
Abstract
A peptide heterodimer comprises two different receptor-targeting peptide ligands. Molecular imaging probes based on dual-receptor targeting peptide heterodimers exhibit improved tumor targeting efficacy for multi-receptor expressing tumors compared with their parent single-receptor targeting peptide monomers. Previously we have developed bombesin (BBN)-RGD (Arg-Gly-Asp) peptide heterodimers, in which BBN and RGD are covalently connected with an asymmetric glutamate linker (J Med Chem 52:425–432, 2009). Although 18F-labeled heterodimers showed significantly better microPET imaging quality than 18F-labeled RGD and BBN monomers in a PC-3 xenograft model which co-expresses gastrin-releasing peptide receptor (GRPR) and integrin αvβ3, tedious heterodimer synthesis due to the asymmetric nature of glutamate linker restricts their clinical applications. In this study, we report the use of a symmetric linker AEADP [AEADP = 3,3′-(2-aminoethylazanediyl)dipropanoic acid] for the synthesis of BBN-RGD peptide heterodimer. The 18F-labeled heterodimer (18F-FB-AEADP-BBN-RGD) showed comparable microPET imaging results with glutamate linked BBN-RGD heterodimers, indicating that the replacement of glutamate linker with AEADP linker did not affect the biological activities of BBN-RGD heterodimer. The heterodimer synthesis is rather easy and straightforward. Because tumors often co-express multiple receptors, the use of a symmetric linker provides a general method of fast assembly of various peptide heterodimers for imaging multi-receptor expressing tumors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Molecular imaging probes based on receptor-targeting peptides have attracted intense attention in the last decade due to their fast clearance, excellent tissue penetration and low immunogenicity (Benedetti et al. 2004; Lee et al. 2010a, b). However, one major drawback of peptide probes is their relatively low tumor affinity and short retention time. To improve the binding affinity of peptide based probes, a variety of methods have been developed. Among them, a new approach using peptide heterodimers has recently shown its great potential in developing high affinity molecular imaging probes (Yan and Chen 2010).
A peptide heterodimer is comprised of two different receptor-targeting peptide ligands which are covalently linked. Molecular imaging probes based on peptide heterodimers exhibit improved tumor targeting efficacy compared with their parent single receptor targeting peptide monomers (Yan and Chen 2010; Li et al. 2008; Liu et al. 2009b, c, d, e; Reubi and Waser 2003; Vagner et al. 2008; Yang et al. 2009). The improvement is believed to be a result of the following: increased receptor number for targeting, increased local ligand concentration and increased binding kinetics.
Androgen independent prostate cancer tissues have been discovered to co-express both gastrin-releasing peptide receptor (GRPR) (di Sant’Agnese 1998; Chung et al. 1992; Glover et al. 2003; Vashchenko and Abrahamsson 2005) and integrin αvβ3 (Friedlander et al. 1995; Horton 1997; Bello et al. 2001). Both bombesin (BBN) peptides that target GRPR (Chen et al. 2004b; Zhang et al. 2006a; Yang et al. 2006; Shi et al. 2008; Ananias et al. 2008) and RGD peptides that target αvβ3 (Cai et al. 2005, 2008; Chen 2006; Liu 2009) have been used in prostate cancer imaging. However, their imaging results are not so satisfactory in term of binding affinity and retention time.
Previously we have developed two 18F-labeled BBN-RGD heterodimers, which showed significantly better microPET imaging quality than 18F-labeled RGD and BBN(7–14) monomers in a PC-3 xenograft model (Li et al. 2008; Liu et al. 2009d). However, there are also drawbacks of these BBN-RGD heterodimers. Our first 18F-labeled BBN-RGD heterodimer (18F-FB-Glu-BBN-RGD), is not a pure compound, instead it is a mixture of two isomers as shown in Fig. 1a. These two regional isomers have similar physical properties and cannot be separated by HPLC. Two isomeric products were formed due to the non-regioselective synthetic strategy we initially used for the preparation of peptide heterodimer. Because the two isomers are inseparable, exact determination of the proportion in which component isomers are present in the mixture is impossible. Because of his, the application of our first generation 18F-FB-Glu-BBN-RGD heterodimer in clinical study is restricted. In order to make a clinically translatable imaging tracer, we further developed a strategy to synthesize a PEGylated BBN-RGD heterodimer with definite structure through solid phase synthesis using orthogonal Alloc protected glutamate. Figure 1b shows the structure of our second 18F-labeled heterodimer (18F-FB-PEG-Glu-BBN-RGD). Although the new peptide heterodimer is a pure compound and has the desired definite structure, the synthesis of this peptide is rather lengthy and cannot use standard Fmoc chemistry. This drawback resulted in high cost for the peptide production and may hamper its further translational application in the clinic even though this peptide heterodimer has also been labeled with other isotopes and fluorophores for prostate cancer and breast cancer imaging (Liu et al. 2009b, c, e).

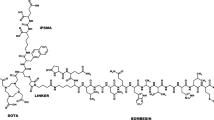
Structures of 18F-labeled BBN-RGD peptide heterodimers
As part of our efforts to develop prostate cancer imaging probes suitable for clinical applications, we designed a new BBN-RGD heterodimer with definite structure by using a symmetric linker AEADP [AEADP = 3,3′-(2-aminoethylazanediyl)dipropanoic acid]. The peptide heterodimer can be easily prepared in solution and the 18F-labeled heterodimer (18F-FB-AEADP-BBN-RGD, as shown in Fig. 1c) showed comparable imaging results with BBN-RGD heterodimers linked with a glutamate linker. Herein, we would like to report its preparation and applications in microPET imaging of PC-3 tumor mice.
Materials and methods
All chemicals purchased were of analytical grade and were used as received without further purification. No-carrier-added 18F-F− was obtained from an in-house GE Healthcare PETtrace cyclotron. Reversed-phase extraction C18 Sep-Pak cartridges were purchased from Waters and were pretreated with ethanol (5 mL) and water (10 mL) before use. The syringe filter and polyethersulfone membranes (pore size, 0.22 mm; diameter, 13 mm) were purchased from Nalge Nunc International. 125I-Echistatin, labeled by the lactoperoxidase method to a specific activity of 74 TBq/mmol (2,000 Ci/mmol) and 125I-[Tyr4]BBN [74 TBq/mmol (2,000 Ci/mmol)] were purchased from PerkinElmer The peptides Aca-BBN(7–14) and c(RGDyK) were purchased from Peptides International. Analytical and semi-preparative reversed-phase high-performance liquid chromatography (RP-HPLC) was performed on a Dionex 680 chromatography system with a UVD 170U absorbance detector and model 105S single-channel radiation detector (Carroll & Ramsey Associates). The recorded data were processed using Chromeleon version 6.50 software. A C18 Vydac protein and peptide column (218TP510; 5 μm, 250 × 10 mm) was used for purification. The flow was set at 5 mL/min using a gradient system starting from 95% solvent A (0.1% trifluoroacetic acid [TFA] in water) and 5% solvent B (0.1% TFA in acetonitrile [ACN]) (0–2 min) and ramped to 35% solvent A and 65% solvent B at 32 min. The flow rate of the analytic HPLC was set at 1 mL/min using the same gradient system, but with a C18 Vydac column (218TP54, 5 μm, 250 × 4.6 mm). The ultraviolet (UV) absorbance was monitored at 218 nm and the identification of the peptides was confirmed based on the UV spectrum acquired using a photodiode array detector and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS).
Synthesis of benzyl 3,3′-(2-(tert-butoxycarbonylamino)ethylazanediyl)dipropanoate
Tert-butyl 2-aminoethylcarbamate (1.6 g, 10 mmol) and benzyl acrylate (16.2 g, 100 mmol) were heated at 70°C under N2 for 7 days. The excess of benzyl acrylate was distilled off at 60°C and the residue was purified by column chromatography to yield 3,3′-(2-(tert-butoxycarbonylamino)ethylazanediyl)dipropanoate as colorless oil (4.3 g, 89%). 1H NMR (CDCl3): δ 7.26–7.36 (m, 10 H), 5.11 (br s, 1 H), 5.04 (bs, 4 H), 3.14–3.15 (m, 2 H), 2.76 (t, J = 6.93 Hz, 4 H), 2.51 (t, J = 5.73 Hz, 2 H), 2.44 (t, J = 6.93 Hz, 4 H), 1.40 (s, 9H).
Synthesis of 3,3′-(2-(tert-butoxycarbonylamino)ethylazanediyl)dipropanoic acid
To a solution of benzyl 3,3′-(2-(tert-butoxycarbonylamino)ethylazanediyl)dipropanoate (1.5 g, 3.1 mmol) in EtOH (100 ml) was added Pd/C (50 mg, 10%). The reaction mixture was bubbled with H2 overnight. The reaction mixture was filtered through a plug of Celite, washed with EtOH (5 mL) and evaporated to dryness to give the crude product as colorless oil. The residue was added CH2Cl2 (5 mL) and the solvent was evaporated to yield 3,3′-(2-(tert-butoxycarbonylamino)ethylazanediyl)dipropanoic acid as a white solid (0.7 g, 74%). 1H NMR (DMSO): δ 2.93 (m, 2 H), 2.65 (t, J = 7.1 Hz, 4 H), 2.41 (t, J = 7.1 Hz, 2 H), 2.29 (t, J = 9.93 Hz, 4 H), 1.38 (s, 9 H).
Synthesis of AEADP-BBN-RGD hetero dimer
To a solution of 3,3′-(2-(tert-butoxycarbonylamino)ethylazanediyl)dipropanoic acid (3.04 mg, 10 μmol) in DMF (300 μL) was added a solution of O-(N-succinimidyl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TSTU) (6.02 mg, 20 μmol) in DMF (600 μL) followed by N,N-diisopropylethylamine (DIPEA) (20 μL). The reaction mixture was stirred for 30 min at room temperature. Aca-BBN(7–14) (abbreviated as BBN) (10.5 mg, 10 μmol) in DMF (1.0 mL) was added and the reaction mixture was stirred for 20 min at room temperature. After adding c(RGDyK) (abbreviated as RGD) (12.4 mg, 20 μmol) in DMF (1.2 mL), the reaction mixture was heated at 60°C for 1 h. The reaction progress was monitored by analytical HPLC. Once the reaction reached completion, HOAc solution (2 mL, 5%) was added to quench the reaction. The Boc protected crude product was purified by preparative HPLC and lyophilized to yield a white powder. The Boc protection group was removed by dissolving the product in 3 mL TFA and stirred for 10 min at room temperature. After removing excess TFA under reduced pressure, the final product was purified by preparative HPLC and lyophilized to afford AEADP-BBN-RGD heterodimer as a white powder (5.9 mg, purity > 95%, 32% overall yield, two steps). Analytical HPLC (R t = 18.1 min) and MS (MALDI-TOF: m/z 1,841.76 for [MH]+ (C84H129N25O20S, calculated [M W] 1,840.96)) analyses confirmed the product identification.
Synthesis of FB-AEADP-BBN-RGD
N-Succinimidyl-4-fluorobenzoate (SFB) (2 mg, 8.4 μmol) in DMF (200 μL), AEADP-BBN-RGD (1.8 mg, 1.0 μmol) in DMF (200 μL) and DIPEA (20 μL) were mixed. The reaction mixture was heated at 60°C for 30 min. The reaction mixture was quenched with 2 mL 5% HOAc. The crude product FB-AEADP-BBN-RGD was purified by preparative HPLC and lyophilized to give a white powder in 85% yield. Analytical HPLC (Rt = 20.3 min) and MS (MALDI-TOF: m/z 1,961.95 for [MH]+ (C91H132FN25O21S, calculated [MW] 1,961.98)) analyses confirmed the product identification.
Radiochemistry
To a mixture of AEADP-BBN-RGD (200 μg) in DMSO (20 μL) and DIPEA (20 μL) was added 18F-SFB which was prepared according to previously reported procedures (Wu et al. 2007; Chen et al. 2004a; Zhang et al. 2006b; Liu et al. 2009a). The reaction mixture was heated for 15 min at 90°C. The reaction was quenched with 800 μL of 5% HOAc. The 18F labeled peptide was purified by semi-preparative HPLC. The desired fractions were combined and the solvent was removed under reduced pressure. The 18F-labeled peptide was then formulated in normal saline and passed through an 0.22-μm Millipore filter into a sterile multi-dose vial for in vitro and in vivo experiments. Yield: 42%.
Cell lines and animal models
The PC-3 human prostate carcinoma cell line was purchased from American Type Culture Collection (ATCC). PC-3 cells were grown in F-12K nutrient mixture (Kaighn’s modification) (Invitrogen) supplemented with 10% (v/v) fetal bovine serum (Invitrogen) at 37°C with 5% CO2. Animal procedures were performed according to a protocol approved by the Stanford University Institutional Animal Care and Use Committee. The PC-3 tumor model was generated by subcutaneous injection of 5 × 106 tumor cells into the front flank of male athymic nude mice (Harlan). Small animal PET studies were performed on the mice 3–4 weeks after inoculation when the tumor volume reached 100–300 mm3.
In vitro cell-binding assay
In vitro integrin αvβ3-binding affinity of FB-AEADP-BBN-RGD was assessed via displacement cell-binding assays using 125I-echistatin as the radioligand. Experiments were performed on U87MG human glioblastoma cells by a previously described method (Wu et al. 2005). In vitro GRPR binding affinity of FB-AEADP-BBN-RGD was assessed via displacement cell-binding assays using 125I-[Tyr4]BBN as the radioligand. Experiments were performed on PC-3 human prostate carcinoma cells by a previously described method (Chen et al. 2004b). IC50 (the best-fit 50% inhibitory concentration) values were calculated by fitting the data with nonlinear regression using Graph-Pad Prism (GraphPad Software, Inc.). The binding of 125I-echistatin on PC-3 and 125I-[Tyr4]BBN on U87MG were performed as negative control. Experiments were performed with triplicate samples.
Cell uptake and efflux studies
Cell uptake and efflux studies of 18F-FB-AEADP-BBN-RGD were performed as follows with PC-3 tumor cells (Li et al. 2008; Yang et al. 2006; Chen et al. 2004b). In the cell uptake experiment, PC-3 cells were seeded into 12-well plates at a density of 5 × 105 cells/well and incubated overnight. PC-3 cells were washed thrice with PBS (phosphate buffered saline) and then 18F-FB-AEADP-BBN-RGD was added in triplicate (~0.3 μCi/well). PC-3 cells were then re-incubated at 37°C. At five different time points (5, 15, 30, 60, and 120 min), cells were rinsed thrice with PBS and lysed with NaOH-SDS (sodium dodecyl sulfate) (0.2 M NaOH, 1% SDS). The cell lysate was collected and measured in a γ counter (Packard, Meriden, CT, USA). In the cell efflux experiment, PC-3 cells were seeded into 12-well plate at a density of 5 × 105 cells/well and incubated overnight. Cells were rinsed thrice with PBS and then 18F-FB-AEADP-BBN-RGD was added in triplicate (~0.3 μCi/well). After incubation at 37°C for 2 h, the cells were washed with PBS, and then re-incubated with serum-free medium. At six different time points (0, 15, 30, 60, 120, 180 min), the cells were washed with PBS and lysed with NaOH-SDS (0.2 M NaOH, 1% SDS). The cell lysate was collected and measured with a γ counter (Packard, Meriden, CT, USA). The cell uptake and efflux values were normalized in terms of added radioactivity and expressed as the percent added radioactivity. All experiments were performed twice with triplicate wells.
MicroPET studies
MicroPET scans and image data analysis were performed using a microPET R4 rodent model scanner (Siemens Medical Solutions) (Li et al. 2008; Liu et al. 2009d). Mice bearing PC-3 tumor were each tail-vein injected with ~3.7 MBq (100 μCi) of 18F-FB-AEADP-BBN-RGD under isoflurane anesthesia. Five-minute static PET images were then acquired at three time points post injection (30 min, 1 h, and 2 h). The image reconstruction was done by two-dimensional ordered subsets expectation maximum (OSEM) algorithm. No attenuation or scatter correction was used. For the integrin and GRPR receptor-blocking experiment, c(RGDyK) (10 mg/kg), Aca-BBN(7–14) (15 mg/kg), or mixture of both RGD and BBN (10 mg/kg RGD and 15 mg/kg BBN) were co-injected with 3.7 MBq of 18F-FB-AEADP-BBN-RGD into PC-3 tumor mice. At 1 h after injection, the 5-min static microPET scans were acquired. For each microPET scan, regions of interest (ROIs) were drawn over the tumor, normal tissue, and major organs on decay-corrected whole-body coronal images using vendor software (ASI Pro 5.2.4.0). The maximum radioactivity concentration (accumulation) within a tumor or an organ was obtained from mean pixel values within the multiple ROI volume, which were converted to MBq/mL/min by using a conversion factor. Assuming a tissue density of 1 g/mL, the ROIs were converted to MBq/g/min and then divided by the administered activity to obtain an imaging ROI-derived %ID/g.
Statistical analysis
Quantitative data are expressed as mean ± SD. Means were compared using 1-way ANOVA and the Student’s t test. P values of <0.05 were considered statistically significant.
Results
Chemistry and radiochemistry
The synthesis of AEADP-BBN-RGD peptide heterodimer was depicted in Fig. 2. The AEADP linker was synthesized in two steps: (1) Michael addition of the mono-Boc-protected diamine with excess benzyl acrylate at 70°C for 7 days afforded the dipropanoate in 89% yield. (2) Deprotection of benzyl group by Pd/C catalyzed hydrogenation produced the bisacid in 74% yield. Assembly of RGD and BBN was carried out stepwise and stoichiometrically, which represents the most critical operation of the whole synthesis. The bisacid was first activated with 2 equiv. of TSTU at room temperature for 30 min, then 1 equiv. of BBN was added. After conjugation for 20 min at room temperature, 1 equiv. of RGD was added and the reaction mixture was heated up to 60°C for 1 h to ensure high conversion. The resulting Boc-protected AEADP-BBN-RGD heterodimer was further treated with anhydrous TFA to remove Boc-protection group to afford AEADP-BBN-RGD heterodimer bearing the free amine functionality. The overall yield was 32% for the assembly and de-protection steps. The crude product mixture contained RGD-BBN, RGD-RGD, and BBN-BBN dimers. Only the desired RGD-BBN heterodimer was isolated. The purity and identity of final product was confirmed by HPLC and MALDI-TOF mass spectrometry.

Synthesis of AEADP-BBN-RGD heterodimer
The 18F-radiolabeling of AEADP-BBN-RGD heterodimer with 18F-SFB was performed in DMSO at 90°C for 15 min. The radiolabeling yield was around 42% from 18F-SFB with high radiochemical purity (>95%). Because the unlabeled peptide could be efficiently separated from the product, the effective specific activity was estimated to be 100 TBq/mmol on the basis of the labeling agent 18F-SFB.
Cell binding assay
125I-c(RGDyK) was used as the radioligand for competitive binding assay to determine the integrin αvβ3 receptor-binding affinity of FB-AEADP-BBN-RGD. The heterodimeric peptide inhibited the binding of 125I-c(RGDyK) to integrin expressing U87MG cells in a concentration-dependent manner (Fig. 3a). The IC50 value for FB-AEADP-BBN-RGD was determined to be 553 ± 1.08 nM. The binding affinities of FB-AEADP-BBN-RGD for GRPR was evaluated using 125I-[Tyr4]BBN as the radioligand in GRPR positive PC-3 cells. Results of the cell-binding assay were plotted in sigmoid curves for the displacement of 125I-[Tyr4]BBN from PC-3 cells as a function of increasing concentration of BBN analogues. The IC50 values were determined to be 167 ± 1.41 nM on PC-3 cells (Fig. 3b). Similar as the previously reported FB-Glu-BBN-RGD heterodimer (Liu et al. 2009d), FB-AEADP-BBN-RGD showed good binding affinities for integrin αvβ3 and GRPR receptors.

a Inhibition of 125I-[Tyr3]-BBN binding to GRPR on PC-3 cells by FB-AEADP-BBN-RGD (n = 3). b Inhibition of 125I-echistatin binding to integrin αvβ3 on U87MG cells by FB-AEADP-BBN-RGD (n = 3). c Cell uptake assay of 18F-FB-AEADP-BBN-RGD on PC-3 tumor cells (n = 3). d Cell efflux assay of 18F-FB-AEADP-BBN-RGD on PC-3 tumor cells (n = 3)
Cell uptake studies
The cell uptake of 18F-FB-AEADP-BBN-RGD was evaluated in PC-3 tumor cells that coexpress GRPR and integrin αvβ3. The results are shown in Fig. 3c. During the first 15 min of incubation, 18F-FB-AEADP-BBN-RGD was taken up by tumor cells rapidly. After 15 min, the uptake was slow and finally reached a plateau. The PC-3 cell uptake of 18F-FB-AEADP-BBN-RGD was 2.64 ± 0.22, 3.90 ± 0.16, 4.38 ± 0.17, 4.68 ± 0.26, and 4.72 ± 0.36% at 5, 15, 30, 60, and 120 min, respectively. The cell uptake values of 18F-FB-AEADP-BBN-RGD are comparable to those of our previously reported 18F-FB-Glu-BBN-RGD tracer in which RGD and BBN were connected by a glutamate linker (Liu et al. 2009d; Li et al. 2008). Efflux of radioactivity from PC-3 tumor cell was almost linear during the 2 h incubation time (Fig. 3d). After 2 h incubation, about 50% of the radioactivity was released from the tumor cells, which was slightly less than what we observed with 18F-FB-Glu-BBN-RGD.
MicroPET imaging
Around 3.7 MBq (100 μCi) of 18F-FB-AEADP-BBN-RGD was intravenous injected into PC-3 tumor-bearing mice (n = 3/group). Figure 4a shows representative coronal microPET images at different time points post-injection. Tumor can be clearly seen at all time points (30, 60 and 120 min). The tumor to contralateral background contrast is also very good. The regions of interest (ROIs) that encompass the entire organ on the microPET coronal images were measured to determine the tumor and major organ activity accumulation. The tumor uptake of 18F-FB-AEADP- BBN-RGD was calculated to be 5.20 ± 1.04, 4.56 ± 0.87 and 4.00 ± 0.59 %ID/g at 30, 60, and 120 min, respectively. The kidneys showed high uptake, indicating that the tracer is mainly excreted via renal route. The liver uptake was relatively low, with the uptake values calculated to be 2.58 ± 0.53, 1.93 ± 0.21 and 1.21 ± 0.10 %ID/g at 30, 60 and 120 min, respectively. As the tracer is cleared rapidly from normal nontargeted organs, the tumor/nontumor (T/NT) ratio increased with time. At 120 min postinjection, the T/NT ratios reached highest points (1.18 ± 0.19, 3.31 ± 0.36 and 5.98 ± 0.39 for kidneys, liver and muscle, respectively). The T/NT ratios of 18F-AEADP- BBN-RGD are comparable to previously reported 18F-FB-Glu-BBN-RGD (Liu et al. 2009d; Li et al. 2008) indicating that the structure of AEADP linker did not have negative effects on tumor targeting in vivo.

Representative coronal microPET images (a) and the radioactivity accumulation quantification (b) in selected organs of the PC-3 tumor-bearing mice at 30, 60 and 120 min after injection of 3.7 MBq of 18F-FB-AEADP-BBN-RGD (n = 3/group). All microPET images were decay-corrected to the injection time. c Calculated tumor/nontumor (T/NT) ratios from (b)
In vivo blocking study also demonstrated that dual-receptor binding ability of 18F-FB-AEADP-BBN-RGD for integrin αvβ3 and GRPR was not changed when symmetric AEADP linker was used. Illustrated in Fig. 5a are the representative coronal images of PC-3 tumor mice at 1 h postinjection of 18F-FB-AEADP-BBN-RGD when co-injected with RGD (10 mg/kg of mouse body weight), BBN (15 mg/kg of mouse body weight), or mixture of both RGD and BBN (10 mg/kg for RGD and 15 mg/kg for BBN). The tumor uptake of 18F-FB-AEADP-BBN-RGD (Fig. 5b) decreased from 4.56 ± 0.87 %ID/g to 1.76 ± 0.17 and 1.40 ± 0.14 %ID/g in the presence of only RGD or BBN, respectively, indicating partial inhibition. The biggest decrease in tumor uptake was observed when the tracer was co-injected with mixture of both RGD and BBN, the tracer uptake values decreased to 1.06 ± 0.20 %ID/g.

a Decay-corrected whole-body coronal microPET images of PC-3 tumor-bearing mice at 1 h after injection of 18F-FB-BBN-RGD and a blocking dose of c(RGDyk) (10 mg/kg of mouse body weight), BBN (15 mg/kg mouse body weight), or mixture of RGD (10 mg/kg) + BBN (15 mg/kg). b Comparison between the uptake of 18F-FB-AEADP-BBN-RGD in PC-3 tumor with or without co-injection of blocking dose of c(RGDyk) (10 mg/kg of mouse body weight), BBN (15 mg/kg mouse body weight), or mixture of RGD (10 mg/kg) + BBN (15 mg/kg)
Discussion
Androgen-independent prostate cancer cells coexpress both gastrin-releasing peptide receptor (GRPR) and integrin αvβ3. Previously, we developed two 18F-labeled BBN-RGD peptide heterodimers, which target both GRPR and integrin and showed significant improvement over BBN or RGD monopeptides. However, their application in the clinic is restricted due to either the mixture nature or inefficient synthesis of the tracers.
A PET tracer of potential large scale clinical applications should be capable of providing high quality images and should be easy to prepare. With this goal in mind, we designed a new BBN-RGD heterodimer based on an AEADP linker. The linker is comprised of one amino and two carboxylic acid functional groups. The two carboxylic acid groups were used to link RGD and BBN peptides by forming amide bonds with the free amino groups of the peptides, the free amino group in AEADP linker was used for 18F labeling through SN2 nucleophilic reaction with 18F-SFB. Activation of both carboxylic acid functional groups of the linker followed by sequential addition of BBN and RGD peptides produced the desired heterodimer. Due to the symmetric nature of the linker, only one heterodimer product with definite structure was formed. In Glu-BBN-RGD heterodimer, the free amino group was attached directly to a secondary carbon atom of the glutamate linker. While in AEADP-BBN-RGD, the protruding free amino group which attached to the AEADP backbone through two methylene groups encounters less steric hindrance.
Our receptor-binding assay results demonstrated that, similar to our previous developed Glu-BBN-RGD heterodimer, the new AEADP-BBN-RGD heterodimer also possesses dual receptor binding ability. The replacement of glutamate linker with AEADP linker did not affect the biological activities of BBN-RGD heterodimer.
Comparison of the PET imaging results of 18F-FB-AEADP-BBN-RGD and 18F-FB-Glu-BBN-RGD revealed that they have comparable tumor uptake and nonspecific tissue uptake. The in vivo blocking experiment demonstrated that co-injection of either RGD or BBN could only partially inhibit the PC-3 tumor uptake of 18F-FB-AEADP-BBN-RGD. When one type of receptors was blocked in the presence of blocking amount of non-radiolabeled peptide monomer, there were still unblocked receptors of other type available for tumor targeting. Co-injection of RGD and BBN mixture could further decrease the tumor uptake because both GRPR and integrin αvβ3 receptors were blocked and no receptors were available for targeting. This observation confirmed that the ability of 18F-FB-AEADP-BBN-RGD to bind both GRPR and integrin αvβ3 was retained in vivo.
By using the new AEADP linker, we have demonstrated that a BBN-RGD peptide heterodimer with definite structure and good imaging quality can be easily prepared without tedious synthesis. Such strategy can be generally applied to other peptide heterodimer preparations. Many tumors co-express multiple receptors, and many targeting peptide ligands are now commercially available. With our strategy of using a symmetric linker, assembly of peptide heterodimers from peptide monomers would be very straightforward (one pot synthesis within 2 h).
It also should be noted that although using a peptide heterodimer is beneficial for tumor detection, there are potential limitations of this approach. For instance, the lack of target specificity, i.e., it cannot determine which specific target is imaged. In addition, binding in normal tissue might be increased since multiple targets are imaged.
Conclusion
In this study, we successfully designed and synthesized a dual GRPR and integrin receptor-targeting heterodimeric peptide, AEADP-BBN-RGD, using a symmetric linker. 18F-labeled AEADP-BBN-RGD showed high tumor uptake and good tumor-to-background contrast in a PC-3 tumor model. The peptide heterodimer was easy to prepare, especially from commercially available monomeric peptides. Because tumors often co-express multiple receptors, this strategy thus provides a general method of fast assembly of various peptide heterodimers for imaging dual receptor expressing tumors.
References
Ananias HJ, de Jong IJ, Dierckx RA, van de Wiele C, Helfrich W, Elsinga PH (2008) Nuclear imaging of prostate cancer with gastrin-releasing-peptide-receptor targeted radiopharmaceuticals. Curr Pharm Des 14(28):3033–3047
Bello L, Francolini M, Marthyn P, Zhang J, Carroll RS, Nikas DC, Strasser JF, Villani R, Cheresh DA, Black PM (2001) αvβ3 and αvβ5 Integrin expression in glioma periphery. Neurosurgery 49(2):380–389 discussion 390
Benedetti E, Morelli G, Accardo A, Mansi R, Tesauro D, Aloj L (2004) Criteria for the design and biological characterization of radiolabeled peptide-based pharmaceuticals. BioDrugs 18(5):279–295
Cai W, Sam Gambhir S, Chen X (2005) Multimodality tumor imaging targeting integrin αvβ3. Biotechniques 39(6 Suppl):S14–S25
Cai W, Niu G, Chen X (2008) Imaging of integrins as biomarkers for tumor angiogenesis. Curr Pharm Des 14(28):2943–2973
Chen X (2006) Multimodality imaging of tumor integrin αvβ3 expression. Mini Rev Med Chem 6(2):227–234
Chen X, Park R, Hou Y, Khankaldyyan V, Gonzales-Gomez I, Tohme M, Bading JR, Laug WE, Conti PS (2004a) Micropet imaging of brain tumor angiogenesis with 18F-labeled pegylated RGD peptide. Eur J Nucl Med Mol Imaging 31(8):1081–1089
Chen X, Park R, Hou Y, Tohme M, Shahinian AH, Bading JR, Conti PS (2004b) Micropet and autoradiographic imaging of grp receptor expression with 64Cu-DOTA-[Lys3]bombesin in human prostate adenocarcinoma xenografts. J Nucl Med 45(8):1390–1397
Chung DH, Evers BM, Beauchamp RD, Upp JR Jr, Rajaraman S, Townsend CM Jr, Thompson JC (1992) Bombesin stimulates growth of human gastrinoma. Surgery 112(6):1059–1065
di Sant’Agnese PA (1998) Neuroendocrine cells of the prostate and neuroendocrine differentiation in prostatic carcinoma: a review of morphologic aspects. Urology 51(5A Suppl):121–124
Friedlander M, Brooks PC, Shaffer RW, Kincaid CM, Varner JA, Cheresh DA (1995) Definition of two angiogenic pathways by distinct αv integrins. Science 270(5241):1500–1502
Glover SC, Tretiakova MS, Carroll RE, Benya RV (2003) Increased frequency of gastrin-releasing peptide receptor gene mutations during colon-adenocarcinoma progression. Mol Carcinog 37(1):5–15
Horton MA (1997) The alpha v beta 3 integrin “Vitronectin receptor”. Int J Biochem Cell Biol 29(5):721–725
Lee S, Xie J, Chen X (2010a) Peptide-based probes for targeted molecular imaging. Biochemistry 49(7):1364–1376
Lee S, Xie J, Chen X (2010b) Peptides and peptide hormones for molecular imaging and disease diagnosis. Chem Rev 110(5):3087–3111
Li ZB, Wu Z, Chen K, Ryu EK, Chen X (2008) 18f-labeled bbn-rgd heterodimer for prostate cancer imaging. J Nucl Med 49(3):453–461
Liu S (2009) Radiolabeled cyclic rgd peptides as integrin αvβ3-targeted radiotracers: Maximizing binding affinity via bivalency. Bioconjug Chem 20(12):2199–2213
Liu S, Liu Z, Chen K, Yan Y, Watzlowik P, Wester HJ, Chin FT, Chen X (2009a) 18F-labeled galacto and pegylated rgd dimers for EPT imaging of αvβ3 integrin expression. Mol Imaging Biol. doi:10.1007/s11307-009-0284-2
Liu Z, Li ZB, Cao Q, Liu S, Wang F, Chen X (2009b) Small-animal pet of tumors with 64Cu-labeled rgd-bombesin heterodimer. J Nucl Med 50(7):1168–1177
Liu Z, Niu G, Wang F, Chen X (2009c) 68Ga-labeled NOTA-RGD-BBN peptide for dual integrin and GRPR-targeted tumor imaging. Eur J Nucl Med Mol Imaging 36(9):1483–1494
Liu Z, Yan Y, Chin FT, Wang F, Chen X (2009d) Dual integrin and gastrin-releasing peptide receptor targeted tumor imaging using 18F-labeled pegylated rgd-bombesin heterodimer 18F-FB-PEG3-Glu-RGD-BBN. J Med Chem 52(2):425–432
Liu Z, Yan Y, Liu S, Wang F, Chen X (2009e) 18F, 64Cu, and 68Ga labeled RGD-bombesin heterodimeric peptides for PET imaging of breast cancer. Bioconjug Chem 20(5):1016–1025
Reubi JC, Waser B (2003) Concomitant expression of several peptide receptors in neuroendocrine tumours: Molecular basis for in vivo multireceptor tumour targeting. Eur J Nucl Med Mol Imaging 30(5):781–793
Shi J, Jia B, Liu Z, Yang Z, Yu Z, Chen K, Chen X, Liu S, Wang F (2008) 99mTc-labeled bombesin(7–14)NH2 with favorable properties for spect imaging of colon cancer. Bioconjug Chem 19(6):1170–1178
Vagner J, Xu L, Handl HL, Josan JS, Morse DL, Mash EA, Gillies RJ, Hruby VJ (2008) Heterobivalent ligands crosslink multiple cell-surface receptors: the human melanocortin-4 and delta-opioid receptors. Angew Chem Int Ed Engl 47(9):1685–1688
Vashchenko N, Abrahamsson PA (2005) Neuroendocrine differentiation in prostate cancer: Implications for new treatment modalities. Eur Urol 47(2):147–155
Wu Y, Zhang X, Xiong Z, Cheng Z, Fisher DR, Liu S, Gambhir SS, Chen X (2005) Micropet imaging of glioma integrin αvβ3 expression using 64Cu-labeled tetrameric rgd peptide. J Nucl Med 46(10):1707–1718
Wu Z, Li ZB, Chen K, Cai W, He L, Chin FT, Li F, Chen X (2007) Micropet of tumor integrin αvβ3 expression using 18f-labeled pegylated tetrameric rgd peptide (18F-FPRGD4). J Nucl Med 48(9):1536–1544
Yan Y, Chen X (2010) Peptide heterodimers for molecular imaging. Amino Acids. doi:10.1007/s00726-010-0546-y
Yang YS, Zhang X, Xiong Z, Chen X (2006) Comparative in vitro and in vivo evaluation of two 64Cu-labeled bombesin analogs in a mouse model of human prostate adenocarcinoma. Nucl Med Biol 33(3):371–380
Yang J, Guo H, Gallazzi F, Berwick M, Padilla RS, Miao Y (2009) Evaluation of a novel arg-gly-asp-conjugated alpha-melanocyte stimulating hormone hybrid peptide for potential melanoma therapy. Bioconjug Chem 20(8):1634–1642
Zhang X, Cai W, Cao F, Schreibmann E, Wu Y, Wu JC, Xing L, Chen X (2006a) 18F-labeled bombesin analogs for targeting GRP receptor-expressing prostate cancer. J Nucl Med 47(3):492–501
Zhang X, Xiong Z, Wu Y, Cai W, Tseng JR, Gambhir SS, Chen X (2006b) Quantitative pet imaging of tumor integrin αvβ3 expression with 18F-FRGD2. J Nucl Med 47(1):113–121
Acknowledgments
This work was supported in part by the National Cancer Institute (NCI) grants R01 CA119053, P50 CA114747, and U54 CA119367) and the Intramural Research Program of the National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH).
Author information
Authors and Affiliations
Corresponding author
Additional information
Y. Yan and K. Chen have contributed equally in this work.
Rights and permissions
About this article
Cite this article
Yan, Y., Chen, K., Yang, M. et al. A new 18F-labeled BBN-RGD peptide heterodimer with a symmetric linker for prostate cancer imaging. Amino Acids 41, 439–447 (2011). https://doi.org/10.1007/s00726-010-0762-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-010-0762-5