
Abstract
The microtubule-associated protein tau is known to be post-translationally modified by the addition of N-acetyl-d-glucosamine monosaccharides to certain serine and threonine residues. These O-GlcNAc modification sites on tau have been challenging to identify due to the inherent complexity of tau from mammalian brains and the fact that the O-GlcNAc modification typically has substoichiometric occupancy. Here, we describe a method for the production of recombinant O-GlcNAc modified tau and, using this tau, we have mapped sites of O-GlcNAc on tau at Thr-123 and Ser-400 using mass spectrometry. We have also detected the presence of a third O-GlcNAc site on either Ser-409, Ser-412, or Ser-413. Using this information we have raised a rabbit polyclonal IgG antibody (3925) that detects tau O-GlcNAc modified at Ser-400. Further, using this antibody we have detected the Ser-400 tau O-GlcNAc modification in rat brain, which confirms the validity of this in vitro mapping approach. The identification of these O-GlcNAc sites on tau and this antibody will enable both in vivo and in vitro experiments designed to understand the possible functional roles of O-GlcNAc on tau.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The microtubule-associated protein tau is a neuronal cytoskeletal protein, the normal function of which is to promote and stabilize the assembly of microtubules (Weingarten et al. 1975). The discovery that tau is the major component of paired helical filaments (PHFs), which comprise the neurofibrillary tangles that are a characteristic of Alzheimer disease (AD) (Grundke-Iqbal et al. 1986), has driven considerable research interest in the area of tau biology. The molecular pathology of AD involves the hyperphosphorylation of tau (Kopke et al. 1993), which leads to its detachment from microtubules (Lindwall and Cole 1984a; Sengupta et al. 1998; Schneider et al. 1999), and facilitates its aggregation into PHFs (Alonso et al. 2001; Necula and Kuret 2005). Tau is also known to be modified by O-GlcNAc both in bovine (Arnold et al. 1996) and in human brain (Liu et al. 2004).
The O-GlcNAc modification is a dynamic post-translational modification that involves the attachment of N-acetyl-d-glucosamine (GlcNAc) moieties to the hydroxyl group of serine and threonine residues (Torres and Hart 1984). O-GlcNAc differs from the classic forms of O- and N-glycosylation in that O-GlcNAc modified proteins are found in the nucleus and cytosol and not within the secretory pathway (Torres and Hart 1984). O-GlcNAc transferase (OGT) catalyzes the transfer of GlcNAc from the donor sugar uridine 5′-diphospho-N-acetylglucosamine (UDP-GlcNAc) to target proteins (Kreppel et al. 1997; Lubas et al. 1997) and a glycoside hydrolase referred to as O-GlcNAcase (OGA) removes the sugar from modified proteins (Dong and Hart 1994; Gao et al. 2001).
Recently, we have shown that increasing O-GlcNAc in mammalian brain can lead to decreased levels of tau phosphorylation (Yuzwa et al. 2008). One model by which increased O-GlcNAc levels can decrease tau phosphorylation is that attachment of O-GlcNAc may occur at residues that can also be phosphorylated or at residues close to potential phosphorylation sites. Alternatively, it has been proposed that O-GlcNAc modification of kinases can alter their activities (Dias et al. 2009). Mapping the O-GlcNAc modification sites on tau is an important step toward testing these possible scenarios. Mapping of phosphorylation sites on tau has enabled an understanding of how site-specific phosphorylation can affect the function of this protein (Lindwall and Cole 1984a; Sengupta et al. 1998; Haase et al. 2004; Necula and Kuret 2005); mapping the O-GlcNAc sites on tau will likewise undoubtedly lead to a greater understanding of its possible functional impact on tau. Mapping the O-GlcNAc modification sites of tau has, however, proven difficult because tau is a mixture of isoforms in the mammalian brain (Janke et al. 1999), is challenging to O-GlcNAc modify in vitro, and contains many serine and threonine residues. The O-GlcNAc modification has generally proven difficult to detect on proteins by mass spectrometry because of the low stoichiometry of this modification and the labile nature of the O-glycosidic linkage. Consequently, methods of enrichment or derivatization of O-GlcNAc modified peptides have proven to be of importance (Wells et al. 2002; Khidekel et al. 2007; Wang et al. 2010). During the course of the studies described here, the O-GlcNAc modification of tau at Ser-400 was discovered using a chemoenzymatic approach (Wang et al. 2010) modified from that described earlier (Khidekel et al. 2007). These derivatization methods require multiple sample handling steps and, in the case of beta-elimination followed by Micheal addition (BEMAD) (Wells et al. 2002) does not allow simultaneous analysis of phosphorylation and O-GlcNAcylation. Mass spectrometers using quadrupole time-of-flight technology have been successfully used to detect the O-GlcNAc modification without the need for enrichment or derivatization (Chalkley and Burlingame 2003) and recently electron transfer dissociation (ETD) in combination with Fourier-transform ion cyclotron mass spectrometry (FT-ICRMS) has recently facilitated mapping of O-GlcNAc modification sites on a number of proteins (Viner et al. 2009).
Here, we describe an alternative approach and apply it to tau protein. This method involves the production of milligram quantities of recombinant O-GlcNAc modified tau and subsequent analysis by nanoLC-ESIMS mass spectrometry. We map two O-GlcNAc modification sites on tau and provide evidence for the presence of a third. Using this information we have developed an O-GlcNAc site-specific tau antibody that recognizes O-GlcNAc on tau at Ser-400 in a peptide-dependent context. We use this polyclonal IgG antibody to detect the O-GlcNAc modification of endogenous tau at Ser-400 in mammalian brain tissue.
Materials and methods
Peptide synthesis
Synthesis of the fluorenylmethyloxycarbonyl (Fmoc) protected and peptafluorophenyl (Pfp) activated per-acetylated β-O-GlcNAc modified serine (Fmoc-Ser(Ac3-β-O-GlcNAc)-Pfp) was carried out essentially as described previously (Simanek et al. 1998; Chen et al. 2006). 1H-NMR chemical shifts were identical to the published values. Glycopeptide synthesis was carried out at BioBasic Inc. (Markham, ON, Canada) using solid phase peptide synthesis to generate the following peptides: Ac-CSPVVSGDTS-NH 2 (unmodified, OH-tau-peptide 1) and Ac-CSPVV g SGDTS-NH 2 (O-GlcNAc modified, OG-tau-peptide 2) on Rink Amide resin using standard Fmoc chemistry with O-benzotriazole-N,N,N′N′-tetramethyl-uronium-hexafluorophosphate/N,N-diisopropylethylamine couplings. Biobasic Inc. deprotected and purified OH-tau-peptide 1 by high performance liquid chromatography (HPLC) and assessed its purity to be >97%. Upon receipt of the fully protected OG-tau-peptide 2 still coupled to the Rink Amide resin, the O-acetyl groups on the GlcNAc moiety were deacetylated by resuspending the resin (0.8 g) in 9 mL of dry methanol. The solution was adjusted to a pH > 9 (judged using pH paper) using a solution of 0.5 M sodium methoxide in dry methanol. The solution was shaken gently for 24 h using an orbital shaker at room temperature. The resin was then washed with methanol three times and then resuspended in a solution containing trifluoroacetic acid (TFA), water, 1,2-ethanedithiol (EDT), and triethylsilane in a ratio of (85:2.5:2.5:10) and was allowed to shake for 2 h at room temperature. The resin was then filtered off and washed three times with fresh TFA. The filtrate was then concentrated in vacuo using a high vacuum rotary evaporator. Cold diethylether was added to the filtrate until a white precipitate formed in the flask. The precipitate was centrifuged at 800 rpm in a Sorvall Legend RT centrifuge in a disposable 50 mL conical centrifuge tube using a swinging bucket TTH-750 rotor. The resulting pellet was then washed three times with cold diethylether followed by centrifugation. Finally, the diethylether was removed and the crude peptide pellet was dried under a stream of nitrogen gas, resuspended in water, lyophilized to dryness and stored at −20°C until required.
Purification of OG-tau-peptide by reversed phase HPLC
The crude peptide (10 mg/run) was loaded onto an Agilent XDB-C18 (9.4 mm × 250 mm) semi-preparative HPLC column housed in an 1100 series Agilent HPLC. The peptide was purified using a linear gradient of 5% acetonitrile (ACN) to 60% ACN over 40 min operating at 2 mL/min. Fractions were collected using a Foxy Jr. fraction collector set to collect 500 μL fractions over the entirety of the HPLC run. The major peak eluted at approximately 12.8 min and was collected in fractions 54–59. These fractions were pooled and high-resolution mass spectrometry was carried out to verify the correct identity of the peptide. HRMS predicted: 1195.5108 Da, found: 1195.5204 Da.
Production of the 3925 antibody in rabbits
10 mg of OG-tau-peptide 2 was sent to Pacific Immunology Corp. (Ramona, CA, USA) where it was coupled to keyhole limpet hemocyanin (KLH) through the N-terminal cysteine. Two New Zealand white rabbits each received a 100 μg injection of the final immunogen in Freund’s complete adjuvant (CFA) on day 0, followed by three 100 μg booster injections in Freund’s incomplete adjuvant (IFA) on days 21, 42, and 70. Production bleeds were carried out on days 49, 63, 80, and 94 and the immune response was verified by enzyme-linked immunosorbant assay (ELISA) where the titer was determined to be 1:500,000.
Affinity purification of the 3925 antibody
1.5 mg of OG-tau-peptide 2 was coupled to 1.5 mL of SulfoLink resin (Pierce) according to the manufacturer’s protocol. The resin was placed into a 5 mL column (Biorad) and equilibrated in phosphate buffered saline pH 7.4 (PBS). 5 mL of serum was then loaded onto the column at a flow rate of ~1 mL/min. The column was washed with 30 mL of PBS and then eluted with 20 mL of 100 mM glycine pH 3, 10% ethylene glycol. The eluate was dialyzed against PBS and then concentrated to 0.5–1.0 mg/mL.
Western blotting
Samples were electrophoresed through 10% sodium dodecyl sulfate polyacrylamide gels (SDS-PAGE) and transferred to nitrocellulose (Bio-rad) membranes. Membranes were then blocked for 1 h at room temperature (RT) with 2% bovine serum albumin (BSA) in PBS containing 0.1% Tween-20 (Sigma) (PBS-T) and then subsequently probed with the appropriate primary antibody delivered in 2% BSA in PBS-T overnight at 4°C. Membranes were then extensively washed with PBS-T, blocked again for 30 min with 2% BSA in PBS-T at RT, and then probed with the appropriate HRP-conjugated secondary antibody delivered in 2% BSA in PBS-T for 1 h at RT. Finally, the membranes were washed extensively with PBS-T and then developed with SuperSignal West Pico Chemiluminescence substrate (Pierce) and exposed to CL-XPosure Film (Pierce). Blots pretreated with alkaline phosphatase (Roche) or with Bacteroides thetaiotaomicron OGA (btOGA, a bacterial homolog of human OGA) (Dennis et al. 2006) were incubated with 100 Units of calf intestinal alkaline phosphatase or 500 μg btOGA respectively in 50 mM Tris–HCl, pH 8.5, 0.1 mM EDTA, 0.5 mM MgCl2 for 4.5 h at 37°C following the first blocking step. The blots were then washed briefly in PBS-T and the rest of the Western blot protocol was followed as described above.
Antibodies used in this study
Mouse monoclonal α-Tau-5, which recognizes the central region of tau in a phosphorylation-independent manner, was purchased from Lab Vision Corporation. Mouse monoclonal α-Tau-46, which recognizes the C-terminal region of tau in a phosphorylation-independent manner was purchased from Abcam. Mouse monoclonal α-O-GlcNAc antibodies, CTD110.6 and RL2, were purchased from Covance and Abcam, respectively. Goat α-mouse IgG, goat α-rabbit IgG, and goat α-mouse IgM horseradish peroxidase conjugated secondary antibodies were obtained from Santa Cruz Biotechnology.
Molecular cloning
The human OGT gene was cloned into the pMal-c2X vector (New England Biolabs) using the following primers: 5′-gccgccggatccaagcgtcttccgtgggcaacgtgg-3′ (BamHI cut site shown in bold) and 5′-gccgccgtcgacctatgctgactcagtgacttcaac-3′ (SalI cut site shown in bold). The H558A point mutation of OGT was created using the following primers: 5′-gagttccgactttgggaatgctcctacttctcaccttatgc-3′ and 5′-gcataaggtgagaagtaggagcattcccaaagtcggaactc-3′. These primers were used to introduce the point mutation using QuikChange site-directed mutagenesis. The longest human isoform of the microtubule-associated protein tau gene (MAPT) was cloned into the pET28a vector (Novagen) using the following primers: 5′-gccgcccatatgatggctgagccccgccagg-3′ (NdeI cut site shown in bold) and 5′-gccgccctcgagttacaaaccctgcttggccaggg-3′ (XhoI cut site shown in bold). The S396A point mutation in Tau441 was created with the following primers: 5′-gcggagatcgtgtacaaggcgccagtggtgtctggggac-3′ and 5′-gtccccagacaccactggcgccttgtacacgatctccgc-3′. The S400A point mutation in Tau441 was created with the following primers: 5′-gcaagtcgccagtggtggctggggacacgtctccc-3′ and 5′-gggagacgtgtccccagccaccactggcgacttgc-3′.
Production of recombinant O-GlcNAc modified tau
The gene encoding Tau441, in pET28a, was co-transformed with the gene encoding wild-type OGT (wtOGT) or H558A OGT (mutOGT), in pMal-c2X, into E. coli Tuner cells (Stratagene). For expression, Tau441 co-transformants were induced with IPTG (0.5 mM) for 4 h or overnight at 22°C (~20 h). The bacterial cells were harvested by centrifugation at 5,000 rpm for 10 min in a Sorvall RC-6 plus centrifuge in a FIBERLite F9S-4x1000y rotor. The bacterial pellets were then resuspended in 30 mL of Ni–NTA column binding buffer (20 mM sodium phosphate, 500 mM NaCl, 5 mM imidazole, pH 7.4). wtOGT/Tau441 and mutOGT/Tau441 co-transformants were lysed by the addition of 2 mg/mL lysozyme (Bioshop) in the presence of one Roche protease Complete tablet per 30 mL of resuspended bacterial pellet. Sonication was carried out at ~30% power using a Fischer Scientific sonic dismembrator (model 500) for six cycles consisting of 20 s followed by a 40 s rest period. Cellular debris was removed by centrifugation at 13,000 rpm in an SS-34 rotor in a Sorvall RC-6 plus centrifuge. The supernatants were then loaded onto two separate HisTrap FF Ni–NTA columns (GE Healthcare) using two separate peristaltic pumps. The columns were washed with 90 mL of Ni–NTA column wash buffer (20 mM sodium phosphate, 500 mM NaCl, 60 mM imidazole, pH 7.4) and then protein was eluted with 25 mL of Ni–NTA column elution buffer (20 mM sodium phosphate, 500 mM NaCl, 250 mM imidazole, pH 7.4). The eluates were then dialyzed against PBS for western blot analysis or for mass spectrometry analysis against 18 mΩ water and then lyophilized. Additionally, in some cases, 5 μg of recombinant O-GlcNAc modified Tau441 was mixed with 5 μg of recombinantly expressed and immobilized metal affinity chromatography (IMAC) purified ncOGT (clone provided by Dr. Suzanne Walker) in the presence of 0.5 mM UDP-GlcNAc in the following buffer: 20 mM potassium phosphate, 12.5 mM MgCl2, 1 mM DTT, protease inhibitor cocktail (Roche), pH 7.4. Following over night incubation at room temperature half of the reaction mixture was purified with C18 Zip Tip (Millipore), dried down, and reconstituted in 0.1% formic acid (FA) for LC/MS/MS analysis.
Mass spectrometry methods
For the identification of the Ser-400 O-GlcNAc modification site and its subsequent detection by multiple reaction monitoring (MRM), data was obtained using an AB Sciex 4000 QTrap mass spectrometer equipped with a nano-spray source and custom built nano-spray head. The LC is a Dionex U3000 with a 1:1,000 splitter. Mobile phase A was 100% water with 0.1% FA and mobile phase B was 90% ACN, 10% water, and 0.1% FA. A graphitized carbon enrichment column was loaded with 1 μL then washed for 10 min at a flow rate of 10 μL/min. LC flow rate was calibrated to 300 nL/min with pure methanol added in at a flow rate of 50 nL/min just before the nano-spray tip. Elution began with a linear ramp from 0% B to 90% B at 30 min and then held for 5 min at 90% B before returning rapidly to 100% A. The enrichment column was made in house by packing a 360 μm O.D. and 250 μm I.D. piece of silica tubing with 7 μm graphitized carbon. The primary column was a 5 μm Hypercarb column from Thermo Scientific (HY35005-050065). Mass spectrometer parameters included a declustering potential of 41 V for the unmodified peptide and 39 V for the peptide carrying the O-GlcNAc. Collision energies ranged from 39 to 45 V for fragment ions without modification and 22.5 V for the fragment ion with modification. Exploratory work was carried out using an information-dependant data acquisition (IDA) method, whereby any ions detected between 400 and 2,000 amu in enhanced MS mode at 4,000 amu/s in excess of 10,000 counts had an enhanced resolution scan performed at 250 amu/s. The enhanced resolution scan was used to acquire a charge state and an accurate mass. These ions were subsequently fragmented using an enhanced product ion scan at 1,000 amu/s using rolling collision energy. This was done by direct injection of 1 μL onto a custom packed Reprosil Gold 3 μm C18 column. The flow rate used was 300 nL/min using a gradient over 90 min. Following identification of an O-GlcNAc modified peptide in the MS/MS spectrum as having the sequence SPVVgSGDTSPR, where g denotes the site of glycosylation, several transitions were established that could be used for MRM analysis of samples to evaluate the presence of the modified peptide as well as the unmodified peptide. Three transitions from the unmodified peptide precursor leading to the formation of y4, y6, and y7 ions were all monitored. Three transitions from O-GlcNAc modified peptide precursor leading to the formation of y4, y6, and y7 fragment ions, plus the mass of O-GlcNAc, were all monitored. Unmodified MRM transitions are 551/460, 551/632, and 551/719. Modified MRM transitions are 653/460, 653/632, and 653/922.
For the identification of the other O-GlcNAc modification sites, mass spectrometry analysis was performed on an LTQ linear ion trap mass spectrometer (Thermo Scientific). All in-line LC–MS/MS runs were performed using an Eksigent NanoLC-AS1 auto-sampler (Eksigent) with nano-spray. Samples were injected onto an IntergraFrit Proteopep II (New Objective) C18 sample trap column (300 Å × 75 μm ID × 2.5 cm bed length) for final desalting and concentration at a flow rate of 3 μL/min (0.1% FA) for 15 min. The inline capillary column coupled to mass spectrometry was an IntegraFrit Proteopep II (New Objective) C18 column (300 Å × 75 μm ID × 10 cm bed length). Sample was eluted from the trap column by a 55-min 5–35% linear gradient of buffer B (100% ACN, 0.1% FA), followed by a 25-min 35–80% linear gradient of buffer B to a 5 min isocratic elution at 80% buffer B at a flow rate of 300 nL per minute. Dynamic exclusion was set such that if an ion was detected it was excluded from analysis for 30 s. The electrospray voltage was 1.5 kV. The mass range for MS scans was 400–2,000 Da. Data were acquired using a single microscan and a maximum inject time of 200 ms for MS, MS2, and MS3 scan types. MS3 on MS/MS was specified for precursor ions showing diagnostic neutral loss of O-GlcNAc mass of either 102 (doubly charged precursor) or 68 (triply charged precursor) in MS/MS. A collision-induced dissociation (CID) energy of 33% was used.
Purification of tau from rat brain
Rat brain was homogenized in PBS containing 500 mM NaCl, 1 mM Thiamet-G, and a Roche protease inhibitor tablet in a 1:1 ratio of buffer to frozen tissue weight. The tissue was homogenized using an IKA tissue homogenizer for 1 min, boiled for 7 min, and subsequently centrifuged at 17,900g. Perchloric acid was then added to a final concentration of 2.5% and allowed to incubate on ice for 15 min after which it was centrifuged at 17,900g. To the supernatant was added the appropriate amount of 5× SDS-PAGE loading buffer followed by 1 M Tris pH 8 until the loading buffer was returned to the characteristic blue color of bromophenol blue in loading buffer, indicating that the pH of the solution was greater than five.
Results
In order to map the O-GlcNAc sites on tau it is necessary to obtain a source of modified tau protein. An approach used very recently by the Hart laboratory made use of rat brain lysate to identify the Ser-400 O-GlcNAc modification site on tau. Due to the inherent complexity of rat brain lysates this approach required initial sample enrichment of the O-GlcNAc modified proteins, through enzymatic labeling and chemical derivatization, prior to analysis by mass spectrometry (Wang et al. 2010). It is likely that this elegant approach will prove useful for identification of physiologically relevant sites on many proteins from various tissues. An alternative approach to obtain O-GlcNAc modified proteins that neither involves the use of tissue lysates nor requires the in vitro modification of the target protein has been proposed (Lim et al. 2002). This alternative approach involves the co-expression of OGT in the presence of the target protein, transcription factor Sp1, in E.coli cells. Upon induction of the two proteins, OGT could O-GlcNAc modify Sp1. We have developed a modified form of this initial approach as seen in Fig. 1a for the production of recombinant O-GlcNAc modified tau. We express the longest human isoform of tau (Tau441) with a hexahistidine tag in the presence of wild-type OGT (wtOGT) for 4 h. During this induction period the wtOGT acts to O-GlcNAc modify Tau441. Tau441 is then purified using a Ni–NTA column affording recombinant O-GlcNAc modified tau (Fig. 1b). In parallel, we also express Tau441 in the presence of a catalytically inactive mutant of OGT (mutOGT) that cannot O-GlcNAc modify Tau441, which serves as a negative control for the recombinant O-GlcNAc modification of tau. As shown in Fig. 1b, only Tau441 expressed in the presence of wtOGT exhibits significant immunoreactivity with two anti-O-GlcNAc antibodies, which both recognize a number of O-GlcNAc modified proteins (Snow et al. 1987; Comer et al. 2001). Preincubation of the CTD110.6 anti-O-GlcNAc antibody with 10 mM free GlcNAc results in complete abolishment of immunoreactivity, suggesting that the interaction of the CTD110.6 antibody with the O-GlcNAc modified Tau441 is specific for O-GlcNAc. As mentioned above, during the course of this research Ser-400 of tau was reported to be O-GlcNAc modified in rat brain (Wang et al. 2010). In an attempt to validate our method for producing recombinant O-GlcNAc modified tau we had previously made a serine to alanine mutation in Tau441 at the positions of Ser-400 and Ser-396 and then expressed these tau mutants in the presence of wtOGT or mutOGT. As can be seen in Fig. 1c, mutation of Ser-400 to alanine results in ~2-fold less O-GlcNAc on tau as judged by CTD110.6 immunoreactivity, whereas the mutation of Ser-396 to alanine results in essentially no change in immunoreactivity. This evidence suggests that the recombinantly produced O-GlcNAc modified tau is very likely modified at Ser-400. To be sure the Tau441 produced in this manner has the highest stoichiometry of O-GlcNAc achievable, we expressed Tau441 in the presence of wtOGT and mutOGT overnight (~20 h). This expression method results in O-GlcNAc modified tau that can be detected by the CTD110.6 O-GlcNAc antibody (Fig. 1d) and is modified with roughly three times more O-GlcNAc than when expressed in the presence of wtOGT for 4 h (not shown).

Production of recombinant O-GlcNAc modified tau. a Schematic representation of the production of recombinant O-GlcNAc modified tau. The plasmids encoding OGT and Tau441 are co-transformed into E. coli cells, induced for up to 20 h, and the O-GlcNAc modified tau is purified. b Recombinant tau is O-GlcNAc modified. Western blots using α-O-GlcNAc antibodies CTD110.6 and RL2 indicate that only when expressed with wtOGT, for 4 h, is Tau441 O-GlcNAc modified. Coomassie blue staining demonstrates equal tau loading on the gel. c Mutation of Tau441 at Ser-400 leads to decreased O-GlcNAc on tau as detected by western blot with the CTD110.6 antibody whereas Tau-5 indicates equal tau loading. d α-O-GlcNAc (CTD110.6) western blot of Tau441 expressed either in the presence of wtOGT or mutOGT expressed for 20 h. Equal tau loading is indicated by probing with the total tau antibody, Tau-46
Using O-GlcNAc modified tau expressed in the presence of wtOGT overnight we have mapped an O-GlcNAc modification site on Tau441 at Ser-400 using LC–MS/MS. The approach used involved information-dependent acquisition of data. This led to the identification and sequencing of an O-GlcNAc modified peptide by monitoring the fragment ions resulting from collision-induced fragmentation of a doubly charged precursor ion of mass 652.8. The peptide and sequence was determined to be 396-SPVVgSGDTSPR-406, where g denotes the site of glycosylation (Fig. 2). This peptide spans Ser-396 to Arg-406 of tau in which Ser-400 is the modification site. This site is the same as was reported in a study that was published (Wang et al. 2010) while our immunization experiments reported below were underway. We established several mass transitions that could be used to monitor both modified and unmodified peptides and established that both peptides could be found in the sample of modified tau, while no O-GlcNAc modified peptide was detected in the unmodified sample of recombinant Tau441 (Fig. 3).

MS/MS fragment-ion spectrum of a peptide from digestion of recombinant tau reveals a site of glycosylation at Ser-400. y and b series fragments that were observed are shown above and below the peptide sequence, respectively

Multiple reaction monitoring (MRM) of O-GlcNAc-modified and unmodified peptide from recombinant tau proteins. a and b show MRM experiments for three transitions for the recombinant O-GlcNAc modified tau and c and d show MRM for three transitions for the recombinant unmodified tau. For each sample of tau the first plot represents three concurrent MRM experiments that use the doubly charged unmodified peptide SPVVSGDTSPR for all precursors and the fragment ions used are the y4, y6, and y7 ions. The second plots use the precursors of SPVVSGDTSPR plus a shift corresponding to an O-GlcNAc modification and fragment ions y4, y6, and y7 plus a shift corresponding to an O-GlcNAc modification. Unmodified MRM transitions are 551/460, 551/632, and 551/719. Modified MRM transitions are 653/460, 653/632, and 653/922
The observation that mutation of Ser-400 of tau reduced, but did not abolish, tau O-GlcNAc modification (Fig. 1) suggested that there are additional sites of O-GlcNAc modification. O-GlcNAc modified recombinant Tau441 was glycosylated in vitro using recombinant OGT in an attempt to increase the stoichiometry of O-GlcNAc modification. In addition to proteolytic digests using trypsin, we also carried out proteolytic digestions of in vitro O-GlcNAc modified Tau441 using a combination of trypsin and AspN, in an effort to increase the number of peptides of a size that are amenable to MS/MS analysis. This approach was expected to lead to increased coverage of the Tau primary sequence. These digests were analyzed by LC–MS/MS in an LTQ ion trap mass spectrometer (see “Materials and methods” for details).
A site of tau O-GlcNAc modification was identified at Thr-123 (Fig. 4) within the peptide 110-DTPSLEDEAAGHVTQAR-126, corresponding to N-terminal AspN cleavage and C-terminal tryptic cleavage. In MS/MS, it is unusual that the 899.2 [M + 2H]2+ O-GlcNAc neutral loss ion is not the dominant ion in the spectra (Fig. 4a). A “zoom” scan of the region corresponding to the precursor ion 1000.3 [M + 2H]2+ revealed overlapping isotopic distributions from two distinct ions. Thus, we believe the MS/MS represents a mixture of fragment ions from these two species, complicating interpretation. However, the specific 899.2 [M + 2H]2+ O-GlcNAc neutral loss ion was selected for MS/MS/MS and provided clear evidence of the O-GlcNAc modified peptide sequence (Fig. 4c). A search for O-GlcNAc modified ions in MS/MS corresponding to the peptide 110-DTPSLEDEAAGHVTQAR-126 revealed several pairs of y ions in the unmodified and O-GlcNAc modified state, including the y6 ion (Fig. 4b). Given the complexity of the spectrum, the y6 unmodified and O-GlcNAc modified ions are shown at twice magnification for clarity.

MS/MS and MS/MS/MS spectra from an in vitro O-GlcNAc modified Tau441 tryptic and AspN digest identifies a site of glycosylation at Thr-123. y and b series fragment ions unmodified or retaining O-GlcNAc are shown for a, b MS/MS of 1000.3 [M + 2H]2+ and for c MS/MS/MS of the O-GlcNAc neutral loss ion in MS/MS at 899.2 [M + 2H]2+
A third site of tau O-GlcNAcylation at either serine 409, 412, or 413 was also identified within the peptide 407-HLSNVSSTGSIDMVDSPQLATLADEVSASLAK-438. MS/MS of a triply charged precursor ion at 1150.5 gave rise to characteristic O-GlcNAc neutral loss ions both in a doubly and in a triply charged state (Fig. 5). The triply charged 1082.4 [M + 3H]3+ was selected for MS/MS/MS. Sparse fragment ion information was generated, but all corresponded to the tau tryptic peptide 407-HLSNVSSTGSIDMVDSPQLATLADEVSASLAK-438 (Fig. 5b). Re-examination of the MS/MS revealed the presence of y and b ion series corresponding to the 407-HLSNVSSTGSIDMVDSPQLATLADEVSASLAK-438 peptide (supplementary Figs. 1–3). Additionally, pairs of unmodified and O-GlcNAc modified b7 and y +230 ions indicate that the modification occurs on either serine 409, 412, or 413.

MS/MS and MS/MS/MS spectra from an in vitro O-GlcNAc modified Tau tryptic digest identifies O-GlcNAc modified peptide 407-HLSNVSSTGSIDMVDSPQLADEVSASLAK-438. a MS/MS of precursor 1150.5 [M + 3H]3+ displays O-GlcNAc neutral loss ions. b y and b series ions for MS/MS/MS of the O-GlcNAc neutral loss ion in MS/MS at 1082.4 [M + 3H]3+
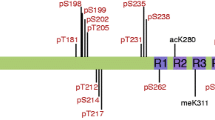
The C-terminal region of tau is one of its most densely phosphorylated regions and has been shown to harbor phosphorylation sites at Ser-396, Ser-400, Ser-404, Ser-409, Ser-412, Ser-413, Ser-416, and Ser-422 (Buee et al. 2000). It has been shown previously that elevation of O-GlcNAc leads to reductions in tau phosphorylation at Ser-396 both ex vivo (Liu et al. 2004) and in vivo (Yuzwa et al. 2008). The presence of O-GlcNAc at Ser-400 could thus explain why increased O-GlcNAc could lead to reduced phosphorylation at Ser-396. The presence of a second O-GlcNAc site on tau between Ser-409 and Ser-413, which is within the region of tau that is heavily phosphorylated, suggests that O-GlcNAc in this region may be important for the regulation of the phosphorylation sites mentioned above. It should be noted that these identified sites may not be the only O-GlcNAc modification sites on tau, further efforts to identify additional sites may prove informative. Previous data suggest that the mechanism for reduction of tau phosphorylation caused by increases in O-GlcNAc could be due to direct competition between these two modifications for the same sites, or for sites which are close to one another. It is also possible, however, that the reductions in tau phosphorylation caused by increased O-GlcNAc could arise from an indirect mechanism. For example, increased O-GlcNAc on a kinase or a phosphatase may decrease or increase the activity of these enzymes, respectively, and would lead to similar downstream effects as would be seen if a direct mechanism were operative. Future studies should clarify which one of these mechanisms is responsible for the apparent reciprocal relationship.
With these data in hand we synthesized the fluorenylmethyloxycarbonyl (Fmoc) protected and pentafluorophenyl (Pfp) activated per-acetylated O-GlcNAc modified serine (Fmoc-Ser(Ac3-β-O-GlcNAc)-Pfp). This Fmoc-Ser(Ac3-β-O-GlcNAc)-Pfp was used to synthesize the peptide which surrounds the Ser-400 O-GlcNAc modification site (Ser-396 to Ser-404) to generate the following sequence: Ac-CSPVV g SGDTS-NH 2 (OG-tau-peptide 2) (Fig. 6a). An unmodified peptide with the following sequence was also synthesized: Ac-CSPVVSGDTS-NH 2 (OH-tau-peptide 1) (Fig. 6a). The OG-tau-peptide 2 was coupled to keyhole limpet hemocyanin (KLH) and used to immunize two New Zealand white rabbits. The serum from one of these animals was affinity purified over an affinity column bearing OG-tau-peptide 2. As shown in Fig. 6b, this antibody (referred to by the number 3925) specifically detects Tau441 only when it is expressed in the presence of wtOGT and not when expressed in the presence of mutOGT. The detection of 3925 with a goat α-rabbit IgG secondary antibody indicates that this antibody is of the IgG isotype. Preincubation of 3925 with 2 μM of OG-tau-peptide 2 completely abolishes the immunoreactivity whereas preincubation with 2 μM of OH-tau-peptide 1 has no effect on the immunoreactivity (Fig. 6b). 50 mM free GlcNAc also has no effect on the immunoreactivity, which indicates that 3925 requires a peptide determinant as well as the pendent O-GlcNAc residue for binding (Fig. 6b). Using decreasing concentrations of O-GlcNAc modified Tau441 we have determined the detection limit of 3925 to be in the range of 20–50 ng O-GlcNAc modified Tau441 when using 2 μg of 3925 per blot (Fig. 6c).

Production a site-specific O-GlcNAc tau antibody. a Peptides used in this study. b Western blot using 3925 to detect Tau441 expressed either in the presence of wtOGT or mutOGT. 3925 specifically recognizes Tau441 only when expressed in the presence of wtOGT. OG-Tau-peptide 2 blocks binding of 3925 to O-GlcNAc modified Tau441 whereas OH-Tau-peptide 1 does not. 50 mM GlcNAc does not block the interaction between 3925 and O-GlcNAc modified tau. Probing with the CTD110.6 antibody confirms that this Tau441 preparation is O-GlcNAc modified and Tau-46 confirms equal Tau441 loading. c The detection limit of the 3925 antibody was found to be in the range of 20–50 ng O-GlcNAc modified Tau441 when using 2 μg of 3925 per blot
To probe the value of this antibody for detecting O-GlcNAc at Ser-400 in tau from brain tissue, we prepared a tau enriched fraction from rat brain by making use of two properties of tau; its heat-stability (Weingarten et al. 1975) and its resistance to perchloric acid treatment (Lindwall and Cole 1984b). The rat tau protein preparation was electrophoresed through SDS-PAGE gels and subjected to western blot analysis using the 3925 antibody. As can be seen (Fig. 7a) 3925 detects protein bands in the range of 43–72 kDa, which are likely attributable to tau because they overlap well with the bands detected (Fig. 7a) using Tau-5 (an antibody recognizing tau) and they are detected both in the boiled lysate as well as in the lysate that was both boiled and treated with perchloric acid. Also evident in the boiled lysate samples are unknown protein bands that appear above the 72 kDa marker, which indicates that 3925 recognizes O-GlcNAc on other proteins. Interestingly, treatment of the western blot membrane with calf intestinal alkaline phosphatase (AP) prior to addition of the 3925 antibody results in a notable increase in immunoreactivity with essentially the same banding pattern as is detected by the Tau-5 antibody (Fig. 7a). This observation suggests that phosphorylation of tau (presumably near Ser-400) blocks the binding of the 3925 and that treatment with AP relieves this blockage. This phosphorylation dependence is reminiscent of a commercially available antibody recognizing tau phosphorylated at Ser-400 (Singer et al. 2005). Singer et al. showed that the binding of the phospho-Tau400 antibody is reduced by 10-fold when a second phosphate is present at Ser-396. To verify specificity of the interaction when detecting tau from rat brain, we pretreated 3925 with OG-Tau-peptide 2. Complete elimination of immunoreactivity was observed (Fig. 7b), which supports the specificity of 3925 for glycopeptide epitopes, such as Ser-400 O-GlcNAc tau. Furthermore, by treating the membrane with a bacterial homolog of OGA, Bacteroides thetaiotaomicron OGA (Dennis et al. 2006) (btOGA) prior to incubation with the 3925 antibody, results in a reduction of the immunoreactivity (Fig. 7b, right most panel). This observation again supports the O-GlcNAc group being a critical determinant for binding of the 3925 antibody to O-GlcNAc-modified tau. Incomplete digestion of the O-GlcNAc from tau under the conditions used likely accounts for the residual 3925 immunoreactivity after treatment with btOGA.

3925 detects the Ser-400 O-GlcNAc modification in the rat brain. Tau protein was enriched from rat brain by treating boiled lysates with perchloric acid. a Western blot using the 3925 antibody (left most panel) detect bands in the range of 43–72 kDa in both the boiled (lane 1) and boiled plus perchloric acid treated lanes (lane 2). These bands appear similar to bands of the same size when using an anti-tau antibody (tau-5) (third panel). Treatment of the blot with calf intestinal alkaline phosphate (AP) results in an increase in 3925 immunoreactivity of approximately 3- to 4-fold (second and fourth panel). The 3925 antibody also appears to detect other proteins in the boiled lysate larger than 72 kDa (lane 1 of the first two panels). b The interaction of the 3925 antibody is specific as indicated by the ability of the OG-Tau-peptide 2 to completely abolish the 3925 immunoreactivity (third panel) and that treatment of the blot with btOGA results in a reduction of 3925 immunoreactivity (fourth panel). Treatment with AP markedly increases 3925 immunoreactivity (second panel)
Discussion
Here, we describe a relatively simple method that allows identification of O-GlcNAc sites of proteins in vitro. It is important to note that O-GlcNAc sites identified in the manner described here using recombinant proteins provide a convenient starting point for probing the physiological role of O-GlcNAc on proteins in vivo as well as for developing useful tools to help understand these roles. We have used this recombinant expression strategy, in conjunction with mass spectrometry techniques, to map O-GlcNAc modification sites on tau. The two O-GlcNAc modification sites identified on recombinant tau are located at Ser-400 and Thr-123. One of these sites, Ser-400, was shown to be O-GlcNAc modified during the generation of the 3925 antibody, which supports the validity of this recombinant method for producing physiologically relevant O-GlcNAc modified tau. We have also detected the presence of a third O-GlcNAc modification site at one of Ser-409, Ser-412, or Ser-413. It will prove interesting to assess whether these other O-GlcNAc modification sites that we have discovered on tau can be detected using tau purified from mammalian brain and these studies are underway. Furthermore, having mapped O-GlcNAc modification sites on tau should enable an understanding of the functional roles of O-GlcNAc on tau.
Using the insights gained from site mapping we were successful in raising a polyclonal antibody toward tau that is O-GlcNAc modified at Ser-400. The most commonly used anti-O-GlcNAc antibody, CTD110.6, is a mouse IgM antibody (Comer et al. 2001) and is therefore of limited use for immunoprecipitation because protein A/protein G do not bind IgM antibodies. Very recently, mouse IgG O-GlcNAc antibodies have been raised using a novel antigen (Teo et al. 2010) and these should prove to be useful reagents. The fact that the 3925 O-GlcNAc tau antibody is an IgG antibody will likewise make this antibody useful in immunoprecipitation experiments of O-GlcNAc modified tau from tissue or cell lysastes. We find this antibody depends on both the peptide determinant and the O-GlcNAc residue. We also find that this antibody can be used to detect tau modified at Ser-400 in rat brain tissue. It is interesting to note that phosphatase digestion results in exposure of the epitope in the sample derived from rat brain, suggesting that phosphorylation may occur proximal to O-GlcNAc at Ser-400. This is intriguing since O-GlcNAc and phosphorylation are both substoichiometric post-translational modifications and, if they are installed independently or antagonistically of each other, then we would predict little effect from phosphatase digestion. Accordingly, the significant effect that is observed here on phosphatase digestion suggests that one of these modifications may direct preferential installation of the other modification. These scenarios are under current investigation and will be reported in due course. The use of this site-specific antibody, and additional antibodies we are currently generating, should facilitate studying the potential roles of O-GlcNAc in the functioning of tau, the regulation of its phosphorylation state, as well as its potential role in the AD brain.
References
Alonso AD, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K (2001) Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci USA 98(12):6923–6928
Arnold CS, Johnson GV, Cole RN, Dong DL, Lee M, Hart GW (1996) The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine. J Biol Chem 271(46):28741–28744
Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 33(1):95–130
Chalkley RJ, Burlingame AL (2003) Identification of novel sites of O-N-acetylglucosamine modification of serum response factor using quadrupole time-of-flight mass spectrometry. Mol Cell Proteomics 2(3):182–190
Chen YX, Du JT, Zhou LX, Liu XH, Zhao YF, Nakanishi H, Li YM (2006) Alternative O-GlcNAcylation/O-phosphorylation of Ser(16) induce different conformational disturbances to the N-terminus of murine estrogen receptor beta. Chem Biol 13(9):937–944
Comer FI, Vosseller K, Wells L, Accavitti MA, Hart GW (2001) Characterization of a mouse monoclonal antibody specific for O-linked N-acetylglucosamine. Anal Biochem 293(2):169–177
Dennis RJ, Taylor EJ et al (2006) Structure and mechanism of a bacterial beta-glucosaminidase having O-GlcNAcase activity. Nat Struct Mol Biol 13(4):365–371
Dias WB, Cheung WD, Wang Z, Hart GW (2009) Regulation of calcium/calmodulin-dependent kinase IV by O-GlcNAc modification. J Biol Chem 284(32):21327–21337
Dong DL, Hart GW (1994) Purification and characterization of an O-GlcNAc selective N-acetyl-beta-D-glucosaminidase from rat spleen cytosol. J Biol Chem 269(30):19321–19330
Gao Y, Wells L, Comer FI, Parker GJ, Hart GW (2001) Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J Biol Chem 276(13):9838–9845
Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM (1986) Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem 261(13):6084–6089
Haase C, Stieler JT, Arendt T, Holzer M (2004) Pseudophosphorylation of tau protein alters its ability for self-aggregation. J Neurochem 88(6):1509–1520
Janke C, Beck M, Stahl T, Holzer M, Brauer K, Bigl V, Arendt T (1999) Phylogenetic diversity of the expression of the microtubule-associated protein tau: implications for neurodegenerative disorders. Mol Brain Res 68(1–2):119–128
Khidekel N, Ficarro SB et al (2007) Probing the dynamics of O-GlcNAc glycosylation in the brain using quantitative proteomics. Nat Chem Biol 3(6):339–348
Kopke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I (1993) Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem 268(32):24374–24384
Kreppel LK, Blomberg MA, Hart GW (1997) Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J Biol Chem 272(14):9308–9315
Lim KH, Ha CH, Chang HI (2002) Production of O-GlcNAc modified recombinant proteins in Escherichia coli. J Microbiol Biotech 12(2):306–311
Lindwall G, Cole RD (1984a) Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem 259(8):5301–5305
Lindwall G, Cole RD (1984b) The purification of tau protein and the occurrence of two phosphorylation states of tau in brain. J Biol Chem 259(19):12241–12245
Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX (2004) O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc Natl Acad Sci USA 101(29):10804–10809
Lubas WA, Frank DW, Krause M, Hanover JA (1997) O-Linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J Biol Chem 272(14):9316–9324
Necula M, Kuret J (2005) Site-specific pseudophosphorylation modulates the rate of tau filament dissociation. FEBS Lett 579(6):1453–1457
Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow EM (1999) Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 38(12):3549–3558
Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K (1998) Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys 357(2):299–309
Simanek EE, Huang DH et al (1998) Glycosylation of threonine of the repeating unit of RNA polymerase II with beta-linked N-acetylglucosame leads to a turn like structure. J Am Chem Soc 120(45):11567–11575
Singer D, Volke D, Hoffmann R (2005) Characterization of phosphorylation dependent antibodies to study the phosphorylation status of the Tau protein. Int J Pep Res Ther 11(4):279–289
Snow CM, Senior A, Gerace L (1987) Monoclonal antibodies identify a group of nuclear pore complex glycoproteins. J Cell Biol 104(5):1143–1156
Teo CF, Ingale S et al (2010) Glycopeptide-specific monoclonal antibodies suggest new roles for O-GlcNAc. Nat Chem Biol 6(5):338–343
Torres CR, Hart GW (1984) Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem 259(5):3308–3317
Viner RI, Zhang T, Second T, Zabrouskov V (2009) Quantification of post-translationally modified peptides of bovine alpha-crystallin using tandem mass tags and electron transfer dissociation. J Proteomics 72(5):874–885
Wang Z, Udeshi ND, O’Malley M, Shabanowitz J, Hunt DF, Hart GW (2010) Enrichment and site mapping of O-linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer dissociation mass spectrometry. Mol Cell Proteomics 9(1):153–160
Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW (1975) A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA 72(5):1858–1862
Wells L, Vosseller K, Cole RN, Cronshaw JM, Matunis MJ, Hart GW (2002) Mapping sites of O-GlcNAc modification using affinity tags for serine and threonine post-translational modifications. Mol Cell Proteomics 1(10):791–804
Yuzwa SA, Macauley MS et al (2008) A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat Chem Biol 4(8):483–490
Acknowledgments
This work was supported by grants from the Canadian Institutes of Health (CIHR) and the Scottish Rite Charitable Foundation. DJV is a Canada Research Chair in Chemical Glycobiology and a Scholar of the Michael Smith Health Research Foundation. SAY is a recipient of a doctoral award from the Alzheimer Society of Canada. Ramesh Kaul and Garrett Whitworth are thanked for assistance with the peptide deprotection.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Yuzwa, S.A., Yadav, A.K., Skorobogatko, Y. et al. Mapping O-GlcNAc modification sites on tau and generation of a site-specific O-GlcNAc tau antibody. Amino Acids 40, 857–868 (2011). https://doi.org/10.1007/s00726-010-0705-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-010-0705-1