
Abstract
Modification of protein by carbonyl compounds under in vitro physiological conditions is site-directed. There are few reports of the site specificity of glycation of proteins using heating conditions of relevance to food processing. The aim of this study was to determine the site specificity of modification of β-casein (βCN) by glucose and methylglyoxal (MGO). βCN (1.33 M, 3.2%) was heated with either glucose (1.345 M, 4.6%) or MGO (1 mM) at 95°C for up to 4 h. Tryptic digests were prepared and analysed by ultra performance liquid chromatography electrospray ionisation mass spectrometry (UPLC-ES/MS). The sites of formation of the Amadori product, N ε-(fructosyl)lysine (FL), and the advanced glycation end-products, N ε-(carboxymethyl)lysine (CML), MGO-derived dihydroxyimidazolidine (MG-DH) and MGO-derived hydroimidazolone (MG-HI), were located. FL and CML were detected at K107 and K176 residues in βCN/glucose incubations. Indigenous N ε-(lactulosyl)lysine was detected at K107 only. MG-DH and MG-HI were detected at R202 and possibly R183 residues in both βCN/glucose and βCN/MGO incubations. Glycation of βCN by glucose and MGO resulted in similar site specificity for MG-DH and MG-HI formation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
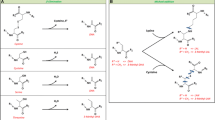
The Maillard reaction is a type of nonenzymic browning reaction that occurs during heating of foods that contain protein and carbonyl compounds such as reducing sugars. Typically, the ε-amino group of a lysine side chain within protein reacts with a carbonyl group to form the Amadori rearrangement product (ARP), which is N ε-(fructosyl)lysine (FL) when glucose is the reactant, as shown in Scheme 1 (Ames 2008). FL undergoes further reaction to form the advanced glycation end-product (AGE) N ε-(carboxymethyl)lysine (CML) (Ames 2008). Methylglyoxal (MGO) is an α-dicarbonyl that is formed during early glycation, either by oxidation of the sugar or fragmentation of the Schiff base or the ARP (Thornalley et al. 1999). MGO reacts with the guanidino group of arginine residues within protein to form a range of AGEs, notably MGO-derived dihydroxyimidazolidine (MG-DH) and MGO-derived hydroimidazolone (MG-HI) (Brock et al. 2007a).

Formation of MGO-derived dihydroxyimidazolidine (MG-DH) and MGO-derived hydroimidazolone (MG-HI) from glucose and MGO
Bovine casein consists of four major proteins αS1-, αS2-, β- and κ-casein. β-Casein (βCN) accounts for 32% of casein in bovine milk (Belitz et al. 2004). βCN is a ~24 kDa protein consisting of 209 amino acids (Fig. 1). The βCN family consists of seven genetic variants (A1, A2, A3, B, C, D and E), with variant A2 predominating (Belitz et al. 2004). The different variants are the result of single or multiple amino acid substitutions. βCN is amphipathic in nature. It has a hydrophobic C-terminus and a hydrophilic N-terminus, which contains five phosphoserine residues residues, at positions 15, 17, 18, 19 and 35 (Swaisgood 2003). Circular dichroism data show that βCN has a secondary structure made up of 9% α-helix and ~25% β-sheet (Belitz et al. 2004).

Amino sequence of β-casein (lysine and arginine residues are indicated in bold)
There is very little information regarding the sites of modification in food proteins although the sites of lactulosylation of αS1-casein (Scaloni et al. 2002) and βCN (Henle and Klostermeyer 1993; Scaloni et al. 2002) have been reported. In contrast, there have been several reports of the site-directed modification of a limited range of proteins, i.e., ribonuclease A, BSA and HSA, incubated under in vitro physiological conditions (Brock et al. 2003; Cotham et al. 2004; Ahmed et al. 2005; Hinton and Ames 2006; Brock et al. 2007a, b). LC-MS is an appropriate tool for this work due to its high sensitivity, reliability and reproducibility.
Ultra performance liquid chromatography (UPLC) operates at pressures of 15,000 psi (~1,000 bar) using columns packed with solid support particles of 1.7 μm diameter. Thus, compared to HPLC, UPLC offers superior resolution, speed and sensitivity of analysis, especially when coupled to a mass spectrometer (Churchwell et al. 2005).
The aim of the present study was to identify the sites of modification in βCN following incubation with glucose or MGO under conditions of relevance to food processing, using UPLC-MS.
Materials and methods
Materials
The following reagents were purchased from Sigma (Gillingham, UK): β-casein (βCN) from bovine milk (≥90%), d-(+)-glucose (ACS grade), methylglyoxal (MGO) solution (40% in H2O), trypsin (sequencing grade), nonafluoropentanoic acid (97%), ethylenediaminetetraacetic acid (EDTA, 99%), ammonium bicarbonate (≥99%), dithiothreitol (99%), 2-mercaptoethanol, 4-vinylpyridine (≥95%), morpholinepropanesulphonic acid hemisodium salt (MOPS, ≥99%), nonafluoropentanoic acid (NFPA, 97%) and HPLC grade water.
Preparation of model systems
Solutions of βCN (1.33 M, 3.2% protein) and d-(+)-glucose (0.1345 M, 4.6% sugar) in phosphate buffer (0.2 M; pH 6.7) were heated at 95°C for 0, 1, 2 and 4 h (βCN/G). A parallel set of solutions in which MGO (1 mM) replaced glucose was heated for 0, 5, 30, 60, and 120 min (βCN/MGO). βCN heated in phosphate buffer without any carbonyl compound was the control. Model systems were prepared in triplicate.
Tryptic digestion
Model systems were ultrafiltrated through a 5 kDa molecular mass cut-off ultrafiltration device (Millipore, Watford, UK) to remove residual sugar/MGO and buffer salts. Protein (0.25 mg, ~10 nmol) was digested according to Brock et al. (2003). Briefly, the protein was reduced with dithiothreitol in MOPS buffer containing urea and EDTA, derivatised with 4-vinylpyridine, and digested with trypsin at an enzyme:substrate ratio of 5:100 (w/w) at 37°C for 5.5 h. Digestion was terminated by freezing at −20°C and digests were analysed by UPLC-MS.
UPLC-MS
Samples were fractioned on a Waters (Manchester, UK) Acquity UPLC system coupled to a Waters Premier triple quadrupole mass spectrometer. Separations were conducted on a Waters UPLC® (2.1 × 50 mm) column. The stationary phase was BEH C18 and the particle size was 1.7 μm. The column was housed in an oven at 50°C. Samples were eluted in a liner gradient running from 90% solvent A (aqueous 5 mM NFPA) to 80% solvent B (acetonitrile) at a flow rate of 0.2 mL/min over 7.5 min. The injection volume was 7.5 μL (equivalent to 25 μg of protein, ~1 nmol). Full scan experiments were conducted by operating the MS in positive ion electrospray mode and using an m/z range of 200–1,800 amu. Other MS operating conditions used were: scan duration, 1 s; capillary voltage, 3 kV; cone voltage, 22 V; source temperature, 130°C; desolvation temperature, 400°C. Data were analysed using MassLynx software (version 4.1), supplied by Waters.
Semi-quantification of modified peptides
Modified and unmodified peptides were located by constructing extracted ion chromatograms at relevant m/z values and semi-quantified based on the percentage contribution of the peak area (% PA) of each peptide to its peptide family (Ames 2005). Briefly, peak areas of each peptide at all detected charge states were summed. The sum of the peak areas of all of the members within each peptide family gave the total peak area. The % PA of each member within a peptide family was calculated by dividing the sum of the PA of all charge states of that peptide by the total peak area and expressing it as a percentage.
Results
Site specificity of FL and CML formation in (βCN/G)
βCN incubated with glucose resulted in two of the eleven lysine residues, i.e., K107, and K176, being modified to FL and/or CML. As an example, Fig. 2 shows the location of the carboxymethylated peptide 170VLPVPQKAVPYPQR183 with K176 modified to form CML (170VK176 CMLR183) within an extracted ion chromatogram. The ions at m/z 826 and 551 corresponded to the 2+ and 3+ charge states of this peptide (Fig. 2a, b). These ions were absent from the corresponding chromatograms for native βCN (Fig. 2c, d). Table 1 lists the peptides that were identified within the K107 and K176 families. Indigenous LL was detected only on K107, i.e., peptide 106H–K107 LL–K113. Peptides 106H–K107 FL–K113 and 106H–K107 CML–K113 indicate modification by glucose on K107. Similarly, peptides 170V–K176 FL–R183 and 170V–K176 CML–R183 indicate modification by glucose on K176.

Location of modified peptide 170VK176 CMLR183 Extracted ion chromatograms at a m/z 826 (2+ charge state) and b m/z 551 (3+ charge state) for βCN incubated with glucose for 4 h show one major peak at 3.28 min. Extracted ion chromatograms at c m/z 826 and d m/z 551 for native βCN show no peak at 3.28 min. The chromatograms are normalised to the same scale
The kinetics of formation of FL and CML on K107 and K176 when βCN is incubated with glucose are depicted in Fig. 3. For the K107 family (Fig. 3a), the % PA of peptide 106H–K107 FL–K113 increased during the first hour of incubation (% PA = 8.8) and then its contribution to the total peak area remained constant up to 4 h. Amounts of the peptide 106H–K107 CML–K113 accounted for 5.4% of the PA after 1 h, and increased to 7.1% at 4 h. As the % PA values for peptides 106HK107 FLK113 and 106HK107 CMLK113CML increased, the contribution made by indigenous LL decreased from 15.3 to ~10% between 1 and 4 h.

K107 and K176 are the main sites of formation of FL and CML in βCN/G incubations. Kinetics of formation of a FL and CML adducts on K-107, peptide 106H–K107–K113 and b FL and CML adducts on K-176, peptide 170V–K176–R183. The amounts are expressed as % PA (see “Materials and methods”). Data points represent the mean of triplicate incubations and the error bars represent the standard deviation
For the K176 family, peptide 170VLPVPQK176 FLAPVYPQR183 (170V–QR183), with K-176 modified to FL (peptide 170V–K176 FL–R183) accounted for 30.5% of the PA after 1 h (Fig. 3b), after which time the value slowly decreased to 22.3% after 4 h. After 4 h of incubation, the % PA for peptide 170V–K176 FL–R183 was ~3-fold higher compared to peptide 106H–K107 FL–K113. Peptide 170VK176 CMLR183 accounted for 5.2% of the PA after 1 h, and increased to 11% after 4 h.
Site specificity of MG-DH and MG-HI formation in βCN/MGO and βCN/G
When βCN was incubated with MGO or glucose, peptide 184DMPIQAFLLYQEPVLGPVRGPFPIIV209 (184D–V209), with R202 modified to MG-DH (184DR202 MG-DHV209) and peptide 184D–V209 with R202 modified to MG-HI (184DR202 MG-HIV209) were formed. Peptide 177AVPYPQRDMPIQAFLLYQEPVLGPVRGPFPIIV209 (177A–V209), containing one MG-DH adduct and peptide 177A–V209 containing one MG-HI adduct, were also detected. Both of these peptides contain arginine at positions 183 and 202 but it was not possible to distinguish which residue was modified using the methodology applied in this study, and the peptide family is designated R183/202. Peptides 177A–V209 with modifications on both R183 and R202 were not detected. Table 2 lists the peptides resulting from modification on R202 and R183/202. Peptides 184DR202 MG-DHV209 and 184DR202 MG-HIV209 indicate modifications on R202. Peptide 177A–R183/202 MG-DH–V209 and 177A–R183/202 MG-HI–V209 indicate modifications on either R183 or R202.
The kinetics of formation of peptides 184D–R202 MG-DH–V209 and 184D–R202 MG-HI–V209 when βCN was incubated with MGO are depicted in Fig. 4a. The % PA of peptide 184D–R202 MG-DH–V209 was 6.4% after 30 min of incubation and decreased to 5% after 2 h. The % PA of peptide 184D–R202 MG-HI–V209 increased during the first 30 min of incubation to 8.1% and to 10% at 2 h. The kinetics of formation of peptides 177A–R183/202 MG-DH–V209 and 177A–R183/202 MG-HI–V209 are depicted in Fig. 4b. Peptide 177A–R183/202 MG-DH–V209 had a % PA of 5.7% after 30 min of incubation and remained at this level to 2 h. The % PA of peptide 177A–R183/202 MG-HI–V209 increased to 9.5% after 30 min of incubation and to 11.5% at 2 h (Fig. 4b).

R202 and R183/202 are the main sites of formation of MG-DH and MG-HI in βCN/MGO and βCN/G incubations. Kinetics of formation of a MG-DH and MG-HI adducts on R-202, peptide 184D–R202–V209 and b MG-DH and MG-HI adducts on R-183/202, peptide 177A–R183/202 MG-DH–V209 in βCN/MGO incubations. c MG-DH and MG-HI adducts on R-202, peptide 184D–R202–V209 and d MG-DH and MG-HI adducts on R-183/202, peptide 177A–R183/202 MG-DH–V209 in βCN/G incubations. The amounts are expressed as % PA (see “Materials and methods”). Data points represent the mean of triplicate incubations and the error bars represent the standard deviation
The kinetics of formation of MG-HI and MG-DH on R202 and R183/202 in βCN/G incubations are depicted in Fig. 4c and d. Peptide 184D–R202 MG-DH–V209 accounted for 2.6% of the PA after 2 h of incubation and increased to 4.6% after 4 h. The % PA of peptide 184D–R202 MG-HI–V209 increased to 5.4% after 2 h of incubation followed by an ~2-fold increase to 4 h (% PA = 12.7) (Fig. 4c). The % PA of peptide 184D–R202 MG-HI–V209 was ~2-fold higher than 184D–R202 MG-DH–V209 after 2 h and 4 h of incubation. The kinetics of formation of peptides 177A–R183/202 MG-DH–V209 and177A–R183/202 MG-HI–V209 are depicted in Fig. 4d. The % PA value for peptide 177A–R183/202 MG-DH–V209 was 5.4% after 1 h and 13.1% after 4 h. The % PA of peptide 177A–R183/202 MG-HI–V209 increased to 8.9% after 1 h of incubation and to 26.1% after 4 h. The % PA for peptide 177A–R183/202 MG-HI–V209 was ~2-fold higher than 177A–R183/202 MG-DH–V209 after 4 h of incubation.
The rates of formation of MG-DH and MG-HI adducts in MGO-modified βCN are highest during the first 30 min of incubation, after which levels remain reasonably constant. In the βCN/glucose incubations, the formation of MG-DH and MG-HI adducts on both peptides increases in an approximately linear fashion over the entire heating time. Regardless of the carbonyl or arginine residue, the ratio, % PA MG-HI peptide: % PA MG-DH peptide was 2–3 (Fig. 4) after 1 h heating for all of the incubations, indicating that these two adducts existed in equilibrium after this heating time.
Discussion
Semi-quantification of tryptic peptides
MS is a very powerful tool in proteomics and several studies in the past have used LC-MS to study sites of modification in different proteins. One means of monitoring levels of modified peptides is by measuring their relative abundance (RA) values. The RA approach is commonly used to account for differences in the amount of protein between different samples. The RA is calculated by dividing the sum of the peak areas of all detected charge states of a modified peptide by the sum of the peak areas of all detected charge states of C-terminal peptide of the protein (Brock et al. 2003). The drawback of this method is that the amounts of different native peptide within a protein cannot be compared due to variability in the MS response. Another approach used to overcome the differences in the MS response involves monitoring the % loss of the unmodified peptides (Hinton and Ames 2006). Alternatively, the modified and unmodified peptides within a peptide group may be expressed as a percentage sum of the PA (% PA) (Ames 2005). The residual standard deviations (RSDs) for the native peptides using the % PA approach are <5% which is better than when using the RA method or monitoring the loss of the unmodified peptide (Ames 2005).
Site specificity of modification
Our previous work investigating the site specificity of formation of FL, CML, glyoxal (GO)-derived-DH, GO-derived HI, MGO-derived DH, MGO-derived HI, tetrahydropyrimidine (THP), argpyrimidine and methionine sulphoxide (MetSO) on ribonuclease (Brock et al. 2003; Cotham et al. 2004; Brock et al. 2007a, b) and formation of FL, the Heyns rearrangement product and CML on BSA (Hinton and Ames 2006) shows modification to be a highly specific process. In the present study, we investigated the site specificity of modification of a food protein glycated under conditions relevant to food processing. Glucose and MGO were the carbonyl reactants employed. Milk is a component of a wide range of processed foods while MGO forms by oxidation of sugar and lipid. Both glucose and MGO are ubiquitous in food.
Our results demonstrate for the first time the sites of formation on βCN of FL and CML from glucose and of MG-HI and MG-DH from MGO and glucose. An earlier study (Henle and Klostermeyer 1993) monitored the reactivity of lysine residues on βCN incubated with lactose. The tryptic peptides of lactosylated βCN revealed K28/29, K32, K99 and K107 to be the favoured sites of glycation with modification occurring within 15 min of incubation at 70°C. Longer incubation times (60 min) led to gradual modification of K105, K113 and K176 while K48, and K169 remained relatively unreactive, even after 60 min at 90°C. A subsequent investigation (Scaloni et al. 2002) reported five lysines on βCN to be modified to LL in different heat-treated milks, with K107 being the favoured site of modification. Lactosylation of βCN was dependent on the degree of heat treatment with only K107 being modified in pasteurised milk, whereas K32, K48, K107, K113 and K176 were lactosylated in UHT and sterilized milk. No previous studies have reported the sites of glycation of βCN by other carbonyl compounds present in food.
In the current study, the kinetics of formation of FL on both K107 and K176 were highest during the first hour of incubation, after which time amounts either remained constant or decreased slowly until 4 h. This could be attributed to formation of CML. FL was formed to a greater extent on K176 compared to K107, while the proportion of CML on both lysines was similar. The reactivity of a lysine residue within a protein depends on various factors including location within the amino acid sequence, nature of adjacent amino acids and charge of the amino acid side chain (Henle and Klostermeyer 1993). Other factors include pH, ionic strength, presence of phosphate ions (Watkins et al. 1987), and reaction temperature (Scaloni et al. 2002). Henle and Klostermeyer (1993) observed that the location of a charged glutamic acid sidechain adjacent to a lysine (as is the case for K107 and K176) promoted lactosylation. It has also been observed that, in haemoglobin, close proximity of an acidic amino acid residue can promote glycation of lysine residues (Shapiro et al. 1980).
In the current study, neither FL nor CML adducts could be detected on the remaining nine lysine residues. This might be due to conformational changes to the protein during heating at 95°C for several hours in the presence of a carbonyl compound. Such changes might limit the accessibility to these unreactive lysine residues.
R202 (and possibly R183) was the main arginine residue undergoing modification in glycated βCN, regardless of whether the carbonyl was MGO added directly to the model system or MGO derived by oxidation of reactant glucose. MG-HI was present in higher amounts than MG-DH, regardless of the carbonyl reactant or time of heating. There are no previous reports of the formation of MG-DH or MG-HI on βCN. In both the current study and our previous work (Brock et al. 2007a) concerning the site-specificity of modification of RNase by MGO, we have shown that certain arginine residues are favoured over others for modification. This may be attributed at least in part to the relative availability of arginine residues and their accessibility to MGO. In addition, the nature of adjacent amino acids might influence reactivity. For example, identification of R410 as the major site of modification of MGO-modified HSA has been attributed to strong electrostatic interaction between R410 and the adjacent polar Y411 (Ahmed et al. 2005). In this respect, R183 is located next to an aspartic acid residue at position 184, and might promote modification in a similar fashion to the proposed effect of glutamic acid residues on glycation of the ε-amino group of lysine residues, discussed above.
Other MGO-derived adducts, i.e., THP, argpyrimidine and CEL, were sought but could not be detected in this study. Also, methionine sulphoxide (MetSO) a marker of oxidation was not detected on any of the 6 methionine residues in either the βCN/glucose or βCN/MGO incubations. These data are in contrast to our previous findings (Brock et al. 2007b), where MetSO was formed in higher amounts than CML on RNase incubated under in vitro physiological conditions. MetSO has been identified on whey protein heated with lactose at 60°C (Meltretter et al. 2007) and in processed milks (Baxter et al. 2007). The lack of detection of MetSO in the current study may be attributed to various factors related to the temperature of heating and the composition of the system investigated. Compared to a temperature of 60°C, a temperature of 95°C may lead to lower levels of oxygen in the system and thus favour the formation of glycation products over MetSO. In processed milks, MetSO may form from oxidised lipid during storage.
In conclusion, we have demonstrated the site specificity of formation of FL, CML, MG-DH and MG-HI on βCN heated under conditions of relevance to food processing. This work has potential application to better understand the relationship between food processing operations and food functional properties and to develop metabolomics procedures for sophisticated monitoring of food processing operations.
Abbreviations
- AGEs:
-
Advanced glycation endproducts
- ALEs:
-
Advanced lipoxidation endproducts
- ARP:
-
Amadori rearrangement product
- CML:
-
N ε-(carboxymethyl)lysine
- FL:
-
Fructoselysine
- MR:
-
Maillard reaction
- NFPA:
-
Nonafluoropentanoic acid
- UPLC-MS:
-
Ultra performance liquid chromatography-mass spectrometry
- TCA:
-
Trichloroacetic acid
- TFA:
-
Trifluoroacetic acid
References
Ahmed N, Dobler D, Dean M, Thornalley PJ (2005) Peptide mapping identifies hotspot site of modification in human serum albumin by methylglyoxal involved in ligand binding and esterase activity. J Biol Chem 7:5724–5732
Ames JM (2005) Application of semiquantitative proteomics techniques to the Maillard reaction. In: Baynes JB, Monnier VM, Ames JM, Thorpe SR (eds) The Maillard reaction: chemistry at the interface of nutrition, aging and disease, vol 1043. Annals of the New York Academy of Sciences, New York, pp 225–235
Ames JM (2008) Determination of N ε-(carboxymethyl)lysine in foods and related systems. In: Schleicher E, Somoza V, Scheiberle P (eds) The Maillard reaction. Recent advances in food and biomedical science, vol 1126. Annals of the New York Academy of Sciences, New York, pp 20–24
Belitz HD, Grosch W, Schieberle P (2004) Food chemistry, 3rd edn. Springer, Berlin
Baxter JH, Lai C-S, Phillips RR, Dowlati L, Chio JJ, Luebbers ST, Dimler SR, Johns PW (2007) Direct determination of methionine sulfoxide in milk proteins by enzyme hydrolysis/high performance liquid chromatography. J Chromatogr A 1157:10–16
Brock JWC, Hinton DJS, Cotham WE, Metz TO, Thorpe SR, Baynes JW, Ames JM (2003) Proteomic analysis of the site specificity of glycation and carboxymethylation of ribonuclease. J Proteome Res 2:506–513
Brock JWC, Cotham WE, Thorpe SR, Baynes JW, Ames JM (2007a) Detection and identification of arginine modifications on methylglyoxal-modified ribonuclease by mass spectrometric analysis. J Mass Spectrom 42:89–100
Brock JWC, Ames JM, Thorpe SR, Baynes JW (2007b) Formation of methonine sulfoxide during glycoxidation and lipoxidation of ribonuclease A. Arch Biochem Biophys 457:170–176
Churchwell MI, Twaddle NC, Meeker LR, Doerge DR (2005) Improving LC-MS sensitivity through increases in chromatographic performance: comparisons of UPLC-ES/MS/MS to HPLC-ES/MS/MS. J Chromatogr B 825:134–143
Cotham WE, Metz TO, Ferguson PL, Brock JW, Hinton DJ, Thorpe SR, Baynes JW, Ames JM (2004) Proteomic analysis of arginine adducts on glyoxal-modified ribonuclease. Mol Cell Proteomics 12:1145–1153
Henle T, Klostermeyer H (1993) The reactivity of the individual protein-bound lysine residues of β-casein A1 during the initial stages of the Maillard reaction. IDF Spec Iss 9303:183–189
Hinton DJ, Ames JM (2006) Site specificity of glycation and carboxymethylation of bovine serum albumin by fructose. Amino Acids 4:425–434
Meltretter J, Seeber S, Humeny A, Becker C-M, Pischetsrieder M. (2007) Site-specific formation of Maillard, oxidation and condensation protducts from whey proteins during reaction with lactose. J Agric Food Chem 55:6096–6103
Scaloni A, Valentina P, Franco P, Fedele E, Froio R, Ferrara L, Bergamo P (2002) Characterization of heat-induced lactosylation products in caseins by immunoenzymatic and mass spectrometric methodologies. Biochim Biophys Acta 1598:30–39
Shapiro R, McManus MJ, Zalut C, Bunn HF (1980) Sites of nonenzymatic glycosylation of human haemoglobin A. J Biol Chem 255:3120–3127
Swaisgood HE (2003) Chemistry of the caseins In: Fox PF, McSweeney PLH (eds) Advanced dairy chemistry, vol 1, Proteins part A 3rd edn. Kluwer Academic/Plenum Publishers, New York, pp 139–202
Thornalley PJ, Langborg A, Minhas HS (1999) Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of protein by glucose. Biochem J 344:109–166
Watkins NG, Negliafisher CI, Dyer DG, Thorpe SR, Baynes JW (1987) Effect of phosphate on the kinetics and specificity of glycation of protein. J Biol Chem 262:7207–7212
Acknowledgments
This work was supported by a studentship from the Department of Agriculture and Rural Development (DARD), Northern Ireland, and Queen’s University Belfast. Davinia Mills (University of Reading) is thanked for her assistance and helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lima, M., Moloney, C. & Ames, J.M. Ultra performance liquid chromatography-mass spectrometric determination of the site specificity of modification of β-casein by glucose and methylglyoxal. Amino Acids 36, 475–481 (2009). https://doi.org/10.1007/s00726-008-0105-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-008-0105-y