
Abstract
This article provides an overview of the application of electron paramagnetic resonance (EPR) and related hyperfine spectroscopies for the investigation of the electronic and geometric structure of the tetramanganese cluster that constitutes the oxygen-evolving complex (OEC) of photosystem II. Starting from the spin physics of Mn ions, a quantum-mechanical description of exchange-coupled oligonuclear Mn systems is given. Then, the focus shifts to the characterization of the OEC with emphasis on the two half-integer spin states of its catalytic cycle, namely the S0 and S2. The development of electronic models based on EPR and related spectroscopies, such as electron nuclear double resonance (ENDOR), is described in detail. The role of broken-symmetry density functional theory (BS-DFT) is also outlined. The characterization of Mn ligands and bound substrate waters via electron nuclear magnetic interactions is illustrated. Implications for the mechanism of water oxidation catalysis are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Light-driven water oxidation is performed by a single enzyme in nature, photosystem II (PS II). Despite its importance and recent advancements in its structural characterization, critical details about the molecular mechanism of water oxidation by the catalytic center of PS II, the oxygen-evolving complex (OEC), are still not resolved. In view of increasing demands for clean, cheap and renewable energy, understanding nature’s unique water-oxidizing enzyme will be of invaluable use for the development of synthetic biomimetic water-splitting catalysts (see [1–3]).
Electron paramagnetic resonance (EPR) spectroscopy is a powerful tool for elucidation of the electronic structure and function of biomacromolecules that contain metallocofactors, e.g., metalloproteins. Manganese-containing cofactors provide a particularly rich playground as the naturally abundant nucleus (55Mn, 100 %) has an intrinsic magnetic moment (nuclear spin I = 5/2, nuclear g factor g n = +1.3819). Thus, spectroscopies that probe the hyperfine interaction, such as electron nuclear double resonance (ENDOR), electron spin echo envelope modulation (ESEEM) and electron–electron double resonance (ELDOR)-detected nuclear magnetic resonance (NMR), are well suited to study these systems. In this article, we review the evolution of the electronic model of the tetramanganese core of the OEC. As a starting point, we introduce the structure and function of the OEC, leading to a description of relevant simpler (model) Mn spin systems and their EPR-spectroscopic investigations. Then, the review focuses on current electronic models of the OEC as determined from the most recent multi-frequency EPR and hyperfine spectroscopic studies combined with density functional theory (DFT) calculations. The subsequent identification of magnetically coupled exchangeable substrate waters provides information on the possible mechanism of the photosynthetic water oxidation reaction.
2 The Oxygen-Evolving Complex
2.1 Photosystem II
Photosystem II is a multi-subunit pigment-protein super-complex found in the thylakoid membrane of organisms that perform oxygenic photosynthesis. It uses visible light (400–700 nm) to drive the oxidation of water, transferring its electrons to bound plastoquinone, reducing it to plastoquinol. Concomitant H+ translocation across the membrane results in a proton gradient. Plastoquinol and the H+ gradient are subsequently used to generate the energy carriers that are used in carbon fixation, nicotinamide adenine dinucleotide phosphate in its reduced form (NADPH) and adenosine triphosphate (ATP), respectively. Four main processes constitute PS II function: (i) Light harvesting and exciton transfer from its antenna complexes to the reaction center of PS II, (ii) excitation and charge separation of the reaction center, (iii) reduction and protonation of acceptor side plastoquinone and (iv) accumulation of oxidizing equivalents on the donor side and H2O oxidation by the tetramanganese core of the OEC. For further details, see [4, 5].
2.2 The Catalytic Reaction Cycle of the Oxygen-Evolving Complex: the S State Cycle
As outlined above, the OEC is driven by the PS II reaction center. Photon absorption leads to an excited pigment state (P680*), which initiates charge separation across the membrane. The first radical pair state P680+/Pheo −D1 rapidly undergoes further electron/hole migration, leading to a cascade of subsequent radical pair states that act to stabilize the charge-separated system and thus allow the comparably slow water oxidation chemistry of the OEC to occur. The oxidation of water to dioxygen is a four-electron process,
whereas photooxidation of P680 is a one-electron process. Thus, the role of the OEC can be thought of as that of an electron hole accumulator. P680+ oxidizes the OEC repeatedly, one electron at a time, until the OEC has been oxidized four times. The OEC then converts water to dioxygen and resets itself to its most reduced state [6, 7]. The various net oxidation states of the Mn4O5Ca core of the OEC are known as the S n states, where the index n = 0, 1, 2, 3, 4 represents the number of stored oxidizing equivalents in the ligated metal cluster proper (Fig. 1). S0 represents the most reduced state, whereas the S4 state is the most oxidized in the catalytic cycle. S1 is the dark-stable state. The S0 state is of sufficient reduction potential that it can be slowly oxidized via a redox-active tyrosine residue of the D2 protein, YD (D2-Tyr160) [8]. YD does not participate further in the S state cycle. Apart from the spontaneous [S4] → S0 reaction step, all of the S n → S n+1 transitions are induced by the photochemical oxidation of P680+, which in turn oxidizes the OEC via the redox-active tyrosine YZ residue of the D1 protein (D1-Tyr161); each S n → S n+1 transition passes through an intermediate S n YZ · state, wherein the oxidized tyrosine is poised to oxidize the metal cluster of the OEC. An [S4] state, different from the S3YZ · state, has not yet been identified. For further information, see [4, 5].

Light (hν)-driven catalytic reaction cycle of the OEC with the putative valence state configurations of the Mn ions in the individual Sn states [9]. S1 is stable in the dark, while S4 decays spontaneously into S0, under the release of O2, allowing (a) new water molecule(s) to bind. Arrows indicate abstraction of an electron by YZ · and proton release from the OEC
2.3 The Mn4O5Ca Core
The OEC is comprised of four manganese ions and one calcium ion [10–12]. Extended X-ray absorption fine structure (EXAFS) spectroscopy was the first technique to provide robust structural constraints for the cluster, including Mn–Mn and Mn–Ca interatomic distances and angles (for a review, see [13]). X-ray crystallography followed these initial studies, taking a decade of effort before an atomic resolution structure was obtained. The first X-ray crystallographic models were of insufficient resolution (3.8 Å [14] and 3.7 Å [15]) to provide a detailed picture; they, nevertheless, allowed many structural motifs to be discarded. These structures resolved a ‘Y-shaped’ electron density map of the tetramanganese cluster; three Mn ions were placed in the corners around one central Mn ion, while the Ca2+ ion and the bridging atoms could not be identified. This basic model was refined in the subsequent ‘London’ structure, at a resolution of 3.5 Å [16]. Here, a trinuclear cubane-like Mn3O4Ca cluster was proposed with the fourth, outer Mn attached directly to one of the O vertices, similar to the cubane Mn4O4 core suggested by Brudvig and Crabtree [17]; in the cubane proper, the four Mn ions form the corners of the cuboid forming a tetramer. Subsequent structures from the Berlin groups, at 3.0 [18] and 2.9 Å [19] resolution, confirmed a ‘3 + 1’ topology, but still did not allow for an assignment of the precise Mn and Ca positions and their connectivity. The same PS II crystals were also studied by polarized EXAFS which led to the development of models based on a common motif of Mn4 connectivity, later termed the ‘fused twist’ topology. Finally, in 2011, after more than 10 years of crystallographic efforts, a PS II X-ray structure with atomic (1.9 Å) resolution was reported [20]. Its Mn4O5Ca cluster clearly resolves the four Mn ions being linked via a network of five μ-oxo bridges. The cluster exhibits a distorted cuboidal topology with three Mn ions and the Ca2+ cofactor as well as four bridging oxygens forming the eight vertices (Fig. 2a, b). The fourth, outer Mn ion is connected to the cuboidal unit via another μ-oxo bridge to one of the Mn corners (‘distorted chair’). The crystal structure allowed for an unambiguous identification of ligating amino acid residues and surrounding H2O/OH− molecules (Fig. 2a). However, corresponding X-ray absorption spectroscopy (XAS) measurements suggest the X-ray radiation dose the PS II crystal experiences during X-ray crystallographic measurement is sufficient to lead to reduction of the high-valent Mn ions and a distortion of the catalytic core [21, 22]. It is therefore, not entirely clear what S state the crystal structure represents. While the group of Pace suggests it to be in the S1 state [23], with an overall valence state (MnIII)4, which on average is 0.5 units lower than generally assumed [9, 13, 24–27], most theoretical studies support a non-physiological oxidation state below S0 [28–30]. As a consequence, geometric models of the physiological states of the OEC have been developed by means of DFT [31–33], also based on the crystal structure [28, 34, 35]. These optimized structures of the OEC poised in the S2 state are more compact, displaying shorter Mn–Mn and Mn–O distances, consistent with earlier EXAFS results. The models typically contain either 4:1 (fused twist) or 3:2 (cubane-like) ratios of μ2-oxo to μ3-oxo bridges, rendering one of the Mn ions of the tetramer, the MnIII, pentacoordinate (Fig. 2c, d). Both the X-ray crystallographic and DFT structures exhibit two H2O/OH− ligands bound to the remote Mn and two H2O ligands bound to the Ca2+ ion.

a X-ray crystallographic structure of the OEC in dark-adapted PS II at 1.9 Å resolution [20]. b The Mn4O5Ca inorganic cluster from the crystal structure [20], as in a. c Fused twist geometry-optimized DFT structure of the Mn4O5Ca core in the S2 state [35]. d Cubane-like geometry-optimized DFT structure of the Mn4O5Ca core in the S2 state [35]. Structures c and d both reproduce the Mn–Mn distance constraints from EXAFS and the magnetic resonance data; see [32, 34, 35] for further details. e The ‘Y coupling’ scheme for the S2 state developed from EPR/55Mn ENDOR [9, 36, 37] (see Sect. 5.2.2). f, g Mn4O5 connectivities of the fused twist (c) and the cubane-like (d) core topologies. The nomenclature of the Mn ions in a, e and f combines the lettering used in earlier EPR and EXAFS spectroscopic work and the numbering from the crystal structures, as in b. W1–W4 represent H2O/OH− ligands. Amino acids, except CP43-Glu354, are from PS II subunit D1. Mn, Ca, N, O, and C atoms are depicted in purple, yellow, blue, red, and gray, respectively
Ca2+ is an essential cofactor in oxygen evolution [38–42]; its removal inhibits water splitting by blocking the OEC early in its reaction cycle and leads to a perturbed EPR signal (See Sect. 2.4). It has been hypothesized that (i) calcium acts in water splitting by binding a substrate water molecule [43–46], as evidenced by the mass spectrometric measurements of 18O-labeled dioxygen release [47, 48], and (ii) it modifies the redox potential of the OEC, perhaps by controlling proton-coupled electron transfer to YZ · [13, 49, 50]. Sr2+ is the only surrogate that is capable of restoring water splitting after Ca2+ removal [38, 51–53]. This may be due to a combination of two important requirements that the surrogate must match: (i) the approximate size and (ii) the Lewis acidity [54] of Ca2+. Chloride ions are also essential for photosynthetic oxygen evolution [48, 55, 56]. Two Cl− binding sites have been identified at, or near the site of water splitting. The Cl− ions, however, do not directly coordinate the OEC [57–61]. Cl− depletion blocks OEC turnover, just as Ca2+ depletion does [59, 62–64]. Cl−, like Ca2+, has been proposed to influence proton transfer from the OEC as part of a hydrogen-bonding network, based on oxygen evolution activity measurements and EPR spectroscopy [65–67].
Two substrate water molecules must bind to the OEC during its reaction cycle. Currently, the sites of substrate binding are unknown. ESEEM and hyperfine correlation spectroscopy (HYSCORE) measurements have demonstrated that the OEC interacts with numerous exchangeable hydrogens, most probably belonging to waters and/or hydroxide groups [68–70]. EPR data [71–76] have indicated that substrate analogues, such as small alcohols and ammonia, are also able to bind to the OEC. In addition, non-EPR techniques have confirmed the presence of water at the OEC, too, e.g., near-infrared (IR) Raman spectroscopy [77], Fourier transform IR (FTIR) spectroscopy [78–80] and mass spectrometric measurements of 18O-labeled dioxygen evolution [81, 82].
2.4 EPR Signals of the Oxygen-Evolving Complex
The four Mn ions that make up the OEC are magnetically coupled in all S states that have been isolated thus far (S0–S3), and all display characteristic EPR signals [74, 83–95]. The S2 state, which has a ground spin state of total spin S t = 1/2, is the best characterized in current literature and gives rise to the well-known S2 state EPR multiline signal [83]. It is centered at g ≈ 2 and contains 18–21 spectral lines with a peak-to-peak spacing of ~7.5–9 mT (Fig. 3a). Depending on the conditions used, the S2 state of higher plant PS II also exhibits broad EPR signals, centered at g ≥ 4.1, which have been assigned to an S t = 5/2 spin state [35, 96–100]. The g ≥ 4.1 signals can also be induced by near-IR illumination of the S2 multiline state at temperatures ≤160 K [96, 97]. Similar high-spin state (g ≥ 4.1) signals have been observed in the S2 state OEC from cyanobacterial PS II [101, 102]. In plant PS II, the presence of small alcohols prevents the formation of the g ≥ 4.1 signal [103–105]. Amongst all the alcohols, methanol (MeOH) has a specific effect in that it modifies the S2 multiline signal [104–106], which is then also no longer sensitive to near-IR illumination [105, 107]. The modified S2 multiline signal is narrower than that seen for the non-treated plant PS II preparation, resolving fewer spectral lines. MeOH also affects the other S states. None of these effects are observed in cyanobacteria; for a comparison see Su et al. [108].

Q-band EPR spectra of the OEC and related model systems. a S2 state of the OEC in PS II from spinach (4 % MeOH) [9]. b S0 state of the OEC in PS II from spinach (4 % MeOH) [9]. c MnII-(μ-OH)-MnIII (PivOH) [MnIIMnIII(μ-OH)-(μ-piv)2(Me3tacn)2](ClO4)2 (piv = (CH3)3CCO2) [109]. d MnIII-(μ-O)2-MnIV (Tacn) [(tacn)2MnIIIMnIV(μ-O)2μ-OAc](BPh4)2 (tacn = 1,4,7-triazacyclononane) [110]. The black solid lines represent the data, the red solid lines represent simulations based on the spin Hamiltonian formalism (see Eqs. 3, 14)
A similar multiline signal centered at g ≈ 2 is observed for the S0 state of the OEC; it contains up to 26 spectral lines with a peak-to-peak spacing of ~8 mT (Fig. 3b) [88, 90, 111]. To observe this signal in spinach, 3–5 % MeOH must be added to the PS II sample. In contrast, an S0 signal in cyanobacteria does not require the addition of MeOH to be observed [112].
Parallel mode signals from integer total spin states have been identified for the S1 and S3 states: in S1 at g ≈ 4.9 [84, 86] and g ≈ 12 [89] and in S3 at g ≈ 4, g ≈ 8 and g ≈ 12 [91, 95]. The temperature dependencies of the g ≈ 4.9 and g ≈ 12 signals indicate that they arise from excited states of effective spins S t = 1 and S t = 2, respectively, and that the S1 state has a ground state of S t = 0 [86]. Recent simulations of the S3 state signals indicated that they originate from an S t = 3 state [95].
‘Split’ EPR signals, which arise from the weak magnetic interaction between the Mn4O5Ca cluster and the YZ · radical are visible in all S n YZ · states [113–120]. This coupling on the order of 10−2 cm−1 results in a broadening of the tetramanganese signals and a splitting of the S = 1/2 YZ · signal.
3 The Mn Oxidation States Relevant to the OEC
3.1 Mononuclear MnII, MnIII, MnIV
3.1.1 Mononuclear MnII Complexes
MnII is a d5 ion (S = 5/2). Its half-occupied valence electron shell (3d) often engenders low g and hyperfine anisotropy and a small fine structure term D. In the circumstance where the fine structure parameter D is significantly smaller than hν (D ≪ hν where h is Planck’s constant and ν is the frequency of the source resonance radiation), only the allowed ΔM S = ±1 transitions, where M S is the electronic magnetic quantum number, are observed in an EPR experiment. The central transitions M S = 1/2 ↔ −1/2, appear at g ≈ 2.0 and manifest as six sharp lines due to the 55Mn hyperfine interaction. When D ≫ hν, the spin manifold splits into three Kramers’ doublets with M S = ±5/2, ±3/2 and ±1/2. The large fine structure term acts to spread the EPR spectrum across a large magnetic field range. These systems typically display much faster spin–lattice relaxation times than for the case D ≪ hν, leading to a broadening of EPR lines, especially of the non-central, outer transitions. Their resonance positions may coincide with magnetic field positions that exceed the sweep range of the magnet. At room temperature, the outer transitions are broadened beyond detection and only the central transitions are observed. This submanifold is effectively a spin S = 1/2 system, with the resonant line being anisotropic due to the zero-field splitting term. Complexes of axial symmetry (symmetry parameter E = 0) exhibit two effective g values, g || = 2.0 and g ⊥ = 6.0. That is, when there is large zero-field splitting, there are turning points in the angular variation of line position at g = 6 and 2. A listing of the spin Hamiltonian parameters—g matrix, D, E, higher order fine structure parameters \( b_{4}^{m} \) (m = 0, 2, 4), and hyperfine matrix a—has been previously tabulated by Misra [121].
3.1.2 Mononuclear MnIII Complexes
MnIII is a d4 ion (S = 2). Unlike MnII, MnIII has an inherently asymmetric electron configuration and thus often displays a large fine structure splitting, of the order of 1–5 cm−1. As a consequence, MnIII is usually not detected at low frequencies (X-, Q-, W-band). The EPR of MnIII has only been detected at very high frequency (VHF) [122]. In principle, the strictly forbidden ΔM S = 0 transition of the MnIII spin manifold can be detected at low frequencies in highly axial systems, i.e. E ≈ 0, by using parallel polarization EPR, wherein B 1 || B, such as for the S1 state signals [84, 86]. (B 1 is the radiation field inducing transitions and B is the external magnetic field.) The fine structure interaction splits the levels into two doublets (M S = ±1, ±2) and a singlet (M S = 0). In addition, the lines are expected to be quite broad. An overview of spin Hamiltonian parameters of monomeric MnIII complexes is listed in Supplementary Table S1 [53].
3.1.3 Mononuclear MnIV Complexes
MnIV is a d3 ion (S = 3/2). Depending on its symmetry, it can display large or small g and hyperfine anisotropy and fine structure terms. In octahedral ligand fields, MnIV ions are characterized by half-filled t2g orbitals and thus usually display small fine structure splittings D, whereas in tetragonal ligand fields, D tends to be larger. As seen for MnII, when D is small, the overlapping electronic transitions are seen at about g ≈ 2. In contrast, when D is large, the spin manifold splits into two Kramer’s doublets M S = ±1/2, ±3/2. The transition 1/2 ↔ −1/2 is observed even with large D, the effective g values being g || = 2.0 and g ⊥ = 4.0. Supplementary Table S2 lists spin Hamiltonian parameters of monomeric MnIV complexes [53].
3.2 Binuclear Mn Complexes
3.2.1 Exchange-Coupled Mn Dimers
The EPR spectra of antiferromagnetically exchange-coupled mixed valence Mn dimers (MnIIMnIII, MnIIIMnIV, Fig. 3c, d) bear characteristics similar to the S2 and S0 multiline signals described above in Sect. 2.4 (Fig. 3a, b). The spin Hamiltonian H for a coupled system consisting of two Mn electron spins S i (i = 1, 2) contains terms representing the electronic exchange interaction −2J S 1 S 2, the dipolar (through-space) term S 1 T S 2 between the electron spins S 1 and S 2 of the two Mn ions, electronic Zeeman terms μ B S i g i B for each Mn ion with μ B being the Bohr magneton, hyperfine interaction terms for each Mn ion with their respective nuclei S i a i I i and the zero-field or fine structure term S i d i S i [110, 123, 124]:
In Eq. (2), J is the isotropic electronic exchange coupling constant, which is negative for antiferromagnetic coupling and positive for ferromagnetic coupling. The magnitude of J depends on the degree of orbital overlap between the two ions and is strongly influenced by the bridging ligand(s). The elements of the dipolar tensor T depend on the orientation of the two Mn ions with the magnitude μ 0 μ 2B g 2/R 3, with μ 0 being the permeability of the free space, and R the inter-spin distance. d i are the fine structure tensors, g i are the g matrices and a i are the hyperfine matrices. S i and I i are the electron and nuclear spin operators, respectively, for the two Mn ions.
The magnitude of J relative to T, d 1 and d 2 predominantly determines the EPR spectrum of a binuclear Mn complex. (Note that the magnitudes of a 1 and a 2 are of the order of hundreds of MHz and thus can always be considered small relative to the magnitudes of J, T, d 1 and d 2.) The EPR spectrum becomes very complex when these terms are of the same order of magnitude. In contrast, when J is large and T, d 1 and d 2 are relatively small, the EPR spectrum is fairly simple to interpret. In the following discussion it is assumed that J is indeed large. In this case, the resultant spin S t = S 1 + S 2 is obtained by the vector coupling rule, and the exchange-coupled system is described by the use of the vector projection model. Thus, S t = |S 1 − S 2|, |S 1 − S 2| + 1, …, |S 1 + S 2| − 1, |S 1 + S 2|. For MnIIMnIII and MnIIIMnIV exchange-coupled systems, the coupling leads to electronic states of total spin S t = 1/2, 3/2, 5/2, 7/2 (and 9/2 in case of MnIIMnIII) in increasing order of energies when the coupling is antiferromagnetic. However, this order reverses when the coupling is ferromagnetic, as shown in Fig. 4 for an exchange-coupled MnIIIMnIV dimer. For a variety of μ-oxo-bridged dimanganese model compounds, the electronic exchange coupling is typically antiferromagnetic. When such a dimer is in a mixed valence state in which the Mn ions differ by one oxidation state, it is usually characterized by a ground state with S t = 1/2. If an antiferromagnetically exchange-coupled dimer is not in a mixed valence state, then the ground state is usually S t = 0. Thus, in strongly antiferromagnetically exchange-coupled Mn dimers, EPR signals are normally observed only for mixed-valence states. The energy levels for an exchange-coupled MnIIIMnIV dimer are shown in Fig. 4.

Energy level diagram for the resulting effective spin states S t of an exchange-coupled MnIIIMnIV dimer as a function of the strength and sign of the J coupling (J < 0: antiferromagnetic, J > 0: ferromagnetic), according to the Heisenberg exchange term −2J S 1 S 2
3.2.2 Exchange-Coupled Dimer States for Large J
In case of a large and negative J, as described in the previous section, the spin Hamiltonian H for the exchange-coupled S t = 1/2 resulting ground state is [110, 124–126]
The effective G matrix of the coupled system is expressed in terms of g 1 and g 2 matrices for the isolated systems as
where
ρ 1 and ρ 2 are referred to as spin projection factors, which can be interpreted as the contributions of the individual electronic spins to the total spin state. For coupled MnIIIMnIV systems (S(MnIII) = 2, S(MnIV) = 3/2), ρ(MnIII) = 2 and ρ(MnIV) = − 1. The third term of Eq. (4) is usually neglected in MnIIIMnIV complexes, since J ≫ D i (i = 1, 2). The effective Mn hyperfine coupling matrices A 1 and A 2 of the coupled system can be expressed in a similar way in terms of the intrinsic (on-site) hyperfine coupling matrices a 1 and a 2:
It is seen that the measured hyperfine anisotropy is in part dependent on the energy spacing of the spin manifold, i.e., on J, d 1 and d 2. This can lead to interesting cases where anisotropy is effectively transferred from one Mn nucleus to the other. Supplementary Table S3 contains a listing of the spin Hamiltonian parameters for MnIIIMnIV mixed-valance complexes [53], as these complexes are of relevance to the recent models for the tetramanganese cluster of the OEC.
3.2.3 Magnetic Coupling of Ligands (e.g. 1H, 13C, 14N, 17O) to a Mn Dimer
The interaction of a ligand nucleus with the electron spin of the Mn dimer is typically small, less than the intrinsic EPR linewidth. As such, it does not need to be explicitly considered when analyzing EPR spectra of these systems. This coupling can, however, be probed by continuous-wave (CW) and pulsed EPR techniques (ESEEM, ENDOR, ELDOR-detected NMR). When measured at the center field, these spectra can be rationalized by considering only the ligand terms of the spin Hamiltonian (hyperfine, quadrupole terms) coupled to an isotropic spin S t = 1/2 state with an effective g value as all powder pattern position are sampled equally; i.e. 55Mn hyperfine terms, etc. do not need to be explicitly included. The ligand hyperfine interaction is typically made up of two components: (i) a through-bond component, the Fermi contact term, which approximates the isotropic hyperfine coupling A iso and (ii) a through-space component, the dipolar coupling A dip, which approximates the anisotropic hyperfine coupling. As the system contains two metals, this latter term is made up of two contributions, representing the interaction of the ligand with each metal center (see Eqs. 9–11), referred to in the literature as the extended point-dipole model [69, 127, 128]. Equation (9) below gives the general expression describing this interaction and, to the right of the second equality sign, gives the product of the constants if the coupling nucleus is 1H.
Here, T h is the 3 × 3 diagonal tensor with the diagonal elements −δ, −Γ + δ/2, Γ + δ/2, with δ and Γ defined by the parameters r 1, r 2 and θ as follows:
Here, for the MnIII–MnIV dimer, r 1, r 2 and d are the MnIII nucleus, the MnIV nucleus and the MnIII–MnIV inter-spin vectors, respectively; and θ, ϕ are the angles between (r 1, r 2) and (r 1, d), respectively, as shown in Fig. 5a.

Geometric representation of the point-dipole model [69, 127, 128] describing the configuration of the various constituents, as shown in [124]. Here, the nucleus could for example be a proton (1H, I = ½), carbon (13C, I = 1/2), nitrogen (14N, I = 1), or oxygen (17O, I = 5/2). a Bridging ligand with non-axial A dip . b Terminal ligand in the extension of the MnIII–MnIV axis exhibiting an axial A dip
Although, in general, A dip is a non-axial tensor, it becomes axial when the proton is located on the line joining the two Mn ions (θ = 0 or π, \( \Upgamma = 3/2\delta \), Fig. 5b), with the diagonal elements being −δ, −δ, 2δ. One can modify Eq. (9) for practical use. The parameters r 2 and θ in Eqs. (10) and (11) can be expressed as
This extended point-dipole model is applicable only if the distances between the magnetic moments are large compared with the size of the spin density distribution. Nevertheless, this model is very helpful in correlating hyperfine matrices to nuclear positions. 1H, 13C, 14N, and 17O couplings in the Mn model complexes [(2-OH-3,5-Cl2-salpn)2MnIIIMnIV] (salpn = N,N′-bissalicylidene-1,3-diamino-2-hydroxypropane) [128], [R4dtneMnIIIMnIV(μ-O)2(μ-OAc)](BPh4)2 (R = H, CH3, dtne = 1,2-bis(1,4,7-triazacyclonon-1-yl)ethane), [(tacn)2MnIIIMnIV(μ-O)2μ-OAc](BPh4)2, [bpy4MnIIIMnIV(μ-O)2](ClO4)3 (bpy = 2,2′-bipyridyl) and [(phen)4MnIIIMnIV(μ-O)2](ClO4)3 (phen = 1,10-phenanthroline) [110, 124, 129–131] were successfully analyzed using this concept. These same principles can be applied to systems with more than two paramagnetic centers and oligonuclear sites in proteins, such as the Mn4O5Ca cluster of the OEC [69, 127], which we will discuss in the following section.
4 The Exchange-Coupled Tetranuclear Manganese Complex
4.1 Effective Spin Hamiltonian and Spin Projection Formalism
A spin Hamiltonian analogous to the dimer system can be built to describe the tetramanganese cluster of the OEC. When poised in the S2 or the S0 state, the four Mn ions couple together to give a ground electronic configuration of total spin S t = 1/2. The oxidation states of the individual Mn ions that make up the OEC in the S2 state are generally considered to be MnIII(MnIV)3 [9, 13, 24–27]. The effective spin Hamiltonian of the ground state can thus be expressed as the sum of Zeeman and hyperfine interactions as [132]:
In Eq. (14) (analogous to Eq. 3), G is the effective tensor for the coupled state with the total spin S t = S 1 + S 2 + S 3 + S 4, as determined using the vector coupling rule, and A i (i = 1, … 4) are the effective hyperfine tensors of the four manganese nuclei. The properties are to be evaluated within the subspace of S because here, the interionic exchange interaction is dominant, i.e. |J| ≫ D i , as observed in MnIIIMnIV complexes. With this coupling, each S i can be replaced by a product of a constant spin projection factor times the total spin operator, i.e. S i = ρ i S, when the Wigner–Eckart theorem is applied. More details about the spin projection formalism are given in the Supplementary Sects. S2 and S3, where the related list of basis vectors and expressions for the calculation of spin projections can be found [53]. The factors ρ i can be expressed as
where the bra-ket notation \( \left| {} \right\rangle \) stands for any state within the S t = 1/2 ground-state wave function given by the Heisenberg–Dirac–van Vleck term \( - 2\sum\limits_{i < j} {J_{ij} {\mathbf{S}}_{i} {\mathbf{S}}_{j} } . \)
As a consequence, in Eq. (14), G becomes
Equation (14) is then transformed, by substituting A i = ρ i a i , into the readily usable form
Equation (17) can be subjected to different approaches to account for the magnetic properties. The obvious approach is to treat each ρ i a i as a single parameter, evaluated by a spectral-fitting procedure, and then find a physical explanation for it. This is, however, a very tedious procedure as it involves four nuclei. There can be as many as 15 principal values for the g and the four 55Mn hyperfine matrices a i , requiring 12 Euler angles for their relative orientations. Instead, this problem can be handled by treating ρ i and a i separately, making full use of the intrinsic Mn hyperfine matrices and their orientations obtained from simulation of model complexe cofactors and proteins. The spin projection factors ρ i are used to scale the intrinsic hyperfine parameters a i to obtain effective hyperfine parameters for the Mn tetramer system. Expressions for ρ i can be derived from an electronic exchange interaction model as a set of pairwise J coupling constants describing the respective spin system, such as the models in Fig. 2e and those presented in detail in Sect. 5.2 [9, 32, 133–135]. These can be used to simulate EPR and hyperfine (ENDOR, ESEEM-type, ELDOR-detected NMR) spectra for the different states of the Mn tetramer; only the relative orientations of the matrix axes need to be established. For this, one makes use of the observation that, for (μ-O)2-bridged MnIIIMnIV dimer systems, the hyperfine and g matrices are essentially axial and nominally collinear. This approach has been successfully applied both in theoretical and experimental studies on the electronic structure and spectroscopic properties of exchange-coupled tetramanganese complexes, such as [MnIIIMn IV3 O6(bpy)6]3+ [134] and [Mn IV3 Ca2O4(O2CBut)8(ButCO2H)4] [136].
4.2 Inclusion of the Fine Structure Term of One or Several Mn Ions
The spin projections calculated above can be corrected for the fine structure terms of the spin Hamiltonian, analogous to the Mn dimer systems. In practice, the fine structure tensor d i of only one sort of Mn ions, that of MnIII, is of a relevant size to be considered. Its valence electron configuration leads to a large Jahn–Teller distortion, described in greater detail in the Supplementary Sect. S4. MnIII always possesses a fine structure constant of significant magnitude D = 1–5 cm−1, whereas MnIV in octahedral environments typically displays fine structure constants an order of magnitude smaller than this [126] (see Supplementary Tables S1 and S2 and the references given therein). For the calculation of anisotropic spin projections, the spin Hamiltonian, which describes the zero-field splitting of the spin states, includes the electronic exchange interaction and the fine structure term of the complex:
In Eq. (18), the spin operator S 1 refers to the MnIII ion, with d 1 being its fine structure tensor, which is assumed to be axial and can thus be expressed in terms of a single parameter D (see [53]):
Inclusion of the fine structure term of the MnIII ion renders the spin projections ρ i as matrices rather than scalars. Since all pairwise exchange couplings J ij have here been assumed to be isotropic, and the fine structure tensor d 1 of the MnIII ion to be axial, the spin projection matrix of the ith Mn ion can be expressed as a diagonal matrix of the form:
5 Electronic Exchange Coupling Models for the Tetramanganese Cluster of the Oxygen-Evolving Complex
In this section, we give an overview of the development of electronic structural models for the Mn4O5Ca cluster of the OEC and the key role EPR spectroscopy played in this exercise. EPR spectroscopy was the first technique to show that the PS II super-complex contained a Mn cofactor that was intimately linked to photosynthetic oxygen evolution. A key finding was the identification of an intermediate displaying an S = 1/2 EPR signal, the so-called multiline signal of the S2 state, first observed in spinach chloroplasts [83, 137]. Based on comparison with Mn model complexes, the S2 state was assigned to either a binuclear MnIIMnIII site or a tetranuclear MnIIIMnIV 3 cluster. The four-flash periodicity of the S2 multiline signal (maximum intensity yield on every fourth light flash in accordance with a four-electron process), together with inhibition studies confirmed that enzymatic water oxidation catalysis was Mn-centered, as had been suggested before (reviewed in [138]). Early proposals for the electronic structure of the oligo-Mn center were developed from spectroscopic data in combination with biochemical experiments already at a time when the geometric structure of the OEC was not known. Outdated models that studies have proven to be physically not relevant are not reviewed here. A description of such earlier models, such as the tetramanganese ‘cubane-like’ core [17], the ‘dimer of dimers’ [132] and the ‘butterfly’ core models possessing simplified electronic exchange coupling schemes and their quantum-mechanical treatment have been reviewed by Misra [139].
Meanwhile, the progress in the X-ray crystallographic structure determination of PS II up to the 1.9 Å structure by Umena et al. [20] in conjunction with new or enhanced spectroscopic methodologies, such as X-ray absorption spectroscopy (XAS) [140–142], electron paramagnetic resonance (EPR/ENDOR) spectroscopy [9, 53, 133], and vibrational spectroscopy [143], now provide a robust picture of the electronic structure of the OEC in its low oxidation states (S0–S2). Just as critical to this process are recent advances in the computational treatment of the OEC, in particular developments in the calculation of EPR-spectroscopic observables using broken-symmetry (BS) DFT [144], providing a connection between the electronic structure as probed by EPR and the molecular structure. The current model of the Mn4O5Ca cluster in the S2 state is described in the following, with particular emphasis on the role that combined EPR and 55Mn ENDOR spectroscopy along with BS-DFT played in its development.
5.1 Calculations of EPR Parameters from Density Functional Theory
5.1.1 Electronic Exchange Coupling and the Heisenberg–Dirac–van Vleck Hamiltonian
For systems containing multiple Mn ions wherein the transition metal centers are antiferromagnetically coupled, the application of the BS formalism is necessary for the correct calculation of the exchange coupling and other EPR parameters from DFT [145–147]. By means of the BS-DFT approach, the exchange coupling constants J and hyperfine coupling constants A iso can be calculated with reasonable accuracy for Mn dimer, trimer and tetramer model systems [134, 148–150]. The pairwise magnetic interactions J ij between the electron spins of the manganese centers are assumed to be described by the isotropic Heisenberg–Dirac–van Vleck (HDvV) Hamiltonian:
The OEC consists of four coupled Mn ions, resulting in six possible exchange coupling pathways J ij . Within the BS-DFT framework, the exact problem is solved without making any assumptions for the magnitude of non-nearest-neighbor interactions. A system of four coupled spin centers contains eight single-determinant representations; one high-spin along with seven BS single-determinant representations from the remaining parallel/antiparallel spin combinations. For example, in the case of one MnIII and three MnIV centers, as in the S2 state, with fixed atomic high-spin d4 or d3 electronic configurations, there are three BS solutions with total M S = 7/2, one BS solution with total M S = 5/2 and three BS solutions with total M S = 1/2. It is noted that the BS energies for each single-determinant solution are not of significance when considered individually. However, considered together, the BS solutions, along with the high-spin solution, allow for the extraction of the exchange coupling constants from the HDvV Hamiltonian. It is only once the analysis of the exchange coupling constants has been made that the energies, spin states, and molecular properties can be related to the experiment [134]. Additionally, it should be noted that individual pairwise exchange couplings are not observables; it is the identity, ordering and spacing of the spin levels derived from a given set of J values that determine the observable energy level spin ladder of a system.
The over-determined equation space from the high-spin and the seven BS solutions for the calculation of the six exchange coupling constants (see Sect. 5.2.3) can be solved using the technique of singular value decomposition (SVD). This method gives the best solution, in the least-squares sense, for the six exchange interactions between four Mn spin centers. Having calculated the six exchange coupling constants for each structure, it is then straightforward to diagonalize the HDvV Hamiltonian to obtain the complete energy level spin ladder, identifying the true ground spin state, and to calculate the associated properties of a given system. The application of the BS-DFT methodology to the calculation of exchange coupling constants has been discussed in previous studies of manganese systems [32, 134, 144, 149, 150].
5.1.2 Hyperfine Coupling Constants from Broken-Symmetry Density Functional Theory
The calculation of hyperfine coupling constants A iso from BS-DFT has been made possible from recent advances in theory, previously described in detail [134], such that now a meaningful comparison of experimental and calculated results can be achieved using this new methodology. It has been used to discriminate between different cluster topologies and ligand environments of Mn ions in oligonuclear clusters [134] and has already been applied to several models of the OEC [32, 34, 35, 135]. For exchange-coupled multinuclear paramagnetic systems, an exact quantum-mechanical treatment would computationally be too demanding for systems of such high complexity. BS-DFT, in combination with the spin projection formalism, described in Sect. 4.1, represents a pragmatic, efficient alternative. For a complete in-depth description of the theory and methodology for the calculation of hyperfine couplings from BS-DFT, the reader is referred to reference [134]. In the present article, only the main concepts will be highlighted and discussed.
For a system composed of metal-centered spin subsystems, with nucleus K belonging to subsystem A, the general equations that directly connect the BS calculation to the observable effective hyperfine coupling constant \( A_{\text{iso}}^{{({\text{K}})}} \) are as follows:
In Eq. (22), S A is the site spin of subsystem A, and the positive or negative sign depends on whether the fragment carries majority (α) or minority (β) spin. \( a_{\text{iso,BS}}^{{({\text{K}})}} \) is the ‘raw’ hyperfine coupling constant calculated directly from the BS formalism, and \( \left\langle {{\mathbf{S}}_{{\mathbf{z}}} } \right\rangle_{\text{BS}} \) is the total electronic magnetic quantum number M S of the appropriate BS wavefunction. In Eq. (23), S t is the fictitious effective total ground-state spin, \( \langle {{\mathbf{S}}_{{\mathbf{z}}}^{{({\text{A}})}} } \rangle \) is the on-site spin expectation value, and \( a_{\text{iso,site}}^{{({\text{K}})}} \) is the on-site isotropic coupling constant. To correctly determine the final projection of the on-site hyperfine coupling constants to a calculated hyperfine constant comparable to experiment, the determination of the site spin expectation value \( \langle {{\mathbf{S}}_{{\mathbf{z}}}^{{({\text{A}})}} } \rangle \) is required. For a subsystem A in a system consisting of subsystems A…N, the site spin expectation value used for the isotropic spin projection coefficients, is given by
where \( | {C_{\text{I}}^{{S_{\text{A}} M_{{S_{\text{A}} }} \ldots S_{\text{N}} M_{{S_{\text{N}} }} }} } |^{2} \) is the weight of the basis state \( \left| {S_{\text{A}} M_{{S_{\text{A}} }} , \ldots ,S_{\text{N}} M_{{S_{\text{N}} }} } \right\rangle \) in the ground-state eigenfunction describing the Kramers’ doublet of lowest energy.
5.2 Current Electronic Model of the Oxygen Evolving Complex in the S2 State
5.2.1 The EPR Experiment
As described above, the four Mn ions that make up the OEC are magnetically coupled in the various S n states; each S n state of the Mn4O5Ca cluster exhibits characteristic EPR signals. Of these, the S2 state, which has a ground state of total spin S t = 1/2, is the best characterized in the current literature, giving rise to the well-known S2 state multiline EPR signal. The S2 multiline signal as observed at 9 GHz (X-band) is centered at g ≈ 2 and contains 18–21 spectral lines with a peak-to-peak spacing of ~7.5–9 mT (Fig. 3a). The multiline is spectrally sparse, since not all of the potential 1,296 allowed transitions within the ground-state S2 manifold are resolved. Thus, no unique fitting of the spectrum can be obtained with the spin Hamiltonian formalism described above. Although multi-frequency EPR (9–94 GHz) provides more details on the spin Hamiltonian parameters, especially with regard to the g matrix, it does not lead to an improved understanding of the hyperfine couplings. This is because of the inhomogeneous linewidth characterizing the multiline spectrum at all these frequencies; in other words, the spectral breadth, which is determined by the 55Mn hyperfine couplings, is such that all powder pattern orientations contribute significantly to all line positions across the spectral envelope in the resultant spectrum, with the exception of those on the extreme edges. Furthermore, the linewidth increases with increasing magnetic field due to strain effects, hampering further the analysis of the hyperfine structure. As a consequence, additional, independent spectroscopic data are required to correctly interpret the multiline spectrum. To this end, 55Mn ENDOR becomes the particular method of choice. More details are described in the following section.
5.2.2 The 55Mn ENDOR Experiment
The 55Mn ENDOR experiment for the S t = 1/2 OEC, or for that matter, for all Mn complexes, is a challenging experiment as the 55Mn-ENDOR spectrum for the OEC is expected to be spread over a large radio frequency range of ~100–300 MHz. The technology to perform such experiments was developed initially in 1994 in the Britt laboratory at the University of California, Davis at X-band [36, 133, 151, 152], and thereafter, in 1995, in the Lubitz laboratory at the Technische Universität, Berlin, and subsequently at the Max-Planck Institute for Bioinorganic Chemistry, Mülheim an der Ruhr, where an extension to higher frequency bands was accomplished [9, 53, 124, 127, 153–157].
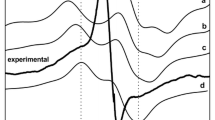
The Q-band 55Mn ENDOR spectrum of the S2 state multiline spectrum of spinach PS II, shown in Fig. 6a, basically resolves four peaks (with additional shoulders) spread over a range of 80–200 MHz. The line shape is virtually invariant when measured at different positions within the spectral envelope. As seen with the multiline spectrum, it also has poor spectral resolution. In contrast to the spectra of the dinuclear model complexes depicted in Fig. 6b, c, the eigenvalues of the individual 55Mn hyperfine matrices (A 1–A 4) are not distinctly resolved for the Mn4O5Ca cluster, but rather overlap each other, such that the turning points in the powder spectrum are not clearly identified. Nevertheless, this experiment yields an important result. As all the eigenvalues of the 55Mn hyperfine matrices are only spread over ~100 MHz, they all are approximately of the same magnitude (Table 1). This then requires all four Mn ions to carry significant electron spin, i.e. possess large spin projection coefficients: |ρ i | ≈ 1 to reproduce both the ENDOR and EPR spectral envelopes simultaneously. This same feature is not observed in mixed valence model complexes (Fig. 6b, c). Here, the two Mn ions carry significantly different spin projections (see Sect. 3.2 about MnIIIMnIV and MnIIMnIII dimers). In these systems, the eigenvalues of the two 55Mn hyperfine matrices (A 1, A 2) are spectrally resolved and thus can be determined by inspection (Fig. 6b, c). While this is not achievable for the OEC, simulations that reproduce satisfactorily both multi-frequency Q- and X-band EPR and ENDOR spectra can be considered to provide a unique solution for the effective spin Hamiltonian parameters (G and the eigenvalues of the 55Mn hyperfine matrices A i ), with the assumption that the principal directions of the G and A i matrices are collinear.

Q-band EPR (vertical) and 55Mn ENDOR (horizontal) spectra of the OEC and related model systems (left) and their atomic structures (right). a The S2 and S0 states of the OEC of in spinach PS II (4 % MeOH) [9]. b MnIII-(μ-O)2-MnIV (Bipy). c MnII-(μ-OH)-MnIII (PivOH) [109]. The black solid lines represent the data, the red dashed lines represent simulations based on the spin Hamiltonian formalism (see Eqs. 3, 14). In the structural models, Mn, Ca, O, N, C and H atoms are shown in purple, yellow, red, blue, black and white, respectively
The eigenvalues of the four 55Mn A i matrices can then be used to evaluate and develop electronic exchange coupling models (J coupling schemes), which define their individual spin projection matrices ρ i . The computed ρ i matrices map the effective A i matrices to the intrinsic (on-site) hyperfine coupling matrices a i according to the relation A i = ρ i a i . For a certain scheme to be valid, the calculated isotropic and anisotropic intrinsic values a iso and a aniso are required to match the known characteristic ranges of MnIII and MnIV ions (see Supplementary Tables S1–S3). For more detailed information, see [32, 37, 53, 123, 133]. In the case of the strongly coupled tetramanganese cluster, in which the electronic exchange coupling is the dominant interaction, the J coupling scheme then also determines the energy level spin ladder. Therefore, a further observable that relates a J coupling model to experimental data is the energy difference Δ between the ground spin state and the first excited spin state [32, 34]. Δ can be estimated from temperature-dependent EPR experiments on the signal of the ground spin state. One method is to measure the temperature dependence of the CW EPR signal intensity [86, 106, 160]. In case an Orbach process is the dominant spin–lattice relaxation mechanism, the temperature dependence of the T 1 relaxation time, which can be determined using the pulse EPR inversion recovery experiment, can be related to Δ [161, 162]. For the S t = 1/2 states of the OEC, the ground-to-first state energy difference has been found to be 3–37 cm−1 in S2 [53, 106, 108, 163, 164] and 22 to ≥30 cm−1 in S0 [90, 165], depending on sample treatment, such as the addition of MeOH or modifications at the Ca2+ site.
In the original work of Peloquin et al. [133], four fitted 55Mn hyperfine matrices, which nominally yield experimental spin projection coefficients (see Sect. 4), were used to develop J coupling schemes for the Mn4O5Ca cluster in the S2 state. They proposed a tetramer topology for the MnIII(MnIV)3 complex, the ‘3 + 1’ model, where three of the four Mn ions are strongly coupled together (J ≥ 50 cm−1), with the fourth, outer Mn ion being more weakly coupled (Fig. 7a), similar to previous theoretical evaluation of EPR and EXAFS data [166]. This ‘dangler’ model was subsequently refined by Charlot et al. [37] and Kulik et al. [9] who favored a Y coupling scheme, where three of the four Mn ions form a trimer unit, with the fourth Mn ion being strongly coupled to one Mn ion of the triangle (Fig. 7b). The position of the only MnIII ion of the S2 state within the Y-shaped core remained ambiguous. Two consistent coupling schemes were developed by Kulik et al. [9], in which the MnIII ion is assigned to either the monomeric MnA or MnC in the triangle. The position of the MnIII ion as part of the trimer unit is supported by the large 14N coupling from the coordinating histidine D1-His332 (see Sect. 5.3) and orientation-dependent EPR/55Mn ENDOR on PS II single crystals [167].

Models for the electronic exchange coupling of the Mn ions in the S2 state of the Mn4O5Ca cluster with pairwise J ij couplings given in cm−1. a The original 3 + 1/dangler model of Peloquin et al. [133]; b Y coupling scheme 3 by Kulik et al. [9]; c Pantazis model 1, [32] based on EXAFS core I [32, 140], and d Pantazis model 11, [32] based on the structure by Siegbahn [33, 158, 159]. In models a and b, the individual Mn spin centers are not assigned to specific Mn positions in a structural model of the OEC; thus, the nomenclature does not contain numbers from X-ray crystallographic structures. In models a and b, the only MnIII ion is MnC, whereas in models c and d, this is MnD1. Models a–c require MnA/A4 to be strongly coupled to MnB/B3. Models b–d require MnC/C2 to be strongly coupled to MnD/D1. The topologies of models a and b on the one hand, and models c and d on the other differ by their connectivity of MnA/A4 to the trimer unit
5.2.3 The Density Functional Approach
The advancement of DFT as a tool to reliably calculate magnetic resonance observables has fundamentally changed the development of OEC models. The interpretation of EPR/55Mn ENDOR data can now be achieved theoretically on the basis of detailed geometric models. First, an atomic structure of the OEC, e.g. based on X-ray crystallographic information, is proposed. BS-DFT then allows one to estimate the theoretical exchange pathways within the specific model of interest. The exchange coupling constants allow the spin projection coefficients and the hyperfine couplings to be calculated and compared against the EPR/55Mn ENDOR experiment. Thus, the EPR/55Mn ENDOR experiment now acts as a filter via which OEC structural candidates are differentiated.
DFT calculations of 55Mn hyperfine coupling constants that were performed by Pantazis et al. [32] for two groups of models of the OEC (Fig. 7c, d), the experimental, polarized EXAFS-based models [140] and the energy-minimized computational model of Siegbahn [33, 159], established for the first time relations between structural, magnetic and spectroscopic properties. Recent work by Cox et al. [53] has shown that only two models are satisfactory with regard to the calculated 55Mn hyperfine coupling constants, one generated from polarized EXAFS core I and the other being a model of Siegbahn, as pointed out in [32, 53]. Additional spin projection analysis indicated that the latter model [33] was the most consistent with all spectroscopic EPR/55Mn ENDOR data to date [53]. These findings agree with those of Schinzel et al. [135] for a similar J coupling scheme derived from this structure. Furthermore, the model has proven to be very robust in that it can be adapted to be valid for several variants of the OEC, such as originating from different species like cyanobacteria and higher plants [108], and explain modifications, such as addition of MeOH [108], substitution of Ca2+ for Sr2+ [53] or removal of Ca2+ [164]. These modifications could all be rationalized on the basis of a modulation of the electronic exchange coupling between MnA4 and the Mn ions of the MnB3MnC2MnD1 moiety and suggest that small molecules, such as small organic alcohols and NH3 [70, 73, 76, 102, 168], bind in the vicinity of the monomer-trimer junction. Similarly, as Ca2+ coordinates one of the oxo-bridges that connect MnA4 with the MnB3MnC2MnD1 moiety, its replacement by Sr2+ or its removal should also perturb the Mn monomer-trimer exchange interaction.
This basic picture of electronic structure perturbation of the OEC can be further extended to include the S > 1/2 spin-state configurations. In a very recent DFT study [35], Pantazis et al. identified a second low-energy conformation for the S2 state Mn4O5Ca cluster, besides the ‘open’ cuboid or fused twist structure. This second form represents a ‘proper’ Mn3O4Ca cuboid, with MnA4 attached as a dangling Mn via a mono-μ-oxo bridge being the unique MnIII ion, such that it resembles a revised model of the original ‘London’ structure [16] proposed by Barber and Murray [169]. It possesses a ground spin S t = 5/2 and supposedly gives rise to the g ≥ 4.1 signals, while the S t = 1/2 open cuboid conformation is the origin of the g ≈ 2 multiline signal.
5.3 Magnetic Coupling of Ligands (e.g. 1H, 13C, 14N, 17O) in the S2 State of the Mn4O5Ca Cluster
The EPR-spectroscopic investigation of the magnetic interaction between the Mn4O5Ca cluster and its ligands, comprising 1H, 13C, 14N, and 17O nuclei, is another experimental handle that has further helped in the development of electronic and structural models of the OEC and to locate potential substrate sites (Fig. 8). Here, the spin projection coefficient of a specific Mn ion can be estimated from the magnitude of ligand couplings to it when compared to corresponding coupling constants measured in model complexes or from DFT calculations.

Detection of various electron nuclear interactions of ligand nuclei with the Mn4O5Ca cluster in the S2 state (center, model 1d2′ from Ames et al. [34]) using different spectroscopic techniques. a 1H Davies ENDOR [131]; b 14N/17O ELDOR-detected NMR [131]; c 13C Mims ENDOR, adapted from [130]; d 14N 3-pulse ESEEM, adapted from [170]. Mn, Ca, N, O, C and H atoms are shown in purple, yellow, blue, red gray and white, respectively. Exchangeable O atoms are depicted in magenta, turquoise and green. For reasons of clarity, only water-derived H atoms are shown. Colored circles indicate contributions from individual nuclei to the respective spectra. Black and blue solid traces represent the data, red and orange solid traces and colored areas represent simulations based on the spin Hamiltonian formalism
This basic approach was applied for the imidazole 14N ligand of histidine D1-His332 to the MnD1 ion. In the S2 state, an effective 14N hyperfine coupling constant A iso ≈ 7 MHz was determined by ESEEM (Fig. 8d) [170, 171] and ELDOR-detected NMR measurements [131] and assigned to D1-His332 by mutagenesis experiments [170, 172]. It is considerably larger than those seen for MnIV-bound histidine ligands in dimeric MnIIIMnIV-complexes, such as in dimanganese-containing catalase (A iso = 2–3 MHz) [173], suggesting that MnD1 is not a MnIV but instead must represent the MnIII ion. The coupling for a nitrogen ligand to the MnIII ion is expected to be larger as it carries a considerably larger spin projection factor (|ρ| ≈ 1.6) as compared with those of the MnIV ions (|ρ| ≈1). Within this framework, the intrinsic hyperfine coupling constant a iso is of the order of 4–5 MHz, midway between on-site couplings seen for equatorial (2–4 MHz) and axial (6 MHz) MnIII ligands. This result compares well to 14N hyperfine and nuclear quadrupole coupling estimates from the BS-DFT studies of Schinzel et al. [135] and Ames et al. [34] for several models of the S2 state of the OEC. Thus, the D1-His332 14N electron nuclear interaction fixes the oxidation state of the MnD1 ion to be MnIII with a spin projection |ρ D1| > 1.5.
13C hyperfine spectroscopy on selectively labeled PS II mutants can identify the binding of amino acid residues to the tetramanganese core. Mims ENDOR experiments on PS II samples with a 13C-labeled C-terminal carboxylate carbon of the D1 protein, from alanine D1-Ala344, revealed an effective isotropic 13C hyperfine coupling constant A iso on the order of 1 MHz (Fig. 8c) [130]. By comparison to the 13C hyperfine coupling of the MnIII-(μ-O)2-MnIV Tacn complex, the COO− group of D1-Ala344 was assigned to be directly Mn-bound, to a MnIV ion carrying a spin projection |ρ| of 1. Taking into account the recent X-ray crystallographic (Fig. 2a) [20] and computational structures (Fig. 8) [32–34, 135], which show the carboxylate group to be ligated to MnC2 and Ca2+, requires MnC2 to have a |ρ C2| ≈ 1 and to be a MnIV ion. Thus, the D1-His332 14N and D1-Ala344 13C electron nuclear couplings both support the tetramer model for the electronic structure of the S2 state of the OEC as derived from 55Mn ENDOR and described in detail in Sect. 5.2 see (Fig. 7d) [32, 34, 53].
Substrate binding to the OEC can be probed using either the 1H (or 2H) or 17O isotopes. Analysis of the coupling model of 1H/2H nuclei to the Mn4O5Ca core is relatively straightforward as they represent more distant nuclei. Thus, their hyperfine interactions with the electronic spin of the tetramanganese cluster can be treated as being dominated by the through-space dipolar interaction, which can be calculated from geometric constraints (see Sect. 3.2.3). The 1H/2H couplings as inferred from CW and pulse (Mims, Davies) ENDOR (Fig. 8a), ESEEM or HYSCORE [68, 69, 76, 131, 174, 175] extend to 8 MHz (A ||). This coupling magnitude is similar to those characterizing terminal water/hydroxo ligands of MnIV ions observed in model complexes, including the dimanganese complexes in [124, 128]. This result can then be readily understood within the tetramer spin projection model for the electronic structure of the S2 state [32, 34, 53]. The largest 1H/2H hyperfine couplings observed represent the terminal H2O/OH− ligands (W1, W2) of the MnA4 ion (|ρ A4| ≈ 1), with a MnA4–H distance of 2.5–3 Å [131, 174, 175]. Protons from Ca2+-bound water (W3, W4) are expected to exhibit smaller couplings as these protons are 1–2 Å further away from any Mn spin center. The absence of any larger coupling precludes the presence of a protonated (μ-hydroxo) bridge in the S2 state [131].
The same approach can be used for the magnetic coupling of 17O nuclei. Recently, Rapatskiy et al. performed W-band ELDOR-detected NMR experiments on PS II (Fig. 8b) [131] and interpreted the measured spectra on the basis of the spin coupling model developed by Pantazis et al. [32] (see Fig. 7d). In 17O-exchanged PS II samples, three distinguishable signals originating from oxygen ligands were detected. Weakly (A iso ≈ 1 MHz) and intermediately (A iso ≈ 5 MHz) coupled 17O species were assigned to terminal H2O/OH− ligands of MnA4 (W1, W2) and of the Ca2+ ion (W3, W4). These results are also consistent with a fused twist core model [131] and the surrounding H2O/OH− ligands present in the structure by Siegbahn [33], which are similar to those in the crystal structure by Umena et al. [20]. Additionally, a strongly coupled 17O species with A iso ≈ 10 MHz was identified. By comparison with the μ-oxo bridged MnIII-(μ-O)2-MnIV BIPY complex (see Fig. 6b), it was assigned to one of the μ-oxo bridges within the cluster. As this putative bridge exchanges with the solvent water on a seconds time scale, it was proposed to serve as one of the two substrates. Based on a comparative analysis of the principal directions of the hyperfine matrix of this 17O and that of the 14N from D1-His332, it was concluded that, in a fused twist core, this μ-oxo bridge represents one of the two μ-oxo atoms bridging MnA4 and MnB3. This has immediate consequences for the possible mechanism of O–O bond formation in the higher oxidation states. According to these findings, the μ-oxo bridge either is attacked by a nucleophilic oxygen atom or undergoes a radical coupling with the another Mn-bound substrate. As proposed by Siegbahn [33], this second substrate would be a water molecule binding in its designated site, supposedly the open coordination site of MnD1, past the S2 state. Whether Mn- or Ca2+-bound, spatial considerations render O5 to be most likely the exchangeable substrate detected (Fig. 1). Unique properties of the O5 μ-oxo bridge are its specific coordination environment, created by direct ligation of the strong Lewis acid Ca2+, and its flexibility, as inferred by the structural equilibrium between being a μ-oxo bridge or a cuboidal corner [35]. These features may serve as the explanation for its unusually rapid solvent exchangeability, at rates much faster than those observed in μ-oxo bridged model complexes.
6 Concluding Remarks
This review article summarizes the necessary theoretical background and the relevant studies making use of EPR and related (hyperfine) spectroscopies to investigate the OEC and the water-splitting reaction. The basic spin physics approach employed to analyze EPR results was introduced, first in terms of manganese models and subsequently with regard to the Mn tetramer of the OEC proper. It is shown how these complicated systems can be treated within the spin Hamiltonian formalism as a coupled entity, with the help of spin projection factors. To obtain these factors, knowledge of a valid electronic model that reproduces the electronic exchange coupling topology effective in the particular system is crucial. State-of-the-art theoretical methodology, namely the BS-DFT formalism, which can be used to relate spectroscopic observables and structural properties, has been described. Through a combination of these techniques, the picture of the OEC in low oxidation states, especially S2 and S0, has been advanced over the past years to an electronic model consistent with an increasing amount of experimental data. These results have helped refine the X-ray crystallography model, which, although having provided an improved structure at near-atomic resolution, still suffers from reductive radiation damage. EPR-based techniques not only help to discriminate between proposed models and to improve these structures, they also provide a number of experiments to directly probe ligand interactions and the binding of substrate molecules. Knowledge of the substrates, their identities, positions and binding kinetics, is key to understand the mechanism of photosynthetic water oxidation. Further investigations of the higher oxidized S states of the Mn4O5Ca cluster, when the O–O bond is formed, will be crucial for its elucidation.
References
A. Magnuson, M. Anderlund, O. Johansson, P. Lindblad, R. Lomoth, T. Polivka, S. Ott, K. Stensjo, S. Styring, V. Sundstrom, L. Hammarstrom, Acc. Chem. Res. 42(12), 1899–1909 (2009)
I. McConnell, G.H. Li, G.W. Brudvig, Chem. Biol. 17(5), 434–447 (2010)
S. Styring, Faraday Discuss. 155, 357–376 (2012)
T. Wydrzynski, S. Satoh, Photosystem II: The Light-Driven Water: Plastoquinone Oxidoreductase (Springer, Dordrecht, 2005)
W. Lubitz, E.J. Reijerse, J. Messinger, Energy Environ. Sci. 1(1), 15–31 (2008)
P. Joliot, G. Barbieri, R. Chabaud, Photochem. Photobiol. 10(5), 309–329 (1969)
B. Kok, B. Forbush, M. Mcgloin, Photochem. Photobiol. 11(6), 457–475 (1970)
S. Styring, A.W. Rutherford, Biochemistry 26(9), 2401–2405 (1987)
L.V. Kulik, B. Epel, W. Lubitz, J. Messinger, J. Am. Chem. Soc. 129(44), 13421–13435 (2007)
G.M. Cheniae, I.F. Martin, Biochim. Biophys. Acta Bioenerg. 197(2), 219–239 (1970)
K. Sauer, Acc. Chem. Res. 13(8), 249–256 (1980)
C.F. Yocum, C.T. Yerkes, R.E. Blankenship, R.R. Sharp, G.T. Babcock, Proc. Natl. Acad. Sci. USA 78(12), 7507–7511 (1981)
V.K. Yachandra, K. Sauer, M.P. Klein, Chem. Rev. 96(7), 2927–2950 (1996)
A. Zouni, H.T. Witt, J. Kern, P. Fromme, N. Krauss, W. Saenger, P. Orth, Nature 409(6821), 739–743 (2001)
N. Kamiya, J.R. Shen, Proc. Natl. Acad. Sci. USA 100(1), 98–103 (2003)
K.N. Ferreira, T.M. Iverson, K. Maghlaoui, J. Barber, S. Iwata, Science 303(5665), 1831–1838 (2004)
G.W. Brudvig, R.H. Crabtree, Proc. Natl. Acad. Sci. USA 83(13), 4586–4588 (1986)
B. Loll, J. Kern, W. Saenger, A. Zouni, J. Biesiadka, Nature 438(7070), 1040–1044 (2005)
A. Guskov, J. Kern, A. Gabdulkhakov, M. Broser, A. Zouni, W. Saenger, Nat. Struct. Mol. Biol. 16(3), 334–342 (2009)
Y. Umena, K. Kawakami, J.-R. Shen, N. Kamiya, Nature 473(7345), 55–60 (2011)
J. Yano, J. Kern, K.D. Irrgang, M.J. Latimer, U. Bergmann, P. Glatzel, Y. Pushkar, J. Biesiadka, B. Loll, K. Sauer, J. Messinger, A. Zouni, V.K. Yachandra, Proc. Natl. Acad. Sci. USA 102(34), 12047–12052 (2005)
M. Grabolle, M. Haumann, C. Muller, P. Liebisch, H. Dau, J. Biol. Chem. 281(8), 4580–4588 (2006)
S. Petrie, P. Gatt, R. Stranger, R.J. Pace, Phys. Chem. Chem. Phys. 14(32), 11333–11343 (2012)
U. Bergmann, M.M. Grush, C.R. Horne, P. DeMarois, J.E. Penner-Hahn, C.F. Yocum, D.W. Wright, C.E. Dube, W.H. Armstrong, G. Christou, H.J. Eppley, S.P. Cramer, J. Phys. Chem. B 102(42), 8350–8352 (1998)
M. Haumann, C. Müller, P. Liebisch, L. Iuzzolino, J. Dittmer, M. Grabolle, T. Neisius, W. Meyer-Klaucke, H. Dau, Biochemistry 44(6), 1894–1908 (2005)
J. Messinger, J.H. Robblee, U. Bergmann, C. Fernandez, P. Glatzel, H. Visser, R.M. Cinco, K.L. McFarlane, E. Bellacchio, S.A. Pizarro, S.P. Cramer, K. Sauer, M.P. Klein, V.K. Yachandra, J. Am. Chem. Soc. 123(32), 7804–7820 (2001)
T.A. Roelofs, W. Liang, M.J. Latimer, R.M. Cinco, A. Rompel, J.C. Andrews, K. Sauer, V.K. Yachandra, M.P. Klein, Proc. Natl. Acad. Sci. USA 93(8), 3335–3340 (1996)
S. Luber, I. Rivalta, Y. Umena, K. Kawakami, J.R. Shen, N. Kamiya, G.W. Brudvig, V.S. Batista, Biochemistry 50(29), 6308–6311 (2011)
A. Grundmeier, H. Dau, Biochim. Biophys. Acta Bioenerg. 1817(1), 88–105 (2012)
A. Galstyan, A. Robertazzi, E.W. Knapp, J. Am. Chem. Soc. 134(17), 7442–7449 (2012)
M. Kusunoki, Biochim. Biophys. Acta Bioenerg. 1767(6), 484–492 (2007)
D.A. Pantazis, M. Orio, T. Petrenko, S. Zein, W. Lubitz, J. Messinger, F. Neese, Phys. Chem. Chem. Phys. 11(31), 6788–6798 (2009)
P.E.M. Siegbahn, Acc. Chem. Res. 42(12), 1871–1880 (2009)
W. Ames, D.A. Pantazis, V. Krewald, N. Cox, J. Messinger, W. Lubitz, F. Neese, J. Am. Chem. Soc. 133(49), 19743–19757 (2011)
D. Pantazis, W. Ames, N. Cox, W. Lubitz, F. Neese, Angew. Chem. Int. Ed. 51(39), 9935–9940 (2012)
J.M. Peloquin, K.A. Campbell, R.D. Britt, J. Am. Chem. Soc. 120(27), 6840–6841 (1998)
M.-F. Charlot, A. Boussac, G. Blondin, Biochim. Biophys. Acta Bioenerg. 1708(1), 120–132 (2005)
D.F. Ghanotakis, G.T. Babcock, C.F. Yocum, FEBS Lett. 167(1), 127–130 (1984)
T.-a. Ono, Y. Inoue, FEBS Lett. 227(2), 147–152 (1988)
M. Sivaraja, J. Tso, G.C. Dismukes, Biochemistry 28(24), 9459–9464 (1989)
A. Boussac, J.L. Zimmermann, A.W. Rutherford, Biochemistry 28(23), 8984–8989 (1989)
A. Boussac, J.-L. Zimmermann, A.W. Rutherford, FEBS Lett. 277(1–2), 69–74 (1990)
A.W. Rutherford, Trends Biochem. Sci. 14(6), 227–232 (1989)
V.L. Pecoraro, M.J. Baldwin, M.T. Caudle, W.-Y. Hsieh, N.A. Law, Pure Appl. Chem. 70(4), 925–929 (1998)
J. Limburg, V.A. Szalai, G.W. Brudvig, J. Chem. Soc. Dalton Trans. 9, 1353–1361 (1999)
J.S. Vrettos, J. Limburg, G.W. Brudvig, Biochim. Biophys. Acta Bioenerg. 1503(1–2), 229–245 (2001)
G. Hendry, T. Wydrzynski, Biochemistry 42(20), 6209–6217 (2003)
H.J. van Gorkom, C.F. Yocum, in Photosystem II: The Light-Driven Water: Plastoquinone Oxidoreductase, ed. by T. Wydrzynski, K. Satoh (Springer, Dordrecht, 2005), pp. 285–305
P.J. Riggs-Gelasco, R. Mei, D.F. Ghanotakis, C.F. Yocum, J.E. Penner-Hahn, J. Am. Chem. Soc. 118(10), 2400–2410 (1996)
W. Junge, M. Haumann, Biochim. Biophys. Acta Bioenerg. 1411(1), 121–133 (1999)
A. Boussac, A.W. Rutherford, Biochemistry 27(9), 3476–3483 (1988)
A. Boussac, F. Rappaport, P. Carrier, J.M. Verbavatz, R. Gobin, D. Kirilovsky, A.W. Rutherford, M. Sugiura, J. Biol. Chem. 279(22), 22809–22819 (2004)
N. Cox, L. Rapatskiy, J.-H. Su, D.A. Pantazis, M. Sugiura, L. Kulik, P. Dorlet, A.W. Rutherford, F. Neese, A. Boussac, W. Lubitz, J. Messinger, J. Am. Chem. Soc. 133(10), 3635–3648 (2011)
J.S. Vrettos, D.A. Stone, G.W. Brudvig, Biochemistry 40(26), 7937–7945 (2001)
C.F. Yocum, in Manganese Redox Enzymes, ed. by V.L. Pecoraro (VCH Publishers, New York, 1992), pp. 71–84
P.H. Homann, Photosynth. Res. 73(1–3), 169–175 (2002)
S. Izawa, R.L. Heath, G. Hind, Biochim. Biophys. Acta Bioenerg. 180(2), 388–398 (1969)
H. Wincencjusz, H.J. van Gorkom, C.F. Yocum, Biochemistry 36(12), 3663–3670 (1997)
H. Wincencjusz, C.F. Yocum, H.J. van Gorkom, Biochemistry 37(23), 8595–8604 (1998)
K. Hasegawa, Y. Kimura, T.A. Ono, Biochemistry 41(46), 13839–13850 (2002)
K. Hasegawa, Y. Kimura, T. Ono, Biophys. J. 86(2), 1042–1050 (2004)
M. Baumgarten, J.S. Philo, G.C. Dismukes, Biochemistry 29(48), 10814–10822 (1990)
A. Boussac, P. Setif, A.W. Rutherford, Biochemistry 31(4), 1224–1234 (1992)
N. Lydakis-Simantiris, P. Dorlet, D.F. Ghanotakis, G.T. Babcock, Biochemistry 37(18), 6427–6435 (1998)
K. Olesen, L.E. Andreasson, Biochemistry 42(7), 2025–2035 (2003)
R. Pokhrel, I.L. McConnell, G.W. Brudvig, Biochemistry 50(14), 2725–2734 (2011)
I. Rivalta, M. Amin, S. Luber, S. Vassiliev, R. Pokhrel, Y. Umena, K. Kawakami, J.R. Shen, N. Kamiya, D. Bruce, G.W. Brudvig, M.R. Gunner, V.S. Batista, Biochemistry 50(29), 6312–6315 (2011)
A. Kawamori, T. Inui, T. Ono, Y. Inoue, FEBS Lett. 254(1–2), 219–224 (1989)
R. Fiege, W. Zweygart, R. Bittl, N. Adir, G. Renger, W. Lubitz, Photosynth. Res. 48(1–2), 227–237 (1996)
R.D. Britt, K.A. Campbell, J.M. Peloquin, M.L. Gilchrist, C.P. Aznar, M.M. Dicus, J. Robblee, J. Messinger, Biochim. Biophys. Acta Bioenerg. 1655, 158–171 (2004)
W.F. Beck, G.W. Brudvig, Biochemistry 25(21), 6479–6486 (1986)
W.F. Beck, J.C. De Paula, G.W. Brudvig, J. Am. Chem. Soc. 108(14), 4018–4022 (1986)
D.A. Force, D.W. Randall, G.A. Lorigan, K.L. Clemens, R.D. Britt, J. Am. Chem. Soc. 120(51), 13321–13333 (1998)
R.D. Britt, J.M. Peloquin, K.A. Campbell, Annu. Rev. Biophy. Biomol. Struct. 29, 463–495 (2000)
M.C.W. Evans, R.J. Ball, J.H.A. Nugent, FEBS Lett. 579(14), 3081–3084 (2005)
K.A. Åhrling, M.C.W. Evans, J.H.A. Nugent, R.J. Ball, R.J. Pace, Biochemistry 45(23), 7069–7082 (2006)
A. Cua, D.H. Stewart, M.J. Reifler, G.W. Brudvig, D.F. Bocian, J. Am. Chem. Soc. 122(9), 2069–2077 (2000)
T. Noguchi, M. Sugiura, Biochemistry 39(36), 10943–10949 (2000)
T. Noguchi, M. Sugiura, Biochemistry 41(52), 15706–15712 (2002)
Y. Kimura, A. Ishii, T. Yamanari, T.A. Ono, Biochemistry 44(21), 7613–7622 (2005)
K. Burda, K.P. Bader, G.H. Schmid, FEBS Lett. 491(1–2), 81–84 (2001)
K. Burda, K.P. Bader, G.H. Schmid, Biochim. Biophys. Acta Bioenerg. 1557, 77–82 (2003)
G.C. Dismukes, Y. Siderer, Proc. Natl. Acad. Sci. USA 78(1), 274–278 (1981)
S.L. Dexhemier, M.P. Klein, J. Am. Chem. Soc. 114(8), 2821–2826 (1992)
H. Levanon, K. Möbius, Annu. Rev. Biophy. Biomol. Struct. 26, 495–540 (1997)
T. Yamauchi, H. Mino, T. Matsukawa, A. Kawamori, T. Ono, Biochemistry 36(24), 7520–7526 (1997)
J. Messinger, J.H.A. Nugent, M.C.W. Evans, Biochemistry 36(37), 11055–11060 (1997)
J. Messinger, J.H. Robblee, W.O. Yu, K. Sauer, V.K. Yachandra, M.P. Klein, J. Am. Chem. Soc. 119(46), 11349–11350 (1997)
K.A. Campbell, J.M. Peloquin, D.P. Pham, R.J. Debus, R.D. Britt, J. Am. Chem. Soc. 120(2), 447–448 (1998)
K.A. Åhrling, S. Peterson, S. Styring, Biochemistry 37(22), 8115–8120 (1998)
T. Matsukawa, H. Mino, D. Yoneda, A. Kawamori, Biochemistry 38(13), 4072–4077 (1999)
K.A. Campbell, D.A. Force, P.J. Nixon, F. Dole, B.A. Diner, R.D. Britt, J. Am. Chem. Soc. 122(15), 3754–3761 (2000)
J.P. McEvoy, G.W. Brudvig, Chem. Rev. 106(11), 4455–4483 (2006)
A. Haddy, Photosynth. Res. 92(3), 357–368 (2007)
A. Boussac, M. Sugiura, A.W. Rutherford, P. Dorlet, J. Am. Chem. Soc. 131(14), 5050–5051 (2009)
A. Boussac, J.J. Girerd, A.W. Rutherford, Biochemistry 35(22), 6984–6989 (1996)
A. Boussac, S. Un, O. Horner, A.W. Rutherford, Biochemistry 37(12), 4001–4007 (1998)
O. Horner, E. Rivière, G. Blondin, S. Un, A.W. Rutherford, J.-J. Girerd, A. Boussac, J. Am. Chem. Soc. 120(31), 7924–7928 (1998)
A. Boussac, A.W. Rutherford, Biochim. Biophys. Acta Bioenerg. 1457(3), 145–156 (2000)
A. Haddy, K.V. Lakshmi, G.W. Brudvig, H.A. Frank, Biophys. J. 87(4), 2885–2896 (2004)
A. Boussac, H. Kuhl, S. Un, M. Rogner, A.W. Rutherford, Biochemistry 37(25), 8995–9000 (1998)
A. Boussac, M. Sugiura, Y. Inoue, A.W. Rutherford, Biochemistry 39(45), 13788–13799 (2000)
J.L. Zimmermann, A.W. Rutherford, Biochemistry 25(16), 4609–4615 (1986)
Z. Deák, S. Peterson, P. Geijer, K.A. Åhrling, S. Styring, Biochim. Biophys. Acta Bioenerg. 1412(3), 240–249 (1999)
P. Geijer, S. Peterson, K.A. Åhrling, Z. Deák, S. Styring, Biochim. Biophys. Acta Bioenerg. 1503(1–2), 83–95 (2001)
R.J. Pace, P.J. Smith, R. Bramley, D. Stehlik, Biochim. Biophys. Acta Bioenerg. 1058(2), 161–170 (1991)
A. Boussac, J. Biol. Inorg. Chem. 2(5), 580–585 (1997)
J.H. Su, N. Cox, W. Ames, D.A. Pantazis, L. Rapatskiy, T. Lohmiller, L.V. Kulik, P. Dorlet, A.W. Rutherford, F. Neese, A. Boussac, W. Lubitz, J. Messinger, Biochim. Biophys. Acta Bioenerg. 1807(7), 829–840 (2011)
N. Cox, W. Ames, B. Epel, L.V. Kulik, L. Rapatskiy, F. Neese, J. Messinger, K. Wieghardt, W. Lubitz, Inorg. Chem. 50(17), 8238–8251 (2011)
K.O. Schäfer, Exchange Coupled Manganese Complexes: Model Systems for the Active Centres of Redoxproteins Investigated with EPR Techniques. Doctoral thesis, Technische Universität Berlin, 2002
K.A. Åhrling, S. Peterson, S. Styring, Biochemistry 36(43), 13148–13152 (1997)
A. Boussac, H. Kuhl, E. Ghibaudi, M. Rogner, A.W. Rutherford, Biochemistry 38(37), 11942–11948 (1999)
J.H.A. Nugent, S. Turconi, M.C.W. Evans, Biochemistry 36(23), 7086–7096 (1997)
J.H.A. Nugent, I.P. Muhiuddin, M.C.W. Evans, Biochemistry 41(12), 4117–4126 (2002)
D. Koulougliotis, J.R. Shen, N. Ioannidis, V. Petrouleas, Biochemistry 42(10), 3045–3053 (2003)
D. Koulougliotis, C. Teutloff, Y. Sanakis, W. Lubitz, V. Petrouleas, Phys. Chem. Chem. Phys. 6(20), 4859–4863 (2004)
V. Petrouleas, D. Koulougliotis, N. Ioannidis, Biochemistry 44(18), 6723–6728 (2005)
J.H. Su, K.G.V. Havelius, F. Mamedov, F.M. Ho, S. Styring, Biochemistry 45(24), 7617–7627 (2006)
K.G.V. Havelius, J.H. Su, Y. Feyziyev, F. Mamedov, S. Styring, Biochemistry 45(30), 9279–9290 (2006)
N. Cox, F.M. Ho, N. Pewnim, R. Steffen, P.J. Smith, K.G. Havelius, J.L. Hughes, L. Debono, S. Styring, E. Krausz, R.J. Pace, Biochim. Biophys. Acta Bioenerg. 1787(7), 882–889 (2009)
S.K. Misra, in Handbook of Electron Spin Resonance, ed. by C.P. Poole, H.A. Farach (Springer, New York, 1999), pp. 115–150
D.P. Goldberg, J. Telser, J. Krzystek, A.G. Montalban, L.-C. Brunel, A.G.M. Barrett, B.M. Hoffman, J. Am. Chem. Soc. 119(37), 8722–8723 (1997)
A. Bencini, D. Gatteschi, EPR of Exchange Coupled Systems (Springer-Verlag, Berlin, 1990)
K.O. Schäfer, R. Bittl, W. Zweygart, F. Lendzian, G. Haselhorst, T. Weyhermuller, K. Wieghardt, W. Lubitz, J. Am. Chem. Soc. 120(50), 13104–13120 (1998)
J.T. Sage, Y.M. Xia, P.G. Debrunner, D.T. Keough, J. Dejersey, B. Zerner, J. Am. Chem. Soc. 111(18), 7239–7247 (1989)
M. Zheng, S.V. Khangulov, G.C. Dismukes, V.V. Barynin, Inorg. Chem. 33(2), 382–387 (1994)
W. Zweygart, PhD Thesis, Technische Universität Berlin, 1995
D.W. Randall, A. Gelasco, M.T. Caudle, V.L. Pecoraro, R.D. Britt, J. Am. Chem. Soc. 119(19), 4481–4491 (1997)
O.M. Usov, V.M. Grigoryants, R. Tagore, G.W. Brudvig, C.P. Scholes, J. Am. Chem. Soc. 129(39), 11886–11887 (2007)
J.A. Stull, T.A. Stich, R.J. Service, R.J. Debus, S.K. Mandal, W.H. Armstrong, R.D. Britt, J. Am. Chem. Soc. 132(2), 446–447 (2010)
L. Rapatskiy, N. Cox, W. Ames, A. Savitsky, J. Sander, M. Nowaczyk, M. Rögner, A. Boussac, F. Neese, J. Messinger, W. Lubitz, J. Am. Chem. Soc. 134(40), 16619–16634 (2013)
M. Zheng, G.C. Dismukes, Inorg. Chem. 35(11), 3307–3319 (1996)
J.M. Peloquin, K.A. Campbell, D.W. Randall, M.A. Evanchik, V.L. Pecoraro, W.H. Armstrong, R.D. Britt, J. Am. Chem. Soc. 122(44), 10926–10942 (2000)
D.A. Pantazis, M. Orio, T. Petrenko, S. Zein, E. Bill, W. Lubitz, J. Messinger, F. Neese, Chem. Eur. J. 15(20), 5108–5123 (2009)
S. Schinzel, J. Schraut, A. Arbuznikov, P. Siegbahn, M. Kaupp, Chem. Eur. J. 16(34), 10424–10438 (2010)
S. Mukherjee, J.A. Stull, J. Yano, T.C. Stamatatos, K. Pringouri, T.A. Stich, K.A. Abboud, R.D. Britt, V.K. Yachandra, G. Christou, Proc. Natl. Acad. Sci. USA 109(7), 2257–2262 (2012)
G.C. Dismukes, Y. Siderer, FEBS Lett. 121(1), 78–80 (1980)
R. Radmer, G. Cheniae, in Primary Processes of Photosynthesis, ed. by J. Barber (Elsevier, Amsterdam, 1977), pp. 303–349
S.K. Misra, in Advanced EPR Applied to Biosciences, Satellite Symposium B in APES’01, ed. by A. Kawamori (Kwansei Gakuin University, 2002), pp. 47–64
J. Yano, J. Kern, K. Sauer, M.J. Latimer, Y. Pushkar, J. Biesiadka, B. Loll, W. Saenger, J. Messinger, A. Zouni, V.K. Yachandra, Science 314(5800), 821–825 (2006)
Y. Pushkar, J. Yano, P. Glatzel, J. Messinger, A. Lewis, K. Sauer, U. Bergmann, V.K. Yachandra, J. Biol. Chem. 282(10), 7198–7208 (2007)
Y.L. Pushkar, J. Yano, K. Sauer, A. Boussac, V.K. Yachandra, Proc. Natl. Acad. Sci. USA 105(6), 1879–1884 (2008)
T. Noguchi, Coord. Chem. Rev. 252(3–4), 336–346 (2008)
F. Neese, Coord. Chem. Rev. 253(5–6), 526–563 (2009)
L. Noodleman, J. Chem. Phys. 74, 5737–5743 (1981)
L. Noodleman, E.R. Davidson, Chem. Phys. 109(1), 131–143 (1986)
L. Noodleman, D.A. Case, Adv. Inorg. Chem. 38, 423–470 (1992)
S. Sinnecker, F. Neese, L. Noodleman, W. Lubitz, J. Am. Chem. Soc. 126(8), 2613–2622 (2004)
M. Orio, D.A. Pantazis, T. Petrenko, F. Neese, Inorg. Chem. 48(15), 7251–7260 (2009)
C. Baffert, M. Orio, D.A. Pantazis, C. Duboc, A.G. Blackman, G. Blondin, F. Neese, A. Deronzier, M.-N. Collomb, Inorg. Chem. 48(21), 10281–10288 (2009)
B.E. Sturgeon, J.A. Ball, D.W. Randall, R.D. Britt, J. Phys. Chem. 98(49), 12871–12883 (1994)
D.W. Randall, B.E. Sturgeon, J.A. Ball, G.A. Lorigan, M.K. Chan, M.P. Klein, W.H. Armstrong, R.D. Britt, J. Am. Chem. Soc. 117(47), 11780–11789 (1995)
W. Zweygart, R. Bittl, K. Wieghardt, W. Lubitz, Chem. Phys. Lett. 261(3), 272–276 (1996)
K.O. Schafer, R. Bittl, F. Lendzian, V. Barynin, T. Weyhermuller, K. Wieghardt, W. Lubitz, J. Phys. Chem. B 107(5), 1242–1250 (2003)
L.V. Kulik, B. Epel, W. Lubitz, J. Messinger, J. Am. Chem. Soc. 127(8), 2392–2393 (2005)
L. Kulik, B. Epel, J. Messinger, W. Lubitz, Photosynth. Res. 84(1–3), 347–353 (2005)
C. Teutloff, K.O. Schafer, S. Sinnecker, V. Barynin, R. Bittl, K. Wieghardt, F. Lendzian, W. Lubitz, Magn. Reson. Chem 43(S1), S51–S64 (2005)
P.E.M. Siegbahn, Chem. Eur. J. 14(27), 8290–8302 (2008)
P.E.M. Siegbahn, J. Am. Chem. Soc. 131(51), 18238–18239 (2009)
R.D. Britt, G.A. Lorigan, K. Sauer, M.P. Klein, J.L. Zimmermann, Biochim. Biophys. Acta Bioenerg. 1140(1), 95–101 (1992)
R. Orbach, Proc. R. Soc. London A 264(1319), 458–484 (1961)
A. Schweiger, G. Jeschke, Principles of Pulse Electron Paramagnetic Resonance (Oxford University Press, Oxford, 2001), p. 225
G.A. Lorigan, R.D. Britt, Biochemistry 33(40), 12072–12076 (1994)
T. Lohmiller, N. Cox, J.H. Su, J. Messinger, W. Lubitz, J. Biol. Chem. 287(29), 24721–24733 (2012)
L.V. Kulik, W. Lubitz, J. Messinger, Biochemistry 44(26), 9368–9374 (2005)
K. Hasegawa, T. Ono, Y. Inoue, M. Kusunoki, Bull. Chem. Soc. Jpn. 72(5), 1013–1023 (1999)
C. Teutloff, S. Pudollek, S. Kessen, M. Broser, A. Zouni, R. Bittl, Phys. Chem. Chem. Phys. 11(31), 6715–6726 (2009)
R.D. Britt, J.L. Zimmermann, K. Sauer, M.P. Klein, J. Am. Chem. Soc. 111(10), 3522–3532 (1989)
J. Barber, J.W. Murray, Phil. Trans. R. Soc. B 363(1494), 1129–1137 (2008)
T.A. Stich, G.J. Yeagle, R.J. Service, R.J. Debus, R.D. Britt, Biochemistry 50(34), 7390–7404 (2011)
G.J. Yeagle, M.L. Gilchrist, R.M. McCarrick, R.D. Britt, Inorg. Chem. 47(6), 1803–1814 (2008)
R.J. Debus, K.A. Campbell, W. Gregor, Z.L. Li, R.L. Burnap, R.D. Britt, Biochemistry 40(12), 3690–3699 (2001)
T.A. Stich, J.W. Whittaker, R.D. Britt, J. Phys. Chem. B 114(45), 14178–14188 (2010)
C.P. Aznar, R.D. Britt, Phil. Trans. R. Soc. B 357(1426), 1359–1365 (2002)
S. Milikisiyants, R. Chatterjee, C.S. Coates, F.H.M. Koua, J.R. Shen, K.V. Lakshmi, Energy Environ. Sci. 5(7), 7747–7756 (2012)
Acknowledgments
S. K. M. is grateful to the Natural Sciences and Engineering Research Council (NSERC) of Canada for partial financial support and to Professor Wolfgang Lubitz for inviting him to spend 3 months of his sabbatical leave at the Max Planck Institute in Mülheim an der Ruhr, Germany. T. L. is supported by the Federal Ministry of Education and Research of Germany (BMBF) in the framework of the Bio-H2 project (03SF0355C).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Lohmiller, T., Ames, W., Lubitz, W. et al. EPR Spectroscopy and the Electronic Structure of the Oxygen-Evolving Complex of Photosystem II. Appl Magn Reson 44, 691–720 (2013). https://doi.org/10.1007/s00723-012-0437-3
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-012-0437-3