
Abstract
Light-induced charge separation in an organic photovoltaic (OPV) composite DTS(FBTTh2)2:PC71BM was studied. DTS(FBTTh2)2 or DTS is a non-polymer electron donor and PC71BM is a fullerene-based electron acceptor. Electron spin echo (ESE) technique has been developed to separate the signal of interfacial charge transfer state (CTS) from that of trapped charges. Pronounced out-of-phase ESE signal was observed within first few microseconds after a laser flash exciting the composite at cryogenic temperatures. This implies correlation of unpaired electron spins of DTS+ and PC71BM– species constituting CTS. The distribution of distances between these species is derived from out-of-phase ESE envelope modulation (ESEEM). Out-of-phase ESEEM traces were numerically simulated by the model assuming both magnetic dipolar and electron–hole exchange interactions within CTS. The most probable distance between DTS+ and PC71BM– within CTS increases from 4.9 nm and 5.7 nm with delays after the laser flash increase from 200 ns to 30 µs at the lowest temperature studied 20 K. This is caused by faster recombination of CTS with shorter electron–hole distance. The electron–hole exchange interaction is about J/h = 1.15 MHz for the smallest interspin distance obtained r0 = 2.5 nm. The overall similarity of the initial electron–hole distance and CTS recombination rate for DTS:PC71BM and polymer/fullerene OPV composites studied previously points to similar photoinduced charge separation mechanisms for these systems.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The donor–acceptor composites based on organic semiconducting π-conjugated molecules are promising materials for active layers of advanced thin-film solar cells with a photoelectric conversion efficiency up to 13–14% [1,2,3]. Some benchmark composites based on conjugated polymers as electron donors and fullerenes as electron acceptors has been widely investigated to clarify the question: which parameters of the composite are essential for efficient photoelectric conversion and how can they be improved [4]? Small molecule (non-polymer) donors are emerging organic photovoltaic (OPV) materials. Important advantage of non-polymer donors is well-defined molecular structure and better reproducibility of OPV devices. Although the efficiency for small molecule donors approaches to that for polymer donors [5, 6], the mechanism of photoelectric conversion is less studied for the former materials.
Insights into the photocurrent generation stages have been gained using various time-resolved techniques for different polymer:fullerene composites [7]. The first step is very fast exciton formation due to light harvesting by donor or acceptor molecule. The second step is exciton diffusion into a donor/acceptor interface and charge separation resulting in a charge transfer state (CTS) [8]. Note that for some composites based on polymers or small molecules, the efficiency of light conversion into CTS is extremely high (for the best systems that exceeds 90%) [9, 10]. The last crucial stages of photocurrent generation are charge dissociation from CTS to free charge carriers and their extraction to electrodes.
Despite large number of studies the details of the dissociation of CTS into charge carriers are unclear. In part, a reason of this is very short CTS lifetime at room temperature which typically does not exceed 1 ns [11]. Therefore, characterisation of electron–hole distances over ensemble of such an elusive species as CTS is still a challenging and important problem [12]. Electron Spin Echo (ESE) technique is quite sensitive method of pulse Electron Paramagnetic Resonance (EPR) spectroscopy. It allows not only measuring g-factors, but also determining the strength of weak magnetic interactions between radicals and spin relaxation times [13, 14]. From EPR viewpoint the positive and negative charges constituting CTS can be treated as organic radicals. The parameters obtained by pulse EPR could provide information about the structure of CTS. However, pulse EPR techniques usually require a low-temperature to reduce CTS lifetime down to the microsecond range. Previously time-resolved EPR and ESE techniques were applied to characterize light-induced radicals in the composite P3HT/PC61BM (poly(3-hexylthiophene)/[6]-phenyl C61 butyric acid methyl ester). The characteristic signal of spin-correlated radical pairs (SCRP) was detected immediately after the photoexcitation and was assigned to CTS [15, 16]. The strength of the magnetic interaction between radicals within CTS of P3HT/PC61BM was estimated from the effect of instantaneous spin diffusion in ESE decay and the average distance of about 4 nm between the electron and hole within CTS was derived [17].
There are advanced pulse EPR techniques which were developed to determine spin interactions within a SCRP [14]. If EPR lines of two radicals are superimposed, the out-of-phase ESE envelope modulation (ESEEM) contains the information about magnetic interactions between their spins. For the weak spin coupling limit the analytical expression of two-pulse out-of-phase ESE dependence on the separation between two mw pulses τ (Fig. 1a) has been derived [18,19,20]:
where T2 is the transversal relaxation time of the radicals, and the modulation frequency.
is determined by the energies of the spin exchange interaction J and the magnetic dipolar interaction. Here r is the distance between the radicals in the point dipole approximation and θ is the angle between the directions of the interspin vector r and the external magnetic field B0. The ratio c/r3 ≡ D, so-called D-value of the tensor of magnetic dipole interaction, is well-known D(MHz) = 77.8 × 103/(r3(A3)). Note that in-phase ESE of a radical pair also should be modulated with exactly the same frequency [21]. However, this modulation is superimposed with a strong in-phase ESE signal of long-living thermalized spins while out-of-phase ESEEM is produced solely by SCRPs.

Pulse sequences which were applied to obtain ESE signal: the two-pulse (a), two-pulse ESE with pre-saturation (b), ESE inversion-recovery three-pulse sequence (c)
The out-of-phase ESEEM has provided the important information about the distances between light-induced spin-correlated radical of a photosynthetic bacterial reaction center [22] and about the structure of spin-correlated CTS in the composite P3HT:PC71BM (PC71BM is a fullerene C70 derivative, [6]-Phenyl C71 butyric acid methyl ester, Fig. 2), at different delays after flash photoexcitation (DAF values) [23]. This approach assumed the distribution of interspin distance within CTS and the averaging of the theoretical out-of-phase ESEEM dependence Eq. (1):
where G(r) is the density probability function of the distance distribution between radicals in CTS.

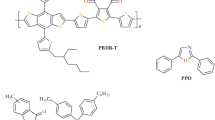
Chemical structures of PC71BM and DTS compounds
Recently, a series of small donor molecules has been successfully applied instead of semiconducting polymers as active layer components of organic solar cells to improve their chemical and morphological stability [3]. Therefore, to determine the feasibility of application of small molecules in active layers of stable and effective organic solar cells, all reliable information on the CTS is of special interest. An example of such advanced non-polymer donor is DTS (DTS(FBTTh2)2, 7,7′-[4,4-Bis(2-ethylhexyl)-4H-silolo[3,2-b:4,5-b′]dithiophene-2,6-diyl]bis[6-fluoro-4-(5′-hexyl-[2,2′-bithiophen]-5-yl)benzo[c][1, 2, 5]thiadiazole], Fig. 2), the solar cell based on DTS:PC71BM composite has demonstrated quite high efficiency of about 7% [10, 24]. Spin Hamiltonian parameters of DTS+ cation-radical and its spin density distribution were determined by EPR and quantum-chemical calculations by Thomson et al. [25]. The principal values of g-tensors of DTS+ and PC71BM− were experimentally determined as (2.0035, 2.0024, 2.0017) [25] and (2.0060, 2.0028, 2.0021) [26], correspondingly.
The spin interactions within CTS in the composites based on small donor molecules have not been investigated yet. The spin interactions within light-induced SCRPs in donor–acceptor composites could be obtained from simulations of time-resolved (transient) EPR spectra [27, 28]. However, analysis of EPR spectrum lineshape is extremely complicated when g-values of electron and hole are close and their EPR lines are superimposed. Therefore, in the present work we developed the out-of-phase ESEEM technique to characterize electron–hole spin interactions within CTS at different times after light absorption. The problem for DTS:PC71BM is intense in-phase ESE signal of thermalized charges trapped in the composite, which is much stronger than out-of-phase ESE signal of CTS. To overcome this problem pre-saturation of in-phase ESE and precise calibration of ESE phase relative to that of thermalized spins are essential. The protocol that complements both of these approaches was applied for the first time.
2 Experimental Section
The chlorobenzene solution of PC71BM (Aldrich) and DTS [DTS(FBTTh2)2, Aldrich] with weight ratio of 1:1.4 was prepared with total concentration of 20 mg/ml. The solution was put in the EPR tube of 4.8 mm outer diameter, and three freeze–pump–thaw cycles were performed. Chlorobenzene was evaporated in the vacuum of about 0.1 torr, which resulted in the formation of the DTS:PC71BM composite on the inner wall of the EPR sample tube. The thickness of the composite was slightly inhomogeneous over the sample, with the estimated average value of 2 μm. Since the typical absorption coefficient of the DTS at 527 nm is about 104 cm−1 [24], the thickness of the composite ensures complete absorption of the laser light in our setup.
CW EPR and ESE measurements were carried out on an X-band ELEXSYS ESP-580E EPR spectrometer equipped with an ER 4118 X-MD-5 dielectric cavity inside an Oxford Instruments CF 935 cryostat.
Laser flashes from TECH-laser (Laser-export Co. Ltd., Russia) with the wavelength of 527 nm, the pulse duration of about 5 ns, and the pulse repetition rate of 1 kHz illuminated the sample. The laser light was directed to the refractive lens along its optical axis and transmitted inside the EPR tube through a quartz light guide. The estimated energy absorbed by the sample is 15 μJ per flash.
ESE signal and ESEEM were obtained using a two-pulse mw pulse sequence applied after a laser flash, Flash – DAF – π/4 – τ – π – echo (Fig. 1a), where DAF is the delay after laser flash, the π/4- and π-pulses were of 8 ns and 24 ns duration, respectively, the initial τ delay was 160 ns. Such mw pulse sequence is optimal to detect out-of-phase ESE signal from SCRP [20] that is why the π/4-pulse was applied instead of the most common π/2-pulse. The major part of ESE signal in the time domain was integrated within an integration time window 140 ns centered at the echo maximum.
The quadrature detection was used: in-phase and out-of-phase ESE signals were measured simultaneously. The phase for the ESE was adjusted to obtain zero out-of-phase ESE signal at DAF = 990 μs. The three-pulse sequence (Fig. 1b) was applied to saturate ESE signal of the long-living photoaccumulated radicals and to measure the out-of-phase ESE signals of CTS. The delay between the pre-saturating π/2-pulse of 20 ns duration and laser flash did not exceed 10 μs and this mw pulse affected only the accumulated radicals.
The ESE inversion-recovery dependences were measured using another three-pulse sequence (Fig. 1c) at dark conditions, before this experiment the radicals were accumulated by the laser irradiation.
All time domain experiments (the ESEEM, the ESE dependences with DAF variation, the ESE inversion-recovery traces) were obtained at the magnetic field B0 corresponded to the maximum of the in-phase echo-detected EPR spectrum.
Temperature was kept at 70 K by cold nitrogen gas flow. Lower temperatures were stabilized by cold helium gas flow.
3 Results and Discussion
3.1 Echo-Detected EPR Spectra: In-Phase ESE and Out-of-Phase ESE
Two-pulse echo-detected (ED) EPR spectra of the light-induced radicals in DTS:PC71BM are shown in Fig. 3. The strong ED EPR spectrum at DAF = 990 μs (which is long compared to CTS lifetime) coincides with the similar ED EPR spectrum obtained without synchronization between laser flashes and the mw pulse sequence (data not shown). This ED EPR spectrum is attributed to the ESE signal from long-living radicals DTS+ and PC71BM− which were accumulated within the composite and were detected by continuous wave EPR (Fig. S1). Because of similar values of g-tensors of DTS+ and PC71BM− radicals, this spectrum represents a single line with characteristic broadening of 5 G. Similar long-living radicals were observed previously in polymer:fullerene composite P3HT/PCBM [15, 29] and were assigned to trapped charges P3HT+ and PCBM− on the basis of their EPR lineshape and very slow recombination. Therefore, the observed long-living radicals DTS+ and PC71BM− could be also attributed to trapped charges.

Two-pulse ED EPR spectrum of light-induced radicals in the DTS:PC71BM composite: in-phase (thick line) and out-of-phase (thin line) ESE components at DAF = 0.2 μs (a), DAF = 990 μs (b), the delay τ = 600 ns. The ED EPR spectrum of CTS (c) was obtained by subtraction of the spectrum (a) and (b). Temperature 40 K
Immediately after photoexcitation, at DAF = 0.2 μs, flash-induced ED EPR spectrum (the difference between ED EPR spectra obtained at small and large DAF values) consists of both in-phase and out-of-phase components (Fig. 3). The presence of out-of-phase ESE is a signature of spin-correlated CTS. Note that the decrease of in-phase ED EPR component at small DAF could be caused by contribution of non-equilibrium populations of the CTS with a negative spin polarization [30]. The dependences of ESE on DAF are presented in Fig. 4. Long after the laser flash the in-phase ESE recovered into its maximum intensity, and the out-of-phase ESE vanished due to CTS decay or spin relaxation. The dependences were fitted by exponential (for 70 K) and stretched exponential (for 40 K and 20 K) functions with the parameters summarized in Table 1. Note that for all temperatures investigated the characteristic times of the in-phase ESE recovery and the out-of-phase ESE decay are very similar. Therefore, the single time Td was used to describe both in-phase and out-of-phase ESE dependence on DAF for each temperature. These values are assigned to a characteristic CTS decay time and they did not exceed 15 μs which is much shorter than the flash repetition time. This implies that essentially all CTSs recombine before the next laser flash.

Two-pulse in-phase (a, c, e, thin lines) and out-of-phase (b, d, f, dash lines) ESE dependences on DAF obtained at temperatures 20 K (a, b), 40 K (c, d) and 70 K (e, f), and their numerical simulations by stretched exponentials (thick lines) with parameters summarized in Table 1. The delay τ = 600 ns
In principle, heating of the sample by laser pulse can induce drift of the ESE phase and its amplitude for thermalized spins. However, for the present case it is unlikely, since very low laser flash intensity was used. During our previous study we noted that ever hundred time stronger laser pulses did not cause noticeable drift of ESE phase for trapped charges [31].
To compare the CTS decay time with longitudinal spin relaxation times T1 the latter values were obtained for long-living radicals in the composite in dark after from ESE inversion-recovery dependences (Fig. 5). The dependences were also fitted by stretched exponential dependences, their parameters are summarized in Table 1. The T1 characteristic value is much longer than CTS lifetimes. Therefore, out-of-phase ESE decay with DAF increase should not be attributed to the spin–lattice relaxation process. Rather it is caused by charge recombination.

ESE inversion-recovery dependences of long-living photoaccumulated radicals measured at dark (thin lines). The mw pulse sequence repetition times were 10 ms (for 20 K), 5 ms (for 40 K) or 1 ms (for 70 K). The parameters of their approximations by stretched exponential dependences (thick lines) are summarized in Table 1
3.2 Out-of-Phase ESEEM
In the out-of-phase ESEEM experiments an artifact of the EPR spectrometer created a technical problem. The phase of two-pulse ESE signal has an instrumental offset depending on τ delay: the out-of-phase ESE depended on τ even for the strong and stable ESE signal of long-living thermalized radicals. However, it is well-known that the out-of-phase ESE could appear only from the radical pairs with non-equilibrium populations of spin states, while all other radicals produce only in-phase ESE signal. The main idea to resolve the problem is to measure a phase offset dependence on τ delay for the strong ESE signal of the accumulated radicals and to use the obtained dependence to correct mw phase of the small ESE signal of CTS. To obtain ESE dependences on τ for accumulated radicals and for CTS two- and three-pulse sequences were applied, correspondingly (Fig. 1a, b). The π/2-pulse in the three-pulse sequence saturates only the ESE of the accumulated radicals and does not affect CTSs due to their short decay time. An effect of the pre-saturating π/2 pulse on ED EPR spectrum is presented in Fig. S2: only out-of-phase ESE of CTS remained at short DAF = 0.2 μs (a weak in-phase ESE signal appeared due to incomplete saturation of EPR spectrum by π/2 mw pulse). Similar approach to separate weak ESE signal of non-equilibrium spin states from strong thermalized background was previously applied by Colvin et al. [32] and Lukina et al. [33].
The protocol of ESE transformation to obtain true out-of-phase ESEEM is described in Supplementary Material (Sec. S3). The out-of-phase ESEEM free from instrumental phase shift were obtained after these transformation steps (Fig. 6), the data for temperatures 20 K and 40 K became nearly identical and did not show a pronounced oscillation. The characteristic spin transversal relaxation time T2 = 2.6 μs was found for temperatures 20–70 K, from the approximation of the in-phase ESE dependences on τ delay after mw phase correction (Fig. S3).

Out-of-phase ESE dependences on τ values obtained at 70 K (a), 20 K (b–e, thin solid lines) and at 40 K (b–d, dot lines) and their simulations (thick lines) assuming the exchange interaction (Eq. 3) and the CTS distance distribution. The dependences are normalized to have the same maximal magnitude. The parameters of the simulations are summarized in Table 2
3.3 Distance Distribution Within Radical Pairs of CTS
To calculate the out-of-phase ESEEM dependences both the dipolar interaction D and exchange interaction J between CTSs of DTS+ and PC71BM− were assumed. The strength of the exchange interaction decays exponentially with interspin distance r [34,35,36]:
where J0 is the exchange interaction value when the interspin distance is rJ0, β is the constant of the exchange interaction decay.
The out-of-phase ESEEM could be described by the expression:
This expression is a modification of Eq. (2) in which the modulation frequency has the contribution of the exchange interaction Eq. (3).
The distance distribution function G(r) was chosen in a form of a modified Gaussian distribution (Fig. 7):

Such distribution does not assume any distances smaller than r0, has a steep rise from r0 to r0 + Δr and rapidly decreases at longer distances. Similar Gaussian-like function was used previously to model electron–hole distance distribution function for radical pairs in organic donor–acceptor composites [37, 38].
The calculated ESEEM traces with the parameters J0, β, rJ0, r0 and Δr presented in Table 2 are shown in Fig. 6. Note that the exchange interaction J(r) has been characterized by single values J0/ħ = 1.15 MHz, β = 0.25 Å−1 and rJ0 = 25 Å. Such exchange interaction is rather weak and had been included mostly to reproduce the pronounced change of sign of the out-of-phase ESE at small τ delays. The β = 0.25 Å−1 value obtained in the present work is similar to that obtained previously for conjugated organic semiconductor systems [35, 36]. The observation of non-zero exchange integral for CTS in DTS:PC71BM blend is in line with similar observations for polymer/fullerene OPV blends [28, 33].
To estimate a possible influence of fast fluctuations of the magnetic interaction within CTS on spin relaxation the Redfield theory [39] could be applied. The contribution into spin longitudinal relaxation rate due to a fluctuation of local magnetic field components ΔBx and ΔBy experienced by the spin of electron or hole is described by the expression [40]:
where τc is correlation time, γ is gyromagnetic ratio for the radicals investigated, ω0 ≈ 6·1010 rad/s is the spectrometer frequency. This expression has a minimum with respect to τc when τc = 1/ω0 ≈ 10−11 s. The scale of local magnetic field fluctuations caused by modulation of electron–hole magnetic dipolar interaction by lattice phonons is much smaller than this strength of this dipolar interaction itself < Δω2 > ≪ ω 2d . For the shortest interspin distance of 25 Å we have < Δω2 > ≪ 1015 rad2/s2. Therefore, the longitudinal spin relaxation time caused by interspin interactions should exceed the value T1d > 10−4 s which exceeds the maximum DAF value. Thus, the spin relaxation can not affect noticeably the dynamics of spin sublevel populations of CTS DTS+:PC71BM− on our experimental timescale.
Since out-of-phase ESE dependences for the given DAF were very similar for temperatures 20 K and 40 K they were modeled with the same parameter sets (Fig. 6b–d). This allows to suggest that at temperatures of 40 K and lower diffusion of electron and hole constituting CTS is frozen on microsecond timescale. Thus, the electron–hole distance obtained in the present work reflects the thermalization length of charges generated upon exciton splitting at DTS:PC71BM interface. CTSs with smaller size recombine faster, as can be derived from the increase of the average electron–hole distance with DAF. The distance of primary charge separation in the DTS:PC71BM composite is close to that for the benchmark photovoltaic composite P3HT:PC71BM obtained early [23]. This allows to suggest that the mechanisms of the interfacial CTS formation in polymer:fullerene and small molecule:fullerene composites are similar.
The increase of r0 with TDAF can be interpreted in frame of tunneling recombination theory. It is usually assumed that electron tunneling rate W(r) decreases exponentially with the distance between the donor and acceptor r [41]:
where νe is frequency factor, a is attenuation factor which depends on the overlap of wavefunctions of donor and acceptor. Therefore, the time-dependent tunneling distance rt, for which the condition W(rt) is fulfilled, scales linearly with logarithm of t:
For our system this means that for the a certain TDAF all CTSs with electron–hole distance smaller than a certain value have recombination probability close to unity. Thus, r0 values derived from our simulation can be interpreted as rt. As can be seem from Fig. 8, the dependence of r0 on is TDAF indeed close to linear. The a value derived from linear approximation of this dependence is 3.2 Å. This value is larger than those typically obtained for frozen solutions of donor and acceptor molecules in insulating organic matrices [41, 42]. This difference can be explained by high degree of conjugation of DTS molecule. Indeed, the observed attenuation factor for DTS/PCBM composite is close to that obtained for donor–acceptor diads covalently linked by π-conjugated bridge [42].

Dependence of the distance r0 on the value of ln(DAF/0.2 μs), DAF = 0.2 μs is minimal DAF value, and its linear approximation
4 Conclusion
An advanced ESE technique has been developed to determine structure and dynamics of CTS in a small molecule:fullerene organic photovoltaic composite. Pre-saturation of relatively strong in-phase ESE signal of trapped charges allows to detect weak out-of-phase ESE signal, which was attributed to light-induced spin-correlated DTS+/PC71BM− CTS. Low-temperature CTS lifetime of about 15 μs was obtained at 20 K. It decreases when temperature increases. Non-exponential ESE decays of CTS with DAF increase are explained by a distribution of CTS lifetimes, which is in turn caused by a distribution of a primary charge separation distance. The characteristic transversal spin relaxation time T2 = 2.6 μs was found for both DTS+ and PC71BM−. In the assumption of the weak but non-zero exchange interaction and in the point dipole approximation the electron–hole distance distribution was fitted by the broad distribution function with maxima between 49 and 57 Å, depending on delay after the laser flash. The CTS with smaller electron–hole distance recombine faster. The DTS+/PC71BM− CTS size distribution and its evolution with DAF increase are similar to those obtained previously for the benchmark polymer:fullerene composite P3HT:PC71BM.
References
B. Guo, W. Li, G. Luo, X. Guo, H. Yao, M. Zhang, J. Hou, Y. Li, W.-Y. Wong, ACS Energy Lett. 3, 2566 (2018)
S. Li, L. Ye, W. Zhao, H. Yan, B. Yang, D. Liu, W. Li, H. Ade, J. Hou, J. Am. Chem. Soc. 140, 7159 (2018)
S.D. Collins, N.A. Ran, M.C. Heiber, T.-Q. Nguyen, Adv. Energy Mater. 7, 1602242 (2017)
S. Koul, N. Hakim, Trans. Electr. Electron. Mater. 19, 319 (2018)
B. Kan, M. Li, Q. Zhang, F. Liu, X. Wan, Y. Wang, W. Ni, G. Long, X. Yang, H. Feng, Y. Zuo, M. Zhang, F. Huang, Y. Cao, T.P. Russell, Y. Chen, J. Am. Chem. Soc. 137, 3886 (2015)
J. Wan, X. Xu, G. Zhang, Y. Li, K. Feng, Q. Peng, Energy Environ. Sci. 10, 1739 (2017)
K. Vandewal, Annu. Rev. Phys. Chem. 67, 113 (2016)
C. Deibel, T. Strobel, V. Dyakonov, Adv. Mater. 22, 4097 (2010)
K. Vandewal, S. Albrecht, E.T. Hoke, K.R. Graham, J. Widmer, J.D. Douglas, M. Schubert, W.R. Mateker, J.T. Bloking, G.F. Burkhard, A. Sellinger, J.M.J. Fréchet, A. Amassian, M.K. Riede, M.D. McGehee, D. Neher, A. Salleo, Nat. Mat. 13, 63 (2014)
J.A. Love, C.M. Proctor, J. Liu, C.J. Takacs, A. Sharenko, T.S. Poll, A.J. Heeger, G.C. Bazan, T.-Q. Nguyen, Adv. Funct. Mater. 23, 5019 (2013)
I.A. Howard, R. Mauer, M. Meister, F. Laquai, J. Am. Chem. Soc. 132, 14866 (2010)
C.R. Poelking, The (Non-)Local Density of States of Electronic Excitations in Organic Semiconductors (Springer, Switzerland, 2018), pp. 105–123. https://doi.org/10.1007/978-3-319-69599-0
L.V. Kulik, S.A. Dzuba, J. Struct. Chem. 45, 298 (2004)
S.A. Dzuba, Russian. Chem. Rev. 74, 619 (2005)
M.N. Uvarov, L.V. Kulik, Appl. Magn. Reson. 44, 97 (2013)
J. Niklas, S. Beaupré, M. Leclerc, T. Xu, L. Yu, A. Sperlich, V. Dyakonov, O.G. Poluektov, J. Phys. Chem. B 119, 7407 (2015)
M.N. Uvarov, A.G. Popov, E.A. Lukina, L.V. Kulik, J. Struct. Chem. 55, 644 (2014)
K.M. Salikhov, Yu.E. Kandrashkin, A.K. Salikhov, Appl. Magn. Reson. 3, 199 (1992)
J. Tang, M.C. Thurnauer, A. Kubo, H. Hara, A. Kawamori, J. Chem. Phys. 106, 7471 (1997)
A.J. Hoff, P. Gast, S.A. Dzuba, C.R. Timmel, C.E. Fursman, P.J. Hore, Spectrochim. Acta A 54, 2283 (1998)
L.V. Kulik, S.A. Dzuba, I.A. Grigoryev, Yu.D. Tsvetkov, Chem. Phys. Lett. 343, 315 (2001)
S.A. Dzuba, P. Gast, A.J. Hoff, Chem. Phys. Lett. 236, 595 (1995)
E.A. Lukina, A.A. Popov, M.N. Uvarov, E.A. Suturina, E.J. Reijerse, L.V. Kulik, Phys. Chem. Chem. Phys. 18, 28585 (2016)
Y. Sun, J. Seifter, L. Huo, Y. Yang, B.B.Y. Hsu, H. Zhou, X. Sun, S. Xiao, L. Jiang, A.J. Heeger, Adv. Energy Mater. 5, 1400987 (2015)
S.A.J. Thomson, J. Niklas, K.L. Mardis, C. Mallares, I.D.W. Samuel, O.G. Poluektov, J. Phys. Chem. C 121, 22707 (2017)
K.L. Mardis, J.N. Webb, T. Holloway, J. Niklas, O.G. Poluektov, J. Phys. Chem. Lett. 6, 4730 (2015)
Y. Kobori, R. Noji, S. Tsuganezawa, J. Phys. Chem. C 117, 1589 (2013)
F. Kraffert, J. Behrends, Mol. Phys. 115, 2373 (2017)
E.A. Lukina, M.N. Uvarov, L.V. Kulik, J. Phys. Chem. C 118, 18307 (2014)
A.A. Popov, E.A. Lukina, L. Rapatskiy, L.V. Kulik, J. Magn. Reson. 276, 86 (2017)
E.A. Lukina, A.A. Popov, M.N. Uvarov, L.V. Kulik, J. Phys. Chem. B 119, 13543 (2015)
M.T. Colvin, R. Carmieli, T. Miura, S. Richert, D.M. Gardner, A.L. Smeigh, S.M. Dyar, S.M. Conron, M.A. Ratner, M.R. Wasielewski, J. Phys. Chem. A 117, 5314 (2013)
E.A. Lukina, E. Suturina, E. Reijerse, W. Lubitz, L.V. Kulik, Phys. Chem. Chem. Phys. 19, 22141 (2017)
R. Calvo, R.A. Isaacson, E.C. Abresch, M.Y. Okamura, G. Feher, Biophys. J. 83, 2440 (2002)
M.L. Kirk, D.A. Shultz, D.E. Stasiw, G.F. Lewis, G. Wang, C.L. Brannen, R.D. Sommer, P.D. Boyle, J. Am. Chem. Soc. 135, 17144 (2013)
M. Shinomiya, K. Higashiguchi, K. Matsuda, J. Org. Chem. 78, 9282 (2013)
T.E. Goliber, J.H. Perlstein, J. Chem. Phys. 80, 4162 (1984)
T. Kirchartz, B.E. Pieters, K. Taretto, U. Rau, Phys. Rev. B 80, 035334 (2009)
A.G. Redfield, Adv. Magn. Opt. Reson. 1, 1 (1965)
C.P. Slichter, Principles of Magnetic Resonance, 3rd edn. (Springer, Berlin, 1996), pp. 145–215
K.I. Zamaraev, R.F. Khairutdinov, Top. Curr. Chem. 163, 1 (1992)
O.S. Wenger, Acc. Chem. Res. 44, 25 (2011)
Acknowledgements
This work was supported by the Russian Science Foundation, project 18-73-00156. We also thank Prof. A. A. Lomov and Dr. A. G. Matveeva for fruitful discussions devoted the out-of-phase ESEEM calculations.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Uvarov, M.N., Kulik, L.V. Charge Transfer State in the Composite DTS(FBTTh2)2:PC71BM: Dynamics of Electron–Hole Distance Distribution After Light Absorption. Appl Magn Reson 50, 1277–1290 (2019). https://doi.org/10.1007/s00723-019-01149-1
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-019-01149-1