET) studies in normal rats in vivo. The cellular association and tissue-specific distribution of 18F-S100A4 in vitro and in vivo correlated well with the protein expression and anatomical localization of RAGE, e.g., in the vascular system and in lung. Compared to other S100 RAGE radioligands, the overall findings of this study indicate that extracellular S100A4 in vivo shows only a moderate interaction with RAGE and, furthermore, exhibits a substantially faster metabolic degradation. On the other hand, the approach allows the use of quantitative small animal PET and provides a novel probe to both delineate functional expression and differentiate multiligand interaction of RAGE under normal and pathophysiological conditions in rodent models of disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
S100A4 is a Ca2+-binding EF-hand protein of the S100 superfamily. S100A4 also known as metastasin or Metastasis Associated Protein is due to its increased expression closely related to tumor progression and metastasis as shown for many cancer entities (Garrett et al. 2006; Helfman et al. 2005; Salama et al. 2008). S100A4 is involved in a broad range of intracellular and extracellular functions which, in particular, mark characteristics of the metastatic cascade. In this regard, S100A4 plays an important role in regulation of cell shape and cell motility and binds to cytoskeletal proteins like non-muscle myosin IIA and IIB, F-actin, and non-muscle tropomyosin (Salama et al. 2008). Furthermore, S100A4 is expressed in highly motile cell types, blood cells (T-lymphocytes, neutrophils, platelets, macrophages), endothelial cells or different types of tumor cells (Li et al. 2010; Tarabykina et al. 2007). Moreover, S100A4 regulates the enzyme activity of the MMP (matrix-metalloproteinases)/TIMP (tissue inhibitors of metalloproteinases) system, which is associated with both the remodeling of the extracellular matrix (ECM) and the induction of angiogenesis (Miranda et al. 2010; Sherbet 2009). Another target of S100A4 is the tumor suppressor p53, which affects p53 oligomerization and cell apoptosis (Berge and Maelandsmo 2010; Fernandez-Fernandez et al. 2005; Grigorian et al. 2001).
Intracellular S100A4 is localized in the nucleus and cytoplasm, and as several other S100 proteins (S100B, S100A8, S100A9, S100A12, S100A13, S100P) it is also secreted into the extracellular space (Boye and Maelandsmo 2010; Logsdon et al. 2007). In this regard, the receptor for advanced glycation endproducts (RAGE) has been suggested as a common extracellular receptor for S100 proteins (Donato 2007; Hsieh et al. 2004; Leclerc et al. 2009b). S100A12 was the first S100 protein identified to bind and activate RAGE and named as extracellular newly identified RAGE binding protein (EN-RAGE) (Hofmann et al. 1999). Interaction between RAGE and S100A4, and, furthermore, S100B, S100A1, S100A2, S100A5, S100A6, S100A7, S100A8/A9, S100A11, S100A13, and S100P at the molecular level has been examined by several groups (Arumugam et al. 2004; Boyd et al. 2008; Dattilo et al. 2007; Gebhardt et al. 2008; Ghavami et al. 2008; Hofmann et al. 1999; Hoppmann et al. 2010; Hsieh et al. 2004; Kiryushko et al. 2006; Leclerc et al. 2009b; Leclerc et al. 2007; Wolf et al. 2008; Yammani et al. 2006). However, studies concerning interaction between recombinant S100A4 and RAGE are rarely, and are only shown for RAGE-bearing cultured chondrocytes using a pull down assay (Yammani et al. 2006). Binding between the two recombinant proteins has only been demonstrated once by surface plasmon resonance spectroscopy (Kiryushko et al. 2006).
Recently, the S100 protein interaction with RAGE has been implicated in many disorders, e.g. in the pathogenesis of diabetes, atherosclerosis, rheumatoid arthritis and other inflammatory disorders, Alzheimer’s disease, and also in cancer (Leclerc et al. 2009b; Logsdon et al. 2007; Stern et al. 2002). Interaction between S100A4 and RAGE is supposed to be a critical factor for inflammation, tumor development, progression and metastasis in several cancers including melanoma (Boye and Maelandsmo 2010; Cunningham et al. 2010; Leclerc et al. 2009a). Malignant melanoma is one of the most highly invasive and metastatic tumors, and RAGE seems to be important in human melanoma tumors and cells (Abe et al. 2004). However, the role of extracellular S100A4 in inflammatory processes and during tumorigenesis is still poorly understood. One reason could be the shortage of sensitive methods for direct assessment of the metabolic fate of circulating S100A4 in vivo.
Preliminary experiments already demonstrated the potential use of fluorine-18 radiolabeled RAGE ligands S100A1, S100B, and S100A12 to follow their metabolic fate by means of small animal positron emission tomography (PET) (Hoppmann et al. 2008a). The present study aimed at advancement of this radiotracer approach using non-carrier-added (n.c.a.) radiolabeling of recombinant human S100A4 with the positron-emitting radionuclide fluorine-18. After chemical and biochemical characterization of the radiolabeled 18F-S100A4 several binding and uptake experiments were performed using primary endothelial cells, melanoma cells, and melanoma cells that overexpress RAGE. Moreover, for radiopharmacological characterization biodistribution experiments and dynamic small animal positron emission tomography (PET) studies were carried out using normal rats.
Materials and methods
Cloning of human S100A4 gene in E. coli expression vector pGEX-6P-1
Total RNA was isolated from the human acute monocytic leukemia cell line THP-1 (ATCC-Nr. TIB-202) using the miRNeasy Mini kit (Qiagen, Hilden, Germany). Complementary DNA (cDNA) was synthesized from 100 ng total RNA using oligonucleotide primers: forward 5′-CTGACTGCTGTCATGGCGT-3′; reverse 5′-ACCACATCAGAGGAGTTTTCAT-3′ with 0.2 μL of SUPERSCRIPT reverse transcriptase (SUPERSCRIPT™ III One-Step RT-PCR Kit with Platinum® Taq DNA Polymerase, Invitrogen, Darmstadt, Germany). The polymerase chain reaction (PCR) product was cloned into the pCR®-TOPO vector (Invitrogen) according to the manufacturer’s instructions. The coding region of S100A4 was amplified by PCR using a 5′ oligonucleotide primer: 5′-GCCCTTGGATCCGCTGTC-3′, containing a BamHI site (underlined) and a 3′ primer: 5′-ACGAATGCGGCCGCACCA-3′, containing a NotI site (underlined). The amplified DNA was cut with the corresponding restriction enzymes and ligated into the BamHI and NotI cloning sites of the expression vector pGEX-6P-1 (GE Healthcare Europe GmbH, Freiburg, Germany). The open reading frame of the plasmid pGEX-S100A4 was confirmed by DNA sequencing. All chemicals, unless otherwise specified, were purchased from Sigma-Aldrich (Munich, Germany).
Production and purification of recombinant S100A4 protein
For bacterial expression, pGEX-S100A4 was transformed into the E. coli strain BL21-CodonPlus(DE3)-RIL (Agilent Technologies, Waldbronn, Germany). Cells grown from an overnight preculture incubated at 37°C were used to inoculate 400 mL Luria–Bertani (LB) medium containing 50 μg/mL ampicillin and were incubated for 2–3 h at 37°C under shaking conditions. At an optical density between 0.8–1.0 at 600 nm the expression of the fusion protein was induced with 1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) at 25°C. Cells were harvested 4 hours after induction by centrifugation at 10,000×g for 5 min at 4°C, then frozen in liquid nitrogen and stored at −70°C until lysis. The cell pellet from a 400 mL culture was resuspended in 10 mL ice cold binding buffer [PBS; 10 mM Na2HPO4, 140 mM NaCl, 2.7 mM KCl, 1.8 mM KH2PO4 pH 7.3, containing 10 μg/mL leupeptin, 1 mM phenylmethyl-sulfonyl fluoride (PMSF), 1 mM dithiothreitol (DTT)] and lysed by sonification. To facilitate solubilization of protein 1% Triton X-100 (v/v), 2 mg/mL lysozyme and 2.5 U/mL benzonase were added and incubated 1 h on ice under shaking conditions. The lysate was centrifuged at 15,500×g for 15 min at 4°C to remove cell debris and denatured protein. The supernatant was subjected to affinity purification using 1 mL bed volume of glutathione–agarose (Biontex, Martinsried/Planegg, Germany) and shaked in a batch on ice for 30 min. The column was washed with 5 bed volumes of ice-cold binding buffer supplemented with 0.25% Triton X-100 and 1 mM DTT to remove unbound protein. For site-specific separation of the GST tag, the S100A4 fusion protein was cleaved with 80 U/mL PreScission™ Protease (GE Healthcare Europe GmbH) on ice for 4 hours with gentle agitation. Finally, S100A4 was eluted in 50 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, pH 7.5. Fractions containing S100A4 were pooled and concentrated via ultrafiltration using a 10 kDa cutoff ultrafiltration membrane (Sartorius AG, Goettingen, Germany). S100A4 protein was subjected to further purification by size exclusion chromatography using an ÄKTA™ FPLC (Fast Protein Liquid Chromatography) system (GE Healthcare, Europe GmbH). A column packed with 24 mL bead volume of Superdex™ 75 10/300 GL was equilibrated with two column volumes of PBS. The sample volume was 1 mL and fractions of 0.5 mL were collected at a flowrate of 0.5–0.8 mL/min. Purified recombinant human S100A4 protein was frozen in liquid nitrogen and stored at −70°C. The concentration of purified recombinant S100A4 protein was measured by the bicinchoninic acid protein assay (BCA, Thermo Fisher Scientific, Bonn, Germany) with bovine serum albumin used as standard. Samples were analyzed by SDS-PAGE followed by Western Blot analysis using polyclonal S100A4 antibody (Dako, Hamburg, Germany) or by Coomassie Blue staining. Electrophoretic band intensities were quantified using advanced image data analyzer (AIDA) software version 4.14 (Raytest, Straubenhardt, Germany).
Mass spectrometry
For identification of recombinant S100A4 protein matrix assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) was used. Therefore, 40 mg/mL sinapinic acid (3,5-dimethoxy-4-hydroxycinnamic acid) was dissolved in TA [0.1% aqueous trifluoroacetic acid (TFA), 33% acetonitrile]. Purified recombinant S100A4 protein was then applied as sandwich sample with 1 μL sinapinic acid plus 1 μL sample plus 1 μL sinapinic acid to the MALDI plate (MTP 384 target plate ground steel TF; Bruker Daltonics, Bremen, Germany) and was allowed to air dry. The molecular weight of each sample was measured on an Autoflex-TOF/TOF mass spectrometer (Bruker Daltonics). The mean molecular mass (m/z) was calculated from multiple independent measurements.
Radiolabeling of recombinant S100A protein
Radiolabeling of recombinant S100 proteins using N-succinimidyl-4-[18F]fluorobenzoate ([18F]SFB) and purification of 18F-fluorobenzoyl-S100 (18F-S100) by size exclusion chromatography were performed as described in detail elsewhere (Hoppmann et al. 2008a). Because of the short half life of 18F (t 1/2 = 109.77 min), it is necessary to adjust radioactivity data in order to compare all the time points. Accordingly, all radioactivity measurements in vitro and in vivo were adjusted and expressed as decay-corrected values.
Cell-free in vitro sRAGE binding assays
Expression and purification of recombinant human soluble RAGE (sRAGE) and the sRAGE binding assay was performed as described previously (Hoppmann et al. 2010). Briefly, Maxisorb™ 96-well plates (Nunc, Langenselbold, Germany) were coated with sRAGE or albumin (each 2.5 μg/well) in bicarbonate/carbonate buffer (50 mM, pH 9.6) and were incubated overnight at 4°C. After washing three times with 250 μL phosphate-buffered saline (PBS) containing 0.05% Tween 20, wells were blocked with PBS containing 2% bovine serum albumin for 2 h. Then, wells with adsorbed sRAGE, albumin, or without protein were incubated with 100 μL radiolabeled 18F-S100A4 (330–400 kBq; in 20 mM HEPES buffer with 150 mM NaCl, 1.2 mM MgCl2, 1.3 mM CaCl2, pH 7.5 supplemented with 2% bovine serum albumin; specific activity at time point of application ranges from 2.0 to 3.5 GBq/μmol; radiochemical purity of 18F-S100A4 was >95%) for 1 h at 37°C with gentle agitation. Subsequently, wells were washed three times with ice cold PBS on ice. Bound protein was dissolved in 100 μL of 0.1 M NaOH containing 1% SDS at 37°C for 30 min under shaking conditions. Fluorine-18 activity (in terms of radioactivity concentration) was measured in a Cobra II gamma counter (Canberra-Packard, Meriden, USA). To further probe the binding of S100A4 to sRAGE, surface plasmon resonance (SPR) measurements were performed on a T100 Biacore system (GE Healthcare Europe GmbH, Freiburg, Germany). Therefore, recombinant sRAGE (20 μg/mL in 10 mM acetate pH 5) was immobilized as ligand on CM5 (carboxymethylated dextran polymer) sensor chips using EDC/NHS chemistry at 25°C (contact time, 500 s; flow rate 10 μL/min). About 5.200 (range 4.300–6.100) arbitrary response (resonance) units (RU) were immobilized. Affinity measurements were carried out with HBS-EP+ (10 mM HEPES, 3.0 mM EDTA-Na2, 0.15 M NaCl (pH 7.4) containing 0.05% surfactant P20) running buffer from Biacore. Various concentrations of S100A4 ranging from 2.5 to 20 μM diluted in running buffer were analyzed. Association and dissociation of all analytes were followed in real-time and measured at 25°C at a flow rate of 30 μL/min (120 s contact time). After each cycle, the surface was regenerated using 10 mM glycine-HCl (pH 3.0). The data were analyzed by Biacore T100 evaluation software version 2.0.1. For calculation of affinity (K D) the response curves of various S100A4 concentrations were fitted to a two-state binding model.
Cell culture and transfection
Human aortic endothelial cells (HAEC; Cascade Biologics C-006-5C) were cultured in EBM-2 medium containing 5% fetal calf serum (FCS) and supplements according to the manufacturer’s instructions. Human melanoma cells (A375; ATCC CRL-1619) were maintained in DMEM, 10% FCS in the presence of Zellshield™ (Minerva Biolabs, Berlin, Germany). For generating stably transfected A375 cell lines, cells were transfected with empty pIRES2-AcGFP1 vectors (Clontech, Saint-Germain-en-Laye, France) or pIRES2-AcGFP1-RAGE using Lipofectamine™ 2000 (Invitrogen), and the transfected cells were selected with 1.2 mg/mL Neomycin (G418; Biochrom, Berlin, Germany). GFP-positive cells were enriched by sorting with a FACSAria flow cytometer (BD Bioscience, Heidelberg, Germany).The resulting transfected cell lines were designated as follows: A375 cells transfected with empty expression vector, A375-mock; A375 cells transfected with human RAGE expression vector, A375-RAGE. Expression levels were determined by Western blotting.
SDS-PAGE and Western blotting
For preparation of cell extracts, cells were washed with PBS, suspended in lysis buffer [50 mM Tris–HCl, 2% sodium dodecyl sulfate (SDS), pH 6.8 supplemented with 1% Triton X-100, 10 μg/mL leupeptine, 1 mM PMSF] and left on ice for 30 min. After sonification, cell suspensions were centrifuged at 15,500×g for 15 min at 4°C to remove cell debris. The protein concentration in each supernatant was quantified using the BCA protein assay (Thermo Fisher Scientific). 40–50 μg of protein were mixed with sample buffer, denatured at 99°C for 10 min and separated by 10% SDS-polyacrylamide gels (PAGE). Then, proteins were transferred to a 0.45 μm polyvinylidine difluoride (PVDF) membrane (Whatman, Dassel, Germany) by semi-dry electroblotting. After blocking of non-specific binding sites with 5% non-fat dry milk in TBST (50 mM Tris–HCl, 150 mM NaCl, pH 8.0, 0.05% Tween 20) for 1 h at room temperature, blots were incubated with primary antibodies RAGE (1:800, sc-8230, Santa Cruz Biotechnology, Inc., Heidelberg, Germany) and β-actin (1:100, A5060, Sigma-Aldrich Chemie GmbH, Munich, Germany) at 4°C overnight. After washing, the blots were incubated for 1 h at room temperature with peroxidase-conjugated secondary antibody anti-goat IgG (1:10,000, A5420, Sigma-Aldrich) or anti-rabbit IgG (1:10,000, A0545, Sigma-Aldrich). All antibodies were diluted in TBST with 5% non-fat dry milk. After washing with TBST, proteins were visualized by chemiluminescence using SuperSignal® West Pico and Femto Chemiluminescent Substrate (Thermo Fisher Scientific) and imaged using a MF-ChemiBIS Bio-Imaging System (Biostep GmbH, Jahnsdorf, Germany).
In vitro cellular association experiments
Cellular association experiments were performed as described elsewhere (Hoppmann et al. 2010). Here, HAEC were seeded in 96-well plates 72 h (5 × 103 cells per well) and 24 h (1 × 104 cells per well) prior to the experiment. The assay was performed in the presence (24 μg per well) or absence of non-radioactive S100A4. The A375 melanoma cells were seeded in 96 well plates 72 h (1 × 104 cells per well) and 24 h (1.5 × 104 per well) prior to the experiment. Then the 18F-S100A4 radiotracer (100–200 kBq; 0.5–1 μg; in PBS) was added for 1 h at 37°C. Subsequently, cells were washed three times with ice-cold PBS and dissolved in 0.1 M NaOH containing 1% SDS. Fluorine-18 activity was measured in the Cobra II gamma counter, and afterwards protein concentration was determined using the BCA protein assay. Cellular association of 18F-S100A4 to A375, A375-mock and A375-RAGE cells were carried out only in the absence of non-radiolabeled ligands.
In vivo experiments
Animal experiments were carried out according to the guidelines of the German Regulations for Animal Welfare. The protocol was approved by the local Ethical Committee for Animal Experiments. For biodistribution and dynamic small animal PET studies male Wistar Unilever rats (HsdCpb:WU strain, Harlan Laboratories, Horst, The Netherlands; aged 7–9 weeks; 180–230 g) were used. In vivo experiments were performed as described elsewhere with some minor modifications (Hoppmann et al. 2008a). In brief, rats (n = 6) under desflurane anesthesia were positioned prone with thorax and abdominal region (organs of interest: heart, lung, liver, kidneys, large vessels) in the center of field of view of a dedicated PET scanner for small animals (microPET P4; CTI Concorde Microsystems, Knoxville, USA). A bolus injection of 500 μL of 18F-S100A4 (10–20 MBq; radiochemical purity >95%; specific activity at time point of injection ranges from 2 to 10 GBq/μmol) was administered via the tail vein within 30 s with (blocking group, n = 3) or without 2.0 μM glycated human low density lipoprotein (glycLDL). Simultaneously with tracer injection, dynamic PET scanning was started for 120 min. Sinogram generation and image reconstruction followed the protocol given by us elsewhere (Pietzsch et al. 2005). Images were analyzed by assigning three-dimensional regions-of-interest (ROI) over the heart region, the liver, and the kidneys using ROVER software (ABX GmbH, Radeberg, Germany). From these ROIs time-activity-curves (TACs) representing the total (decay-corrected) fluorine-18 activity in a defined volume and expressed as radioactivity concentration, percent of maximum were obtained in each rat. TACs from ROIs over the heart region, majorly representing the cardiac blood pool, were used to estimate the pharmacokinetic parameter mean residence time (MRT) (Hoppmann et al. 2010). Therefore, the numerical module of SAAM II program (University of Washington, Seattle, WA, USA) using a derivative-free nonlinear regression analysis was used to fit 18F-activity data from ROIs over the heart region from each animal to multiexponential equations. The individual responses were quite consistent, showing a three-exponential equation A(t) = A 1 e −k1t + A 2 e −k2t + A 3 e −k3t, where A(t) = % of injected dose per cm3, to provide the best fit. The distribution/tissue association (t d/a1/2 ; t 1/2 k1 + t 1/2 k2) and elimination (t e1/2 , t 1/2 k3) half-lifes were calculated as 0.693/k. For estimation of MRT the area under the heart region TAC and the area under the momentum curve were calculated using standard equations according to Wolfe (1992).
For biodistribution experiments, two groups (each n = 4) of rats were administered into the tail vein with 500 μL 18F-S100A4 (1.2–1.5 MBq) and sacrificed at 5 and 60 min post injection (p.i.) by heart puncture under desflurane anaesthesia. Organs and tissues of interest were rapidly excised, weighed, and the fluorine-18 activity was determined using the Cobra II gamma counter crosscalibrated with a dose calibrator (Isomed 2000, MED, Dresden, Germany) by aliquots of the injected activity. The radioactivity concentration in organs and selected tissues was calculated as the percentage of the injected dose per gram tissue (%ID/g). For metabolic investigations, in rats catheters were placed under desflurane anesthesia into the right common carotid artery. A volume of 1 mL 18F-S100A4 containing 5 MBq (radiochemical purity >95%; PBS, pH 7.4; specific activity at time point of injection ranges from 2 to 10 GBq/μmol) was injected into the tail vein. Arterial blood samples (0.5 mL) were taken at 10, 30, and 60 min p.i. The depleted blood volume was compensated by the injection of saline. Blood samples were immediately centrifuged for 5 min at 10.000×g. Arterial plasma samples were boiled using SDS-sample buffer containing β-mercaptoethanol at 95°C for 20 min, subjected to SDS-PAGE and radioluminography. Radioluminographic intensities of the electrophoretic bands were quantified using AIDA software version 4.14 (Raytest) and expressed as percentage of total activity amount.
Statistical analysis
Descriptive data were expressed as mean [standard deviation (SD)]. Statistical analyses were performed using one-way ANOVA coupled with a post hoc Bonferroni correction analysis (Origin Pro 8 GSR1, Northhampton, MA, USA). For all analyses a value of P < 0.05 was considered as statistically significant.
Results
Purification of S100A4 protein
The recombinant S100A4 fusion protein expressed from the prokaryotic expression vector pGEX-6P-1 containing a GST tag region at its amino terminus was isolated by affinity chromatography in a one-step purification scheme using glutathione–agarose. The purity and identity of S100A4 protein were examined by polyacrylamide gel electrophoresis and Western blotting with polyclonal S100A4 antibody (Fig. 1a, b). After cleavage of the S100A4 fusion protein with PreScission™ Protease we obtained a mixture of protein bands which correspond presumably to the fusion protein (s), the cleaved glutathione-S-transferase (r), the cleaved S100A4 monomer (m) and S100A4 homodimers (n), respectively (Fig. 1a, lane 2). The remaining impurities were removed by size exclusion chromatography using a Superdex 75 column. The purified recombinant S100A4 protein is shown as an intensive protein band at 12 kDa (m) that can be assigned to the corresponding calculated molecular weight of the monomer subunit (Fig. 1a, lane 3). In addition, there is a protein band at 14 kDa (p), which could be identified as fragmented GST by protein sequencing (nanoLC-ESI-MSMS, Proteome Factory). The proteins that migrate at 24 kDa are supposed to be SDS-resistant S100A4 dimers because of its detection in the Western blot analysis (Fig. 1b). Densitometric analysis of lane 3 in Fig. 1a showed that the S100A4 monomer represents 87% and the S100A4 dimer represents 5% of the total protein (Fig. 1a). The analysis of the band intensities of the specific Western blot revealed a distribution between the S100A4 monomer and homodimer of 93% and 7%, respectively (Fig. 1b). The correct molecular weight of the recombinant S100A4 protein was verified by MALDI-TOF mass spectrometry. The spectra showed a specific signal at m/z 12,356 Da (Fig. 2a). This value is in good agreement with the corresponding calculated molecular weight of the mature S100A4 protein considering the additional residues of 8 amino acids (sequence GPLGSAVM) at the N-terminal region (Mr 12,338 Da) (Fig. 2b). The two additional peaks in the MALDI spectra represent the fragmented GST (m/z 14,852 Da) and the S100A4 dimer (m/z 24,705 Da).

Bacterial expression and purification of recombinant S100A4. Representative SDS-PAGE analysis (a; 15%; Coomassie Brilliant blue staining) of S100A4 protein at various steps of purification. Lane 1 induced BL21[pGEX-S100A4] cells 1:10 dissolved in loading buffer; lane 2 protein fraction eluted after GST pulldown and cleavage with PreScission™ Protease; lane 3 purified recombinant S100A4 obtained after size exclusion chromatography. Representative Western blot (b) of the purified recombinant S100A4 obtained after size exclusion chromatography. Arrows indicate S100A4 monomer (m) protein band, SDS-resistant dimers (n), fragmented GST (p), cleaved glutathione-S-transferase (r) and the fusion protein (s)

MALDI-TOF mass spectrum and amino acid sequence of recombinant S100A4 protein. Small characters in the MALDI TOF mass spectrum (a) indicate m/z of [M + H]+ ions for S100A4 monomer (m), S100A4 dimer (n), and fragmented GST (p) (l, m/z of [M + 2H]2+ ion of m). The purified recombinant S100A4 protein contains 108 amino acids (b) in total and the additional residues of 8 amino acids (sequence GPLGSAVM) at the N-terminal region are marked in bold letters
Radiolabeling of the recombinant S100A4 protein
The radiolabeling reaction of recombinant S100A4 with non-carrier-added [18F]SFB resulted in selective [18F]fluorobenzoylation at the chemically accessible N-terminal glycine residue. Besides the amino terminus, the recombinant S100A4 contains 11 lysine side chain residues as possible sites for [18F]fluorobenzoylation (Fig. 2b). To minimize the number of different radiolabeled S100A4 species, reaction conditions at pH ≤ 8 were used throughout, thus strictly favoring selective [18F]fluorobenzoylation at the amino-terminal glycine residue of the protein. Under strong basic conditions (pH >> 8.4) the ε-amino group of lysine side chain residues is nearly completely deprotonated and becomes the preferred site for conjugation with [18F]SFB. Radiolabeled S100A4 was then isolated from unreacted [18F]SFB and radioactive by-products, e.g., [18F]fluorobenzoic acid by size exclusion chromatography. Fluorine-18 activity distribution was determined by radioluminography after SDS-PAGE. Figure 3 shows a representative SDS-PAGE pattern of a complete separation of 18F-S100A4. The electrophoretic bands of 18F-S100A4 monomer and dimer recovers >95% of total 18F-activity in the purified fraction. Of note, no aggregation or fragmentation of S100A4 occurred during the labeling procedure.

Radiolabeling of S100A4. Representative SDS-PAGE analysis (8–18%) after 18F-radiolabeling of recombinant S100A4. Radioactivity distribution was determined by radioluminography (top) and protein bands were determined by Coomassie Brilliant blue staining (bottom) of the same gel. Lane 1, unlabeled S100A4; lane 2, the reaction mixture of 18F-SFB and S100A4; lane 3, the purified 18F-S100A4. Arrows indicate the 18F-S100A4 protein bands with the monomer (m) and the SDS-resistant dimers (n)
Binding of S100A4 and 18F-S100A4 to sRAGE
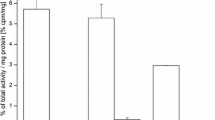
The SPR measurements for interaction of non-radiolabeled recombinant human S100A4 to immobilized human sRAGE revealed a specific but moderate affinity when compared to other S100 proteins (Leclerc et al. 2009b) with a K D of 27.8 μM (Fig. 4). Moreover, the radioligand binding assay confirmed that radiolabeled 18F-S100A4 sustains its overall binding capacity to immobilized sRAGE. In this regard, 96 well microtiter plates with adsorbed sRAGE from human origin bound 24% more 18F-S100A4 than the negative control (wells with bicarbonate/carbonate buffer alone) and 17% more than control wells containing BSA (Fig. 5, P < 0.05).

In vitro binding of S100A4 protein to immobilized sRAGE. Surface plasmon resonance binding experiments of unlabeled S100A4 to sRAGE were performed using the Biacore system. Results of representative experiments (sensorgrams) are shown. For evaluation of S100A4 binding kinetics, concentrations ranging from 2.5 to 20 μM were used. The calculated K D is 2.78 × 10−5 M. Solid lines represent observed data; dotted lines indicate computer-derived fits as calculated by a two-state binding model

In vitro binding of 18F-S100A4 protein to immobilized sRAGE. In vitro sRAGE radioligand binding assay was performed by incubation of immobilized sRAGE with radiolabeled 18F-S100A4, (300 kBq (equals 1.5 μg protein) per well) for 1 h at 37°C. Results of sRAGE- and BSA-binding are expressed as percent of control (wells coated with bicarbonate/carbonate buffer) and represent mean ± SD (n = 8, 3 independent experiments). *P < 0.05 for comparison with both the control and BSA values
18F-S100A4 association to HAEC and A375-RAGE cells
We evaluated the relative contribution of 18F-S100A4 association to RAGE when it was overexpressed on A375 melanoma cells. RAGE expression was verified by Western blot analysis. A375 cells transfected with pIRES2-AcGFP1-RAGE (A375-RAGE) displayed a significant increase in the expression level of RAGE as compared with parental A375 cells and mock-transfected cells (Fig. 6a). In HAEC RAGE expression in homeostasis is considerably present, but is lower compared to A375 cells (Fig. 6c). The cellular binding assay was performed at 4 and 37°C in order to differentiate between ligand binding alone and possible energy-dependent ligand internalization. At 4°C, we observed a significantly higher binding of 18F-S100A4 to A375-RAGE cells compared with the controls (A375, A375-mock) (Fig. 6b). In contrast, 18F-S100A4 showed no significant changes in cellular association to A375-RAGE cells compared with the controls at 37°C. In addition, we investigated the 18F-S100A4 cellular association in HAEC in the presence or absence of non-radiolabeled S100A4. Overall, there is a higher cellular association of 18F-S100A4 in HAEC cells than in A375 melanoma cells at 37°C. Cellular association of 18F-S100A4 in HAEC cells was significantly inhibited in the presence of a 23 molar excess of S100A4 at 37°C (Fig. 6d).

Association of 18F-S100A4 to A375-RAGE melanoma cells and HAEC. a Western blot analyses of A375, A375-mock, A375-RAGE, and c HAEC; b comparison of 18F-S100A4 association to A375, A375-mock and A375-RAGE cells at 37 and 4°C, respectively, and d HAEC at 37°C. Data are expressed as mean ± SD (A375: n ≥ 5, 3 independent experiments; HAEC: n ≥ 5, 2 independent experiments). *P < 0.05, for increased binding of 18F-S100A4 to A375-RAGE compared to the controls b and for lower cellular association to HAEC when cells were preincubated with non-radiolabeled S100A4 (d)
In vivo investigations
In vivo investigations showed a moderate stability of 18F-S100A4 that is similar to stability of 18F-S100B, but substantially lower compared to both 18F-S100A1 and 18F-S100A12, which have been published by us elsewhere in detail (Hoppmann et al. 2008a). After injection of 18F-S100A4 about 90% at 30 min and 67% at 1 h, respectively, of the 18F-activity could be assigned to intact 18F-S100A4. On the other hand, these investigations revealed the formation of two radioactive peptide metabolites I (4 and 20% of the 18F-radioactivity at 30 and 60 min, respectively), and II (6 and 12%) as detected by SDS-PAGE (data not shown in detail). To complement the in vitro results, indicating only a moderate affine association of 18F-S100A4 to RAGE, when compared with other 18F-S100 proteins employed in former experiments (Hoppmann et al. 2008a, 2010), we analyzed the metabolic fate of 18F-S100A4 in rats in vivo. Figure 7b shows a representative small animal PET image (maximum intensity projection) after single intravenous injection of 18F-S100A4. Images were analyzed by assigning three-dimensional regions of interest (ROI) over the heart region (representing the cardiac blood pool), the lung, the liver and the kidneys. From these ROIs time–activity curves (TACs) representing the total 18F-activity in a defined volume (expressed as radioactivity concentration, percent of maximum) were obtained. After the first systemic passage (fast distribution and tissue association phase; t d/a1/2 , 0.5 ± 0.1 min) a considerable fraction of intact 18F-S100A4 remained in the circulating blood (t e1/2 , 57 ± 10 min). This is consistent with the biodistribution data (Fig. 7a). Systemic clearance of 18F-activity after intravenous injection of 18F-S100A4 in the rat is almost completely explained by a very fast uptake in the kidneys through glomerular filtration of intact 18F-S100A4, and the two radioactive proteolytic metabolites I and II, respectively, and excretion into the urine (32 ± 2% of injected dose at 5 min p.i. and 70 ± 7% of injected dose at 60 min p.i.). Uptake of 18F-S100A4 in the liver and excretion into the bile only marginally contribute to blood clearance. At 5 and 60 min after injection only 1.4 ± 0.2% and 1.5 ± 0.1% of the injected dose was associated with intestine. Besides the kidneys, major sites for tissue-association of intact 18F-S100AA4 in rats were the spleen, the liver, the lung, the adrenals, and the vasculature. Comparable to other S100 proteins, rat lung and vasculature show a substantial temporary retention of 18F-S100A4 (Fig. 7c). This retention cannot be explained by the circulating blood pool activity alone, and therefore, may be indicative for a tissue-specific interaction of 18F-S100A4 with RAGE-overexpressing cells. The lung-to-blood pool ratio increases to a maximum value of 1.73. This ratio is lower compared to those observed with other 18F-S100 proteins (Hoppmann et al. 2008b), but is also indicative for a specific lung association of 18F-S100A4. This is supported by the biodistribution data. Blocking PET experiments with molar excess of glycLDL, a well characterized ligand for RAGE, showed a significant decrement of the specific lung association of 18F-S100A4 by 27 ± 4% (Fig. 7c). This indicates that glycLDL can efficiently displace 18F-S100A4 from potential binding sites in the lung. The amount of 18F-S100A4 in the free circulating blood increased in parallel. The results obtained in biodistribution experiments and PET studies, respectively, correlate well with experiments aiming at characterization of expression of RAGE in Wistar rats both on the mRNA and protein level published by us elsewhere (Hoppmann et al. 2010). These experiments revealed highest RAGE expression levels in the lung. In other organs, such as heart, muscle, kidneys, and liver, quantitative RT-PCR showed a significantly lower RAGE expression when compared to the lung. Furthermore, endothelial cells, as a model for vasculature, show a substantial expression of RAGE (Hoppmann et al. 2010).

Biodistribution and small animal PET experiments. a Biodistribution of 18F-S100A4 in male Wistar rats at 5 min and at 60 min p.i. (mean ± SD, n = 4). b Representative coronal images of thoracic region of a male Wistar rat obtained from small animal PET study, showing 18F-radioactivity distribution in vivo as maximum intensity projection (0–120 min) after intravenous injection of 18F-S100A4. H heart, Lu lung, Li liver, K kidney, V blood vessel, S spleen. Black color shows highest radioactivity concentration. c Representative time–activity-curves showing kinetics of the 18F-radioactivity concentration during the entire study period of 120 min after injection of 18F-S100A4 in a male Wistar rat as calculated from ROI analysis of dynamic small animal PET scans over the heart region, representing the cardiac blood pool, as well as lung and kidneys. The insert graph shows the lung-to-blood pool ratio of 18F-radioactivity concentration during the entire study period of 120 min after injection of 18F-S100A4 in rats with (diamonds) and without (circles) blocking of potential binding sites by excess glycLDL
Discussion
The present study reports preliminary experiments on radiolabeling of purified recombinant human S100A4 with the positron emitter fluorine-18 using the acylating reagent [18F]SFB and the subsequent use of [18F]fluorobenzoylated S100A4 (18F-S100A4) in cellular binding experiments in endothelial and melanoma cells and RAGE overexpressing melanoma cells in vitro as well as biodistribution experiments and small animal PET studies in normal rats in vivo. Radiolabeling with [18F]SFB is considered to be a validated and well characterized method for the mild incorporation of 18F into S100 proteins without alteration of their biological activity and functionality, respectively (Hoppmann et al. 2008a, b, 2010). Target protein interactions of S100 proteins have been characterized in the last decade primarily on a biochemical or cellular in vitro level (Arumugam et al. 2004; Hofmann et al. 1999; Hsieh et al. 2004; Huttunen et al. 2000; Leclerc et al. 2009b; Zimmer et al. 2003). On the other hand, in vivo experiments for functional characterization of potential S100 tissue-specific binding sites are still scarce. In this regard, although the in vitro binding of S100A4 to, e.g., RAGE, and stimulation of RAGE dependent signaling pathways clearly have been demonstrated by others, the role of S100A4/RAGE axis or interaction of extracellular S100A4 with other putative receptors in vivo is not understood. In due consideration of the intrinsic properties of small animal PET and the continuous developments of this technique, radiolabeling of S100A4 with [18F]SFB represents a valuable approach for functional characterization of potential tissue-specific retention, receptor binding, accumulation or elimination of S100A4 and other circulating S100 proteins in vivo. In this regard, S100 protein-RAGE interaction in vivo even has been confirmed for S100A1, S100B, and S100A12 (Hoppmann et al. 2008a, 2010). In the present study, we also demonstrate a substantial interaction of S100A4 with organs and tissues showing high expression of RAGE, in particular, lung and large blood vessels. However, the current PET study argued that under physiological conditions in the rat interaction of extracellular S100A4 with the receptor for advanced glycation endproducts (RAGE) is weak to moderate when compared to the other S100-RAGE ligands investigated by radiotracer approaches (Hoppmann et al. 2008a, 2010). The in vivo kinetics of all 18F-radiolabeled S100 proteins investigated up to now is characterized by a relatively long residence time in the circulating blood as well as a sustained tissue-associated temporary retention in the lung and the whole vascular system with S100A12 > S100B > S100A1 > S100A4. Contrastable to other S100 radioligands the tissue-specific interaction of 18F-S100A4 within lung and vasculature could be significantly diminished by excess human glycated low density lipoproteins (glycLDL) that are well characterized to be specific RAGE ligands in vitro and in vivo (Hoppmann et al. 2008b, 2010). Besides the lower affinity to the receptor (as shown in SPR analysis), a substantially faster metabolic degradation of S100A4 should also contribute to this observation. Moreover, like the other S100 radioligands, the recognition of 18F-S100A4 by other receptors, e.g., internalizing scavenger receptors, cannot be excluded. Indicative for this assumption is the high uptake of 18F-S100A4 observed in liver, spleen, and adrenals, organs comprising cells that overexpress scavenger receptors, e.g., CD36, SRBI, etc. (Hoppmann et al. 2010). Furthermore, the cellular binding studies in both an endothelial cell (HAEC) and a melanoma cell (A375) model revealed a substantially higher 18F-S100A4 binding to putative binding sites in endothelial cells than in melanoma cells. It is likely that scavenger receptors and other endothelial surface structures contribute to the higher 18F-S100A4 association (Adachi and Tsujimoto 2006; Hoppmann et al. 2010). Binding of S100A4, S100A8 and S100A9 to heparin sulfate moieties on the cell surface of endothelial cells have also been reported (Kiryushko et al. 2006; Robinson et al. 2002). Furthermore, S100A4 interacts with annexin II on the surface of endothelial cells (Semov et al. 2005). Nevertheless, the significant higher association of 18F-S100A4 to A375 cells overexpressing RAGE compared with parental A375 cells and mock-transfected cells demonstrate the direct interaction of 18F-S100A4 and RAGE. Considering a physiological situation with various competing RAGE ligands available, which exhibit higher concentrations and higher affinity to the receptor, the systemic interaction between extracellular S100A4 and RAGE thus is likely to be of minor importance. On the other hand, a typical attribute of RAGE is that it is expressed at relatively low levels in homeostasis but in situations characterized by enhanced cellular activation or stress, the expression of RAGE is strikingly enhanced, particularly in certain tissue compartments (Schmidt et al. 1999). Under these circumstances, S100A4 could contribute to the overall complex interaction with RAGE. The present approach can contribute to the understanding of the role of extracellular S100A4 in specific pathophysiological situations. Ongoing work aims at investigating in more details the role of S100A4/RAGE axis in animal models of disease, e.g., tumor and metastasis models, in vivo.
References
Abe R, Shimizu T, Sugawara H, Watanabe H, Nakamura H, Choei H, Sasaki N, Yamagishi S, Takeuchi M, Shimizu H (2004) Regulation of human melanoma growth and metastasis by AGE-AGE receptor interactions. J Invest Dermatol 122:461–467
Adachi H, Tsujimoto M (2006) Endothelial scavenger receptors. Prog Lipid Res 45:379–404
Arumugam T, Simeone DM, Schmidt AM, Logsdon CD (2004) S100P stimulates cell proliferation and survival via receptor for activated glycation end products (RAGE). J Biol Chem 279:5059–5065
Berge G, Maelandsmo GM (2010) Evaluation of potential interactions between the metastasis-associated protein S100A4 and the tumor suppressor protein p53. Amino Acids (in press)
Boyd JH, Kan B, Roberts H, Wang Y, Walley KR (2008) S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circ Res 102:1239–1246
Boye K, Maelandsmo GM (2010) S100A4 and metastasis: a small actor playing many roles. Am J Pathol 176:528–535
Cunningham MF, Docherty NG, Burke JP, O’Connell PR (2010) S100A4 expression is increased in stricture fibroblasts from patients with fibrostenosing Crohn’s disease and promotes intestinal fibroblast migration. Am J Physiol Gastrointest Liver Physiol 299:G457–G466
Dattilo BM, Fritz G, Leclerc E, Kooi CW, Heizmann CW, Chazin WJ (2007) The extracellular region of the receptor for advanced glycation end products is composed of two independent structural units. Biochemistry 46:6957–6970
Donato R (2007) RAGE: a single receptor for several ligands and different cellular responses: the case of certain S100 proteins. Curr Mol Med 7:711–724
Fernandez-Fernandez MR, Veprintsev DB, Fersht AR (2005) Proteins of the S100 family regulate the oligomerization of p53 tumor suppressor. Proc Natl Acad Sci USA 102:4735–4740
Garrett SC, Varney KM, Weber DJ, Bresnick AR (2006) S100A4, a mediator of metastasis. J Biol Chem 281:677–680
Gebhardt C et al (2008) RAGE signaling sustains inflammation and promotes tumor development. J Exp Med 205:275–285
Ghavami S, Rashedi I, Dattilo BM, Eshraghi M, Chazin WJ, Hashemi M, Wesselborg S, Kerkhoff C, Los M (2008) S100A8/A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. J Leukoc Biol 83:1484–1492
Grigorian M et al (2001) Tumor suppressor p53 protein is a new target for the metastasis-associated Mts1/S100A4 protein: functional consequences of their interaction. J Biol Chem 276:22699–22708
Helfman DM, Kim EJ, Lukanidin E, Grigorian M (2005) The metastasis associated protein S100A4: role in tumour progression and metastasis. Br J Cancer 92:1955–1958
Hofmann MA et al (1999) RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97:889–901
Hoppmann S, Steinbach J, Pietzsch J (2010) Scavenger receptors are associated with cellular interactions of S100A12 in vitro and in vivo. Int J Biochem Cell Biol 42:651–661
Hoppmann S, Haase C, Richter S, Pietzsch J (2008a) Expression, purification and fluorine-18 radiolabeling of recombinant S100 proteins—potential probes for molecular imaging of receptor for advanced glycation endproducts (RAGE) in vivo. Protein Expr Purif 57:143–152
Hoppmann S, Haase C, Richter S, Strobel K, Steinbach J, Pietzsch J (2008b) Fluorine-18 radiolabeling of S100/calgranulins: potential probes for molecular imaging of receptor for advanced glycation endproducts (RAGE) in vivo. In: Chen X (ed) Recent advances of bioconjugation chemistry in molecular imaging. Research Signpost, Trivandrum, pp 329–351
Hsieh HL, Schafer BW, Weigle B, Heizmann CW (2004) S100 protein translocation in response to extracellular S100 is mediated by receptor for advanced glycation endproducts in human endothelial cells. Biochem Biophys Res Commun 316:949–959
Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R, Rauvala H (2000) Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem 275:40096–40105
Kiryushko D, Novitskaya V, Soroka V, Klingelhofer J, Lukanidin E, Berezin V, Bock E (2006) Molecular mechanisms of Ca(2+) signaling in neurons induced by the S100A4 protein. Mol Cell Biol 26:3625–3638
Leclerc E, Fritz G, Weibel M, Heizmann CW, Galichet A (2007) S100B and S100A6 differentially modulate cell survival by interacting with distinct RAGE (receptor for advanced glycation end products) immunoglobulin domains. J Biol Chem 282:31317–31331
Leclerc E, Heizmann CW, Vetter SW (2009a) RAGE and S100 protein transcription levels are highly variable in human melanoma tumors and cells. Gen Physiol Biophys 28(Spec No Focus):F65–F75
Leclerc E, Fritz G, Vetter SW, Heizmann CW (2009b) Binding of S100 proteins to RAGE: an update. Biochim Biophys Acta 1793(6):993–1007
Li ZH, Dulyaninova NG, House RP, Almo SC, Bresnick AR (2010) S100A4 regulates macrophage chemotaxis. Mol Biol Cell 21:2598–2610
Logsdon CD, Fuentes MK, Huang EH, Arumugam T (2007) RAGE and RAGE ligands in cancer. Curr Mol Med 7:777–789
Miranda KJ, Loeser RF, Yammani RR (2010) Sumoylation and nuclear translocation of S100A4 regulate IL-1beta-mediated production of matrix metalloproteinase-13. J Biol Chem 285(41):31517–31524
Pietzsch J, Bergmann R, Wuest F, Pawelke B, Hultsch C, van den Hoff J (2005) Catabolism of native and oxidized low density lipoproteins: in vivo insights from small animal positron emission tomography studies. Amino Acids 29:389–404
Robinson MJ, Tessier P, Poulsom R, Hogg N (2002) The S100 family heterodimer, MRP-8/14, binds with high affinity to heparin and heparan sulfate glycosaminoglycans on endothelial cells. J Biol Chem 277:3658–3665
Salama I, Malone PS, Mihaimeed F, Jones JL (2008) A review of the S100 proteins in cancer. Eur J Surg Oncol 34(4):357–364
Schmidt AM, Yan SD, Wautier JL, Stern D (1999) Activation of receptor for advanced glycation end products: a mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ Res 84:489–497
Semov A, Moreno MJ, Onichtchenko A, Abulrob A, Ball M, Ekiel I, Pietrzynski G, Stanimirovic D, Alakhov V (2005) Metastasis-associated protein S100A4 induces angiogenesis through interaction with Annexin II and accelerated plasmin formation. J Biol Chem 280:20833–20841
Sherbet GV (2009) Metastasis promoter S100A4 is a potentially valuable molecular target for cancer therapy. Cancer Lett 280:15–30
Stern D, Yan SD, Yan SF, Schmidt AM (2002) Receptor for advanced glycation endproducts: a multiligand receptor magnifying cell stress in diverse pathologic settings. Adv Drug Deliv Rev 54:1615–1625
Tarabykina S, Griffiths TR, Tulchinsky E, Mellon JK, Bronstein IB, Kriajevska M (2007) Metastasis-associated protein S100A4: spotlight on its role in cell migration. Curr Cancer Drug Targets 7:217–228
Wolf R et al (2008) Chemotactic activity of S100A7 (Psoriasin) is mediated by the receptor for advanced glycation end products and potentiates inflammation with highly homologous but functionally distinct S100A15. J Immunol 181:1499–1506
Wolfe RR (1992) Radioactive and stable isotope tracers in biomedicine: principles and practice of kinetic analysis. Wiley Liss, New York
Yammani RR, Carlson CS, Bresnick AR, Loeser RF (2006) Increase in production of matrix metalloproteinase 13 by human articular chondrocytes due to stimulation with S100A4: role of the receptor for advanced glycation end products. Arthritis Rheum 54:2901–2911
Zimmer DB, Wright Sadosky P, Weber DJ (2003) Molecular mechanisms of S100-target protein interactions. Microsc Res Tech 60:552–559
Acknowledgments
The authors are grateful to Mareike Barth, Catharina Heinig, Regina Herrlich, Uta Lenkeit, and Aline Morgenegg for their expert technical assistance. The authors thank Torsten Kniess, Ph.D., and Birgit Mosch, Ph.D. for their expert advice and many stimulating discussions. This work in part was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG; grant no. PI304/1-1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wolf, S., Haase-Kohn, C., Lenk, J. et al. Expression, purification and fluorine-18 radiolabeling of recombinant S100A4: a potential probe for molecular imaging of receptor for advanced glycation endproducts in vivo?. Amino Acids 41, 809–820 (2011). https://doi.org/10.1007/s00726-010-0822-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-010-0822-x