
Abstract
Withania somnifera is one of the most important medicinal plant and is credited with various pharmacological activities. In this study, in vitro multiple shoot cultures were exposed to different concentrations (5–300 μM) of cadmium (Cd) as cadmium sulphate to explore its ability to accumulate the heavy metal ion and its impact on the metabolic status and adaptive responses. The results showed that supplemental exposure to Cd interfered with N, P, and K uptake creating N, P, and K deficiency at higher doses of Cd that also caused stunting of growth, chlorosis, and necrosis. The study showed that in vitro shoots could markedly accumulate Cd in a concentration-dependent manner. Enzymatic activities and isozymic pattern of catalase, ascorbate peroxidase, guaiacol peroxidase, peroxidase, glutathione-S-transferase, glutathione peroxidase, monodehydroascorbate reductase, and dehydroascorbate reductase were altered substantially under Cd exposure. Sugar metabolism was also markedly modulated under Cd stress. Various other parameters including contents of photosynthetic pigments, phenolics, tocopherol, flavonoids, reduced glutathione, nonprotein thiol, ascorbate, and proline displayed major inductive responses reflecting their protective role. The results showed that interplay of enzymatic as well as nonenzymatic responses constituted a system endeavor of tolerance of Cd accumulation and an efficient scavenging strategy of its stress implications.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Plants are affected by different kinds of biotic and abiotic stresses such as drought, salinity, and metal toxicity in their natural environment. Heavy metals occur naturally in soil as rare elements, which poses a serious threat to the environment when present in higher levels by adversely affecting growth and productivity of plants and human health (Kaur et al. 2012). Cadmium (Cd) is one of the most toxic heavy metals for human and plants, entering into the environment mainly through phosphate fertilizers, industrial processes, and farming practices. It has been ranked number seven among the top 20 toxins (Mahmood et al. 2009; Gill and Tuteja 2011). Its absorption coefficient is higher as compared to with other heavy metals like zinc and copper. Absorption of Cd depends on the Cd concentration in soil, the proportion of Cd available as Cd2+, and uptake and sequestering ability of the plants. Cadmium is toxic for most of the plants at concentrations greater than 5–10 μg g−1 dry weight, except for Cd-hyper accumulators which can tolerate even Cd concentration of 100 μg g−1 dry weight (Gill and Tuteja 2011).
Plants possess natural ability to take up non-essential metals, including heavy metals such as Cd. The uptake of Cd triggers a set of complex changes in plant growth attributes as well as modulations at biochemical and physiological levels (Namdjoyan et al. 2012). The toxicity of Cd causes stunted growth, chlorosis, necrosis, inhibition of uptake of many essential elements, inhibition of certain enzymes activities, decrease in water potential, alterations in membrane permeability, and generation of reactive oxygen species (ROS) within subcellular compartments (Mahmood et al. 2009; Nedjimi and Daoud 2009; Beltagi et al. 2010). It is reported in literature that Cd interferes in the net performance of many biochemical and physiological processes like photosynthesis, respiration, protein metabolism, nutrient uptake, and transport (Gill and Tuteja 2011).
To deal with the stress and induced damages by ROS, plants have evolved a well-organized antioxidative enzymatic mechanism involving catalytic activities of antioxidant enzymes to scavenge the ROS and thereby eliminate or reduce their damaging effects (Khatun et al. 2008; Jaleel et al. 2009; Kaur et al. 2012; Sabir et al. 2012). Among these antioxidative enzymes, superoxide dismutase (SOD) constitutes the primary step of cellular defense and dismutates O2 ·− to H2O2 and O2 (Mittler et al. 2004). Peroxidase (POD) decomposes H2O2 by oxidation of co-substrates such as phenolic compounds and/or antioxidants, whereas catalase (CAT) breaks down H2O2 into water and molecular oxygen. Glutathione reductase (GR) catalyzes the NADPH-dependent reduction of oxidized glutathione (GSSG) to the reduced glutathione (GSH). Glutathione-S-transferase (GSTs) has the ability to metabolize a wide range of toxic exogenous compounds via GSH conjugation (Zeng et al. 2009). Glutathione peroxidase (GPX) has an important role in the reduction of lipid peroxides and other hydroperoxide by GSH (Kramer 2010). Thus, a concerted and balanced action of these enzymes is entailed to optimally functionalize their scavenging systems and mechanisms in line with the extent and nature of ROS generated. The responsiveness and efficiency of the scavenging system of plants governs the performance, endurance, and survival capability of plants under stress conditions (Metwally et al. 2005; Mahmood et al. 2009).
Withania somnifera (Ashwagandha) is one of the most valued medicinal plants of Ayurveda and other traditional systems of medicine in India, and is still being commonly used in Indian traditional health care systems. Indeed, W. somnifera has recently gained a lot more attention in research due to emerging strong evidences from diverse research groups across the world in favor of its preventive/protective and curative role in many clinical problems including neurodegeneration and improving interneuron signal communications by repair of synapses between nerve cells (Sangwan et al. 2004). Different parts of W. somnifera are used in Ayurvedic and Unani systems of medicine. Leaves of W. somnifera are used for the treatment of tumor and tubercular glands and also used as antihelminthic (Tuli et al. 2009). Roots of the plant are used in constipation, senile debility, rheumatism, debility, nervous exhaustion, loss of memory, loss of muscular energy, spermatorrhea, syphilis, rheumatic fever etc. (Khan et al. 2007; Tuli et al. 2009). The medicinal properties of W. somnifera have been attributed to the presence/abundance of specialized secondary metabolites mainly of alkaloid and ergostane skeleton steroidal (withanolide) nature (Sangwan et al. 2004, 2007, 2008).
Some earlier reports including ours on in vitro cultures have shown the ability of multiple shoot cultures to grow under diverse and difficult conditions (Khatun et al. 2008; Sabir et al. 2012, 2013). Therefore, objectives of the present study are to explore (1) if the exposure of heavy metals like Cd in the growing medium can influence the accumulation of Cd in W. somnifera, (2) types of various ROS generated under this heavy metal stress, and (3) the interplay of various cellular systems in protecting the tissue against Cd-induced damages. To achieve these objectives and to evaluate the net impact of Cd stress, in vitro shoots of W. somnifera were used as an experimental material. It was aimed at deciphering the modulations in profiles of catalytic levels of antioxidant enzymes under controlled Cd stress and possible scavenging mechanism engaged in dealing with the adversity of Cd toxicity in W. somnifera. The outcome of the study has a bearing on knowing the ability/susceptibility of W. somnifera to accumulate heavy metals. The subject has its obvious relevance in view of the use of the plant and processes for its various preparations and biomolecules for health gains. The study shows, for the first time, that multiple shoot cultures of W. somnifera are capable of accumulating high amounts of Cd when exposed to exogenous levels of Cd in the medium. The study was performed under in vitro conditions to monitor the growth and uptake in a time-bound and controlled manner. The leads of the study not only provide important insights and cues to the metabolic efficiency of the plant to endure performance and survival of the plant under a heavy load of metal stress but also invoke the forthcoming studies needed to develop evaluative package of practices for the herb stipulating the impact of cultivating it in heavy metal-contaminated environments like industrial vicinity. The study also demonstrates various intrinsic responses that occur in shoots as a system in response to heavy metal accumulation for future prospects of its comparison with the plants grown under field conditions. To our knowledge, this is the first report where W. somnifera shoot cultures have been shown to accumulate Cd and exhibit various metabolic and metabolite responses associated with the Cd accumulation.
Materials and methods
Materials
In vitro shoot cultures of W. somnifera var NMITLI 118 were generated and maintained in Murashige and Skoog (MS) medium (Murashige and Skoog 1962) supplemented with 30 g l−1 sucrose, 8 g l−1 agar, 1 mg l−1 BAP and 0.5 mg l−1 kinetin, and pH was adjusted to 5.8, as reported earlier (Sabir et al. 2012). Shoots of uniform size were selected for Cd treatment. All other chemicals and reagents used for biochemical estimation and enzymatic assays were purchased from Sigma (St. Louis, MO).
Treatment with Cd and growth conditions
Shoot cultures were transferred to MS medium supplemented with different concentrations of cadmium sulphate, mild Cd concentration (5, 10, 20 μM), moderate Cd concentration (50, 100 μM), toxic Cd concentration (150, 200, 300 μM), and lethal Cd concentration (500, 750, 1,000 μM). For biochemical analysis, shoots treated with mild to toxic Cd concentrations including control shoots were taken. Lethal Cd concentration-treated shoots were not taken for various biochemical analyses because shoots were unable to grow. Cultures were incubated at 24 ± 2 °C at 100 μmol m−2 s−1 with white fluorescent light illumination for 16-h light and 8-h dark period. After 30 days of incubation on Cd-supplemented media, shoots were harvested, either directly used or frozen in liquid nitrogen and stored at −80 °C for various analyses.
Determination of Cd, nitrogen (N), phosphorus (P), and potassium (K) contents
For determining the amount of Cd accumulated by W. somnifera, shoots were dried in an oven at 65 °C for 3 days until constant weight. Dried samples were ground to fine powder in pestle mortar and digested using HNO3/HClO4 (3:2) diacid digestion. Digested samples were dissolved in triple distilled water (TDW), and Cd content was measured through ICP–atomic absorption spectrometry (Perkin–Elmer). The uptake of N was measured by Kjeldahl method, and P uptake was estimated using vanedomolybdophosphoric acid (yellow color) method by flow-injection analysis. The uptake of K was estimated using flame photometer after diacid digestion of the samples.
Measurement of growth and estimation of total photosynthetic pigment contents
Growth parameters such as fresh weight (FW) and dry weight (DW) were measured after 30 days of treatment. For DW analysis, shoots were kept in an oven at 65 °C until constant weight. Chlorophyll a (chl-a), chlorophyll b (chl-b), carotenoid, and total chlorophyll were extracted and estimated from fresh leaves according to method of Hiscox and Israelstam (1979) and calculated by Arnon’s formula.
Estimation of lipid peroxidation
Lipid peroxidation was determined by the measurement of total TBARS content expressed as equivalents of malondialdehyde (MDA; a byproduct of lipid peroxidation) using 2-thiobarbituric acid (TBA) (Heath and Packer 1968). Frozen samples were taken and homogenized with two volumes of ice-cold 0.1 % (w/v) trichloroacetic acid (TCA), and homogenate was centrifuged at 12,000g for 15 min at 4 °C. Assay mixture containing 0.5 ml of aliquot and 1.5 ml of 0.5 % (w/v) TBA dissolved in 20 % (w/v) TCA was heated to 95 °C for 35 min and then rapidly cooled on an ice bath to terminate the reaction. After centrifugation (12,000g for 15 min at 4 °C), absorbance (532 nm) was measured and values corresponding to nonspecific absorption (600 nm) were subtracted. The total MDA content was calculated using the extinction coefficient of 155 mM−1 cm −1 and expressed as nanomole MDA per gram DW.
Estimation of proline content
Proline accumulation was estimated colorimetrically based on proline’s reaction with ninhydrin using l-proline as standard (Bates et al. 1973). Tissue sample (0.5 g) was homogenized with 2 ml of 3 % sulfosalicylic acid. Homogenate was centrifuged at 10,000 rpm for 20 min, and supernatant was taken and added in a 1:1:1 ratio with ninhydrin acid and glacial acetic acid and boiled for 1 h and immediately kept on ice to terminate the reaction. The developed color was extracted with 4 ml of toluene, and absorbance was measured at 520 nm.
Estimation of total phenolic content
The total phenolics of tissues was measured by using the Folin–Ciocalteu reagent using gallic acid as standard (Khatun et al. 2008). Tissue was homogenized in 90 % ethanol and centrifuged at 10,000 rpm for 15 min. A 0.5 ml of aliquot was added to ten-times diluted Folin–Ciocalteu reagent. Assay mixture was shaken and incubated for 6 min at room temperature. After incubation, 1.25 ml of 7 % Na2CO3 solution was added and mixture was diluted with TDW to a final volume of 3 ml and mixed thoroughly. Absorbance was measured at 760 nm after incubation of 90 min at 23 °C.
Estimation of total ascorbate (AsA + DHA), AsA, and DHA
For total ascorbate content analysis, tissue was homogenized in 5 % TCA and centrifuged for 20 min at 12,000g at 4 °C. Supernatant was collected and used for analysis. Total AsA content was determined using 1 ml assay mixture containing 0.2 ml extract, 0.05 M potassium phosphate buffer (KPB; pH 7.4), 3 mM ethylenediaminetetraacetic acid (EDTA), and 1 mM dithiothreitol (DTT), and was incubated for 10 min at 25 °C. After incubation, 0.1 ml of N-ethylmaleimide (NEM), 0.4 ml of 0.61 M TCA, 0.4 ml of 0.8 M orthophosphoric acid, and 0.4 ml of α,α′-bipyridyl were added. A 0.2 ml of FeCl3 was added to assay mixture, and absorbance was measured at 525 nm after incubating the mixture at 40 °C for 1 h. For AsA analysis, same assay mixture was used, except DTT and NEM were replaced by TDW. Total ascorbate (AsA + dehydroascorbate (DHA)) and AsA were estimated using standard curve generated for l-AsA. The DHA content was calculated by the subtraction of AsA from total AsA (Tzure-Meng et al. 2009).
Estimation of total flavonoid
Flavonoid content was estimated by colorimetric method using quercetin as standard (Chang et al. 2002). Tissue was ground in methanol and centrifuged for 20 min at 12,000g. Extract (1 ml) was added to 2 ml of 2,4-dinitrophenylhydrazine (DNPH) and incubated for 50 min at 50 °C. After cooling to room temperature, 5 ml of 1 % potassium hydroxide in 70 % methanol was added to the reaction mixture and incubated for 2 min. After incubation, 1 ml of the mixture was mixed with 5 ml of methanol and centrifuged for 10 min at 5,000g. Absorbance was measured at 495 nm.
Estimation of tocopherol
For tocopherol measurement, tissue was ground in methanol and centrifuged for 10 min at 12,000 rpm. Extract (1 ml) was added to dipyridyl solution (0.125 g dipyridyl dissolved in 25 ml of ethanol, stored in dark bottle at 4 °C). Absorbance was measured at 522 nm and α tocopherol was used as standard for computing concentrations (Kivack and Akay 2005).
Estimation of GSH and NPSH
For GSH and nonprotein thiol (NPSH) assay, shoot samples were homogenized in 3 ml of 5 % sulfosalicylic acid under cold conditions and centrifuged for 10 min at 10,000g. Supernatant was used for both GSH and NPSH analysis. For GSH analysis, 0.5 ml of supernatant was added to 1 ml of reaction buffer (0.5 mM EDTA, 0.05 ml of 3 mM 5-5'-dithiobis-(2-nitrobenzoic acid) (DTNB)) and incubated for 5 min at room temperature. Absorbance was recorded at 412 nm. For NPSH analysis, test extract was added to 1 ml of reaction buffer (0.1 M KPB, pH 7), 0.5 mM EDTA, and 0.5 ml of 1 mM DTNB and incubated for 10 min. Absorbance was measured at 412 nm and GSH was used as standard for computing concentrations (Namdjoyan et al. 2012).
Estimation of invertase enzyme activity
Fresh shoots were homogenized in 0.1 M KPB (pH 6.5) containing 10 % polyvinylpolypyrrolidone (PVPP). Homogenate was centrifuged at 12,000 rpm for 30 min at 4 °C. Assay systems for invertase activity contained alkaline (0.1 M potassium citrate, pH 8.5) or acidic (0.2 M sodium acetate, pH 4.8) or neutral (0.1 M KPB, pH 7.0) buffer, substrate (10 mM sucrose), and enzyme extract. Assay mixture was incubated at 37 °C for 30 min, and reaction was terminated by keeping the mixture in boiling water bath for 5 min. One milliliter of Nelson’s solution (A + B) was added to assay mixture and boiled for 20 min. Absorbance was measured at 620 nm after adding Nelson’s solution C (Nelson 1944). Total protein content of the extracts was determined according to Bradford method (Bradford 1976), using BSA as a standard.
Estimation of sugar content
Total sugar contents were measured according to Nelson method, using glucose as standard (Nelson 1944). Fresh shoot tissue was boiled in 80 % ethanol for 15 min; supernatant was collected and used for total sugar as well as total reducing sugar estimations. For total reducing sugar estimation, 1 ml of Nelson’s solution (A: B) in ratio (25:1) was added to supernatant and mixture was boiled for 20 min. Absorbance was measured at 620 nm after adding 1 ml of Nelson reagent C. For total sugar estimation, 0.2 ml of aliquot was taken and volume made up to 1 ml with TDW. Four milliliters of anthrone reagent was added to aliquot and heated for 8 min. Absorbance was measured at 630 nm.
Metabolism-related enzyme activities
For determination of glucose-6-phosphate dehydrogenase (G6PDH; EC 1.1.1.49), shikimate dehydrogenase (SKDH; EC 1.1.1.25), cinnamyl alcohol dehydrogenase (CAD; EC 1.1.1.195), phenylalanine ammonia lyase (PAL; EC 4.3.1.5), and polyphenol oxidase (PPO; EC 1.10.3.2) activities, young leaves were homogenized in 0.1 M KPB (pH 6.5) containing 10 % PVPP, 0.5 mM DTT, and 5 mM β-mercaptoethanol. Homogenate was centrifuged at 12,000 rpm for 45 min at 4 °C. Supernatant was used for enzyme assay. The activity of G6PDH was measured using assay mixture consisting of 50 mM Tris–HCl (pH 7.4), 5 mM MgCl2, 0.5 mM glucose-6-phosphate, and 0.5 mM NADP. The reaction was initiated by the addition of enzyme extract, and increase in absorbance was measured at 340 nm. The activity of SKDH was assayed in 0.1 M Tris–HCl buffer (pH 9) containing 2 mM shikimic acid, 0.5 mM NADP, and enzyme extract. Increase in absorbance was recorded for over 3 min at 340 nm (Kováčik et al. 2009). The enzyme activity of CAD was assayed in 0.1 M Tris–HCl buffer (pH 8.8) containing 1 mM coniferyl alcohol, 1 mM NADP, and enzyme extract. The enzyme activities of G6PDH, SKDH, and CAD were calculated using molar absorption coefficient of 6.22 mM−1 cm−1 (Kováčik et al. 2009). Assay mixture for PAL contained 50 mM Tris–HCl (pH 8.8), 20 mM phenylalanine, and enzyme extract. Reaction mixture was incubated for 30 min at 30 °C. TCA (10%) was added to terminate the reaction, and absorbance was measured at 290 nm (Ali et al. 2007). The assay for PPO enzyme was prepared in 50 mM KPB (pH 7.0) buffer containing 60 mM catechol and enzyme extract. Increase in absorbance was read over for 2 min at 420 nm (Jaleel et al. 2009).
Proline metabolism-related enzymes
The shoot tissues from treated and control samples was extracted using extraction buffer containing 0.1 M KPB (pH 7.4), 1 mM pyridoxal-5-phosphate, 1 mM EDTA, 10 mM β-mercaptoethanol, and 1 % PVPP. The supernatant was used for enzyme assays. The assay mixture of ornithine aminotransferase (OAT) contained 0.1 M KPB (pH 8.0), 50 mM ornithine, 20 mM α-ketoglutarate, 1 mM pyridoxal phosphate, and enzyme extract. After incubating the mixture for 30 min at 37 °C, 10 % TCA was added to terminate the reaction. Absorbance of the developed color was measured at 440 nm after adding 0.5 % o-aminobenzaldehyde. The assay mixture of pyrroline-5-corboxylate reductase (P5C) contained 0.1 M KPB (pH 7.4), 1 mM pyrroline-5-carobxylate, 0.12 mM NADH, and enzyme extract. Decrease in absorbance was measured immediately after addition of pyrroline-5-carobxylate at 340 nm. The assay mixture of proline oxidase (PO) contained 15 mM l-proline, 0.01 cytochrome C, 0.1 M KPB (pH 8.0), 0.5 % (v/v) Triton X-100, and enzyme extract. TCA (10%) was added after incubating the mixture for 30 min at 37 °C. Absorbance of the developed color was measured at 440 nm after adding 0.5 % o-aminobenzaldehyde (Madan et al. 1995).
Antioxidative enzyme activity
Fresh tissue was homogenized in 2 ml of extraction buffer containing 0.1 M KPB (pH 7.8), 0.1 mM EDTA, 1 % Triton X-100, 5 % PVPP, and 2 mM β-mercaptoethanol and centrifuged for 12,000g for 35 min. The activity of SOD (EC 1.15.1.1) was measured by monitoring the inhibition of nitroblue tetrazolium (NBT) reduction at 560 nm. The assay mixture of SOD consisted of 0.1 M KPB (pH 8.0), 0.025 % Triton X-100, 0.1 mM EDTA, 12 mM l-methionine, 75 μM NBT, 2 μM riboflavin, and enzyme extract. The reaction mixture was incubated for 30 min under 15-W fluorescent lamp at 28 °C, and absorbance was measured at 560 nm. Blank and controls were run in the same manner but without illumination and enzyme (Khatun et al. 2008). The enzyme activity of CAT was assayed by using reaction mixture containing 50 mM KPB (pH 7.0), 30 mM H2O2, and enzyme extract. Decrease in absorbance was measured at 240 nm as a consequence of H2O2 consumption (Sabir et al. 2012). The reaction mixture of ascorbate peroxidase (APX;,EC 1.11.1.11) contained 50 mM KPB (pH 7.0), 0.1 mM EDTA, 0.1 mM ascorbic acid, 0.1 mM H2O2, and enzyme extract. The decrease in absorbance at 290 nm for 1 min was measured after adding H2O2 (Sabir et al. 2012). For guaiacol peroxidase (G-POD), assay mixture contained 0.01 M KPB (pH 7), 80 mM guaiacol, 80 mM H2O2, and enzyme extract. The increase in absorbance was measured at 290 nm for 1 min after adding H2O2 (Sabir et al. 2012). For POD (EC 1.11.1.7) activity, reaction mixture contained of 0.1 M KPB (pH 6.8), 0.01 M pyrogallol, 5 mM H2O2, and enzyme extract. The reaction mixture was incubated for 5 min at 25 °C. Absorbance was measured at 420 nm after terminating the reaction by adding 2.5 N H2SO4 to reaction mixture (Khatun et al. 2008). For GR (EC 1.6.4.2) activity, assay mixture contained 0.1 M KPB (pH 7.0), 1 mM EDTA, 0.5 mM DTNB, 2 mM NADPH, 1 mM GSSG, and enzyme extract. Glutathione-dependent oxidation of NADPH at 340 nm was measured (Khatun et al. 2008). The reaction mixture of GST contained 0.1 M KPB (pH 6.5), 1.5 mM GSH, 1 mM CDNB, and enzyme extract. Increase in absorbance was observed at 240 nm (Beltagi et al. 2010). The assay mixture of GPX contained 0.56 M KPB (pH 7), 0.5 M EDTA, 1 mM NaNO3, 0.2 mM NADPH, and enzyme extract. The decrease in absorbance at 340 nm was measured (Khatun et al. 2008). The reaction mixture for monodehydroascorbate reductase (MDHAR) contained 0.1 M KPB (pH 7), 0.0125 % Triton X-100, 0.2 mM NADH, 2.5 mM ascorbate, 0.25 U ascorbate oxidase, and enzyme extract. Increased in absorbance was measured at 340 nm (Khatun et al. 2008). The assay mixture of dehydroascorbate reductase (DHAR) contained 0.1 M KPB (pH 7), 1 mM EDTA, 5 mM GSH, and 0.2 mM dehydro ascorbate. Increase in absorbance was measured at 265 nm (Khatun et al. 2008).
Native polyacrylamide gel electrophoresis
For the determination of profile of antioxidant isozymes, protein extracts in equal amounts were subjected to native polyacrylamide gel electrophoresis (PAGE) with 10 % resolving gel and 4 % stacking gel at 4 °C. The POD isozymes were visualized by incubating the gels in a solution of 0.2 M acetate buffer (pH 5.5), 0.03 % H2O2, and 1 mM o-dianisidine (Sangwan et al. 2003). For APX, gel was incubated in 50 mM KPB (pH 7), 2 mM sodium ascorbate, 4 mM sodium acetate, and 2 mM H2O2 for 20 min. Gel was stained in a solution containing 50 mM KPB (pH 7.8), 28 mM TEMED, and 2 mM NBT for 2 or 3 min after washing the gel with 50 mM KPB (pH 7.8) for 1 min (Chandrashekar and Umesha 2012). For amyl esterase (EST) isozymes, gel was incubated in 25 ml solution of 0.1 M sodium phosphate buffer (pH 6.5) containing 1 mM fast blue RR salt, 2 mM of α-napthyl, and 2 mM β-napthyl acetate (Sangwan et al. 2003). Gel was incubated in 25 ml of 0.1 M KPB (pH 7.5) containing 10.7 mM methionine, 0.97 mM NBT, 0.1 mM EDTA, and 4.2 mM riboflavin for SOD (Sabir et al. 2012). For glutamate oxaloacetate transaminase (GOT) staining, gel was incubated in 25 ml of solution containing 0.2 M Tris–HCl (pH 8.5), 2 mM pyridoxal phosphate, 4 mM l-aspartic acid, 4.3 mM α-ketoglutarate, and 5.4 mM fast blue BB salt (Sangwan et al. 2003). For diaphorase (DIA) staining, gel was incubated in 25 ml of 0.2 M Tris–HCl (pH 8.5) supplemented with 0.12 mM 2,6-dichlorophenolindophenol (DCPIP), 0.5 mM 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), and 0.3 mM NADH (Sangwan et al. 2003). For SKDH, gel was incubated in 12.5 ml of 0.1 M Tris–HCl buffer (pH 9.0) containing 0.25 mM NADP, 1.7 mM shikimic acid, 0.48 mM MTT, and 0.06 mM PMS. The staining for GR activity was performed by incubating gels in a solution containing 0.25 M Tris–HCl buffer (pH 7.5), 3.4 mM GSSG, 3 mM EDTA, 0.50 mM NADPH, and 40 μM DCPIP and MTT (Yannarelli et al. 2007). For CAT staining, gel was washed in TDW and incubated in 50 mM KPB (pH 7.0) containing 50 µ ml−1 horseradish peroxidase for 45 min in the dark. H2O2 (5 μmolar) was added to the gel then again incubated for 10 min after that gel was stained with 0.5 mg ml−1 DAB for 3–4 min. Negative bands were observed on dark orange brown background. The detection for GPX was performed by incubating the gel with 0.1 M KPB (pH 6.5) and 12.5 mM guaiacol and 1.7 mM benzidine and 12 mM H2O2 until the appearance of bands (Yannarelli et al. 2007; Sabir et al. 2012).
Determination of total ROS
For the total ROS measurement, tissue was incubated in 50 mM KPB and 10 μM DCFH-DA for 30 min. Total ROS was measured fluorometrically (Bernstein et al. 2010).
Determination of superoxide radical
Leaf tissue was incubated in 1 ml of 20 mM KPB (pH 6) containing 500 μM XTT in the dark at 25 °C in a shaker for 5 h. Absorbance at 470 nm was observed (Schopfer et al. 2001).
Determination of hydrogen peroxide production
Leaves were incubated in 2 ml of KPB (20 mM, pH 6.0) containing 5 μM scopoletin and 1 U ml−1 (final concentration) horseradish peroxidase in the dark at 25 °C on a shaker. The production of H2O2 was evaluated by fluorescence monitoring (Schopfer et al. 2001).
Determination of hydroxyl radical
Leaf tissue was incubated in 1.5 ml of buffer containing 20 mM 2-deoxy-d-ribose for 4 h and centrifuged for 5 min at 12,000g. A 0.5 ml of supernatant was added to incubation medium containing 0.5 ml of TBA (10 g l−1 in 50 mM NaOH) and 0.5 ml of TCA (28 g l−1). Mixture was boiled for 10 min, and after cooling, hydroxyl radical generation was measured fluorometrically (Schopfer et al. 2001).
Estimation of hydrogen peroxide, hydroxyl, and superoxide radicals’ scavenging activities
Leaves were ground in 2 ml methanol and centrifuged for 10,000g for 15 min. Supernatant was added to 4 mM H2O2 solution which was prepared in 0.1 M KPB (pH 7.4) and incubated for 10 min. The absorbance of the solution was measured at 230 nm against blank solution for calculation of H2O2 scavenging activity. For hydroxyl radical scavenging activity measurement, the above extract was added to the solution of KPB (pH 7.4) containing 1 mM EDTA, 1 mM FeCl3,1 mM l-ascorbic acid, 20 mM H2O2, and 30 mM deoxyribose. The reaction mixture was boiled for 15 min after incubating for 1 h at 37 °C then, 1 ml of 2.8 % (w/v) TCA and 1 ml of 1 % (w/w) TBA were added. The developed color was measured at 532 nm. For superoxide radical scavenging activity measurement, the reaction mixture contained 67 mM KPB (pH 6.8), 2 mM EDTA, 2 μM riboflavin, above extract, and 50 μM NBT. Absorbance was measured at 530 nm before and after the illumination. The percentage inhibition of superoxide generation was measured by comparing the absorbance values of control and treated samples (Vajanathappa and Badami 2009).
Estimation of total antioxidant activity
Total antioxidant activity was measured using ferric-reducing antioxidant power property (FRAP) assay. One hundred microliters of aliquot was added to 2 ml of FRAP reagent (1 mM TPTZ and 20 mM FeCl3 in 0.25 M sodium acetate, pH 5.6) and thoroughly mixed. Absorbance was measured at 595 nm after incubating the mixture for 5 min at 37 °C. Ammonium ferrous sulphate was used as standard (Molan et al. 2012).
Estimation of total reducing power activity
Mixture of methanolic extract and 2.5 ml of 1 % potassium ferric cyanide were incubated for 20 min at 50 °C. A 2.5 ml of 10 % TCA was added and centrifuged for 10 min at 6,500g. Absorbance was measured at 700 nm after adding 1 ml of 0.1 % FeCl3 (Yen Duh 1993).
Statistical analysis
Experiment was carried out with at least three replications, and the data were subjected to one-way ANOVA to assess the significant difference using Duncan’s multiple range test (P < 0.05). Mean values were computed, and standard deviation was indicated except where it was too small to be visible in graphical representation.
Results and discussion
Among heavy metals, Cd is extremely toxic to plants and animals and it persists in the environment due to its long half-life. In the present study, Cd toxicity and tolerance in medicinal plant W. somnifera have been analyzed in terms of responses of various growth-related factors of ROS metabolism, and enzymatic and nonenzymatic antioxidant responses engaged by the plant in the process. Various visual symptoms showing toxicity such as yellowing, necrosis, stunted growth, and side-branching inhibition were noticed in the treated shoots (Supplementary Fig. 1A and B). After 10 days of treatment, necrosis became visible only at 300 μM, while yellowing, shoot inhibition, and side-branching inhibition were quite apparent at toxic Cd (150–300 μM) concentrations (Supplementary Fig. 1A; Supplementary Table 1). These toxic effects were possibly due to alterations in cell division, photosynthesis, respiration, and protein synthesis resulting in the inhibition of normal growth of plant. Yellowing and chlorosis of leaves at toxic and lethal Cd concentrations might be due to the replacement of iron on the active sites by heavy metal resulting to iron chlorosis, as observed earlier in other plants such as Prunus cerasifera (Lombardi and Sebastiani 2005). Necrosis might have resulted from the generation of ROS which eventually leads to programmed cell death. FW and DW of cultured shoots were drastically decreased by 0.13- and 0.35-fold in comparison with control at toxic Cd concentration (150–300 μM), while at mild and moderate Cd concentrations, FW and DW weight was almost equal to control (Supplementary Fig. 2). Growth and photosynthetic pigment contents were severely inhibited at toxic Cd concentration which decreased photosynthetic pigment contents (Fig. 3) and uptake of essential mineral nutrients (N, P, and K) (Fig. 1) and caused necrosis (Supplementary Fig. 1A and B).

Cd accumulation in W. somnifera shoots grown in medium supplemented with Cd and its effect on uptake of N, P, and K. Average values are given. Error bar represents the standard deviation (n = 3)
In shoot cultures, the level of Cd was found to increase by 38- to 2,949-folds from mild to lethal Cd concentrations (5–1,000 μM) after 30 days of treatment in comparison to control (Supplementary Fig. 1A and B). It is reported that Cd can accumulate through different processes of Cd translocation with accumulation occurring in vacuole (Vogeli and Wagner 1990) cell wall and apoplast (Hart et al. 1998). Our results revealed that W. somnifera could accumulate high levels of Cd, up to the Cd concentration of 500 μM but beyond this plants failed to survive.
The N, P, and K are primary nutrient elements which are very essential for plant growth and development (Uchida 2000). There was no significant effect of Cd observed on N uptake at mild to moderate Cd concentrations, while at toxic Cd concentration, N uptake was strongly inhibited. The N content was found to be maximum at 5 μM (1.2-folds) and minimum at 300 μM (0.29-folds) in comparison to control. The study showed that Cd did not affect N uptake efficiently at mild to moderate concentrations, but at toxic concentration, it had adverse effect on N uptake resulting into retarded growth and necrosis of multiple shoots. The other mineral nutrient P is a main constituent of energy currencies (ADP, ATP, NADP, etc.), genetic materials (DNA and RNA), and cell membrane component (phospholipids). It plays a very important role in photosynthesis and respiration and in the development of root, flower, and seed (Uchida 2000). The supplementation of Cd in the medium exhibited negative regulation of P uptake, exhibiting a decrease in it under mild to lethal Cd concentrations by 0.9- to 0.1-folds in comparison to control. This might be due to the fact that Cd at higher concentrations inhibited P uptake by competing with its absorption and/or translocation. Other major nutrient K is known to act as activator of several enzymes as well as coenzyme and has an essential role in protein synthesis, cuticle layer formations, stomatal function maintenance of turgor potential, and disease resistance (Uchida 2000). The uptake of K increased at moderate Cd concentrations by 1.8-folds then sharply decreased at lethal Cd concentration by 0.14-folds in comparison with control, respectively (Fig. 1). These results suggest that Cd interferes with P and N uptake ,while in the case of K uptake, Cd enhanced the K uptake up to moderate Cd concentration, but at toxic and lethal Cd concentrations (200–1000 μM), there was a sharp decrease in K uptake. The uptake and translocation of Cd occurs through the transporters, so this process may be hindered leading to slowed nutrient uptake and translocation besides accompanying changes in the modification of permeability of plasma membrane and alterations in the membrane integrity, i.e., leakage of plasma membrane and by disturbance in transportation mechanism (Nazar et al. 2012). Similarly, inhibition of N, P, and K uptake by Cd supplementation is reported earlier (Metwally et al. 2005; Nedjimi and Daoud 2009).
Total photosynthetic pigment contents enhanced at 50 μM (1.75-folds higher than control) and reduced by 0.13-folds at 300 μM in comparison with control (Fig. 2). Total chlorophyll (chl a + chl b), chl a, chl b, and carotenoids were decreased by 0.1- to 0.2-folds, respectively, in comparison with control at toxic Cd concentration (200 to 300 μM) (Fig. 2). Reduction in total photosynthetic pigment contents might be owing to several reasons such as inhibition of chlorophyll biosynthesis, breakdown of pigments or their precursors, and destruction of chloroplast membrane by lipid peroxidation due to lack of antioxidant such as carotenoids or due to the increase in peroxidase activity (Mishra and Prakash 2010). Loss of pigments could have a direct adverse effect on photosynthesis. This phenomenon itself may be an indication of onset of oxidative stress impinging on the plants (Zheng et al. 2010).

Effect of Cd supplementation on total photosynthetic pigment contents in in vitro grown W. somnifera shoots after 30 days of treatment. Average values are given. Error bar represents the standard deviation (n = 3)
The product of unsaturated fatty acid peroxidation MDA is considered as the general indicator of lipid peroxidation as a result of stress. The MDA content was increased by 3.36- to 15.72-folds as compared to control, respectively upon Cd supplementation (Fig. 3). The increased MDA content indicated that Cd caused severe oxidative stress by stimulating excessive ROS generation which damaged the biological membranes and induced oxidative stress (Kaur et al. 2012).

Effect of Cd supplementation on MDA and on other nonenzymatic antioxidants in in vitro shoots of W. somnifera after 30 days of treatment. Average values are given. Error bar represents the standard deviation (n = 3)
Various nonenzymatic agents related to antioxidant actions such as total phenolics, proline, tocopherol, flavonoid, GSH, total AsA (AsA + DHA), AsA, and DHA were accumulated upon Cd exposure, confirming their active involvement in Cd detoxification and ROS-scavenging mechanism in W. somnifera multiple shoot cultures. Total phenolics, AsA, and NPSH increased in a concentration dependent manner by 4.96-, 5.35-, and 2.30-folds, respectively, in comparison with control upon Cd exposure, while proline, tocopherol, flavonoid, GSH, total ascorbate, and DHA were initially increased by 5.12-, 4.66-, 6.07-, 8.74-, 6.17-, and 6.66-folds, respectively, in comparison with control and then decreased at toxic Cd concentrations (Fig. 3). Our data suggest that nonenzymatic antioxidants like phenolic, AsA, and NPSH have profound role in Cd detoxification at all concentrations of Cd. Further, other nonenzymatic antioxidants such as proline, tocopherol, flavonoid, GSH, total ascorbate, and DHA were invoked in Cd detoxification up to certain concentration at toxic levels, i.e., 150–200 μM (except proline). Whereas at 300 μM, these adaptive responses ceased to account by that extent, allowing Cd adverse effects to reach overall toxic impact (Fig. 7).
Accumulation of proline content has been shown to play an active role in the heavy metal tolerance mechanism. We have investigated the enzyme activities of P5C, OAT, and PO related with proline metabolism. Among these, P5C and OAT are involved in proline biosynthesis, while PO is a proline catabolic enzyme (Madan et al. 1995). These observations suggested that proline content was increased by 1.0- to 5.0-folds, respectively, in comparison with control upon Cd exposure which coincided with the enhanced activity of P5C and OAT (Fig. 4b). At toxic Cd concentrations (200–300 μM), proline content decreased by 0.83-folds in comparison with control due to high activity of PO which possibly degraded the proline (Figs. 3 and 4b). Proline participates in maintaining several cellular functions including reconstruction of chlorophyll, osmoregulation, stabilization of protein, scavenging of hydroxyl radical, quenching of singlet oxygen, regulation of the cytosolic pH, and regulation of NAD/NADH ratio and as a source of nitrogen and energy for growth recovery (Mishra and Prakash 2010; Saha et al. 2010; Sabir et al. 2012).

a Effect of Cd supplementation on antioxidative enzyme activities in shoot cultures of W. somnifera after 30 days of treatment. Average values are given. Error bar represents the standard deviation (n = 3). b Effect of Cd supplementation on the enzyme activities of phenolic metabolism pathway (PAL, SKDH, G6PDH, CAD, and PPO), proline metabolic pathway enzymes (OAT, P5C, and PO), and invertase enzyme in in vitro shoots of W. somnifera after 30 days of treatment. Average values are given. Error bar represents the standard deviation (n = 3)
Active involvement of phenolic accumulation as a response to Cd exposure has been noticed. Phenolic content increased by 0.6- to 5.0-folds, respectively, in comparison with control, indicating its role in Cd detoxification mechanism. It increased due to enhanced activities of phenolic metabolism-related enzymes like G6PDH (provide precursor for the shikimate pathway), PAL (crucial enzyme in the phenylpropanoid pathway, synthesizes trans cinnamic acid), SKDH (providing precursor for PAL), and CAD (providing precursor for lignin biosynthesis) (Ali et al. 2007; Kivack and Akay 2005). Level of their activities enhanced (except CAD activity which decreased by 0.6- to 0.8-folds, respectively, in comparison to control at toxic Cd concentration) by 1.0- to 4.0-folds in comparison with control upon Cd exposure to synthesize phenolic contents to reduce the oxidative stress (Fig. 4b). The PPO activity is not affected by Cd at mild to moderate Cd concentrations (5–100 μM) in W. somnifera (Fig. 4b). Our results also showed that under Cd stress, phenolic contents increased and H2O2 decreased concomitantly, suggesting that phenolics may be participating in H2O2 detoxification process to a considerable extent. Phenolics scavenge H2O2 through a peroxidase action whereby phenolics are oxidized to phenoxyl radicals which can be reduced by AsA (Jaleel et al. 2009). Phenolics act as metal chelators and have the ability to eliminate radical species. Antioxidative activity might be assayed directly or indirectly by using total phenolic contents as a surrogate measure; it is mainly due to their redox properties (Ali et al. 2007). Phenolics have also been shown to be upregulated in response to higher salt concentrations in W. somnifera (Sabir et al. 2012).
Total ascorbate, AsA, and DHA are considered as the most abundant, low-molecular weight antioxidant playing a key role in defense against metal-induced oxidative stress caused by enhanced levels of ROS. These metabolites increased by 6.17-, 5.35-, and 6.66-folds in comparison with control, suggesting their active involvement in Cd detoxification. The MDHAR and DHAR play essential roles in the synthesis of reduced ascorbate. The former MDHAR converts the monodehydroascorbate into DHA (which is harmful to the plant), and DHAR helps in the regeneration of AsA from DHA. The increased activities of these enzymes resulted in higher accumulation of GSH and AsA (Figs. 3 and 4b). Under copper stress, similar observations were made in W. somnifera (Khatun et al. 2008). It has been shown that AsA minimizes the damage caused by oxidative process through synergic function with other antioxidants (Jaleel et al. 2009). The other functions of AsA includes, scavenging of H2O2 by ascorbate–glutathione cycle, elimination of ROS, and maintenance of α-tocopherol in reduced form (Mishra and Prakash 2010).
Elevation in tocopherol content by 4.65-folds in comparison with control (Fig. 3) suggested its active role in Cd detoxification, but this response of enhancement ceased at higher concentration of Cd which might be due to the inhibition of tocopherol synthesis or activation of some enzyme which is involved in degradation of tocopherol. Similar results were reported for safflower under Cd stress (Namdjoyan et al. 2012). α-Tocopherol (vitamin E) is known as natural antioxidant which interacts with the polyunsaturated acyl group of lipids that stabilizes the membranes. It also helps in scavenging and quenching of various ROS and lipid soluble byproducts of oxidative stress (Jaleel et al. 2009).
The levels of flavonoids were altered under Cd treatment, indicating its role in detoxification and chelating mechanism of Cd. Flavonoid content was increased by 1.0–6-folds, respectively, in comparison with control (Fig. 3). Flavonoids are secondary metabolite with significant antioxidant and chelating property. Its antioxidant property depends on the structure and substitution pattern of hydroxyl groups. The GSH and NPSH are also involved in Cd detoxification mechanism as shown by their increased content by 1.2- to 9-folds and 1- to 2.5-folds, respectively, in comparison with control under Cd exposure (Fig. 3). In W. somnifera, GSH content increased with increasing Cd concentration as reported previously for rice and safflower (Namdjoyan et al. 2012). The antioxidant GSH, a low molecular weight, sulphur-containing tripeptide is an essential component of the cellular antioxidative defense system, which keeps ROS level under control, and the GSH levels are constitutively higher in plants adapted to stress conditions. It is reported that GSH is an ideal biochemical parameter for plants to handle cellular situations of oxidative stress and heavy metal stress (Jaleel et al. 2009). Under Cd stress, NPSH also increased, suggesting its protective role against oxidative stress. Increased levels of GSH were also found in safflower and Sesbania drummondi (Zeng et al. 2009). In plants, enhanced level of NPSH may be associated with enhanced S assimilation due to the overexpression of genes involved in this process (Namdjoyan et al. 2012).
Enhancement of total sugar (16-folds), reducing sugar (43-folds), and invertase activity indicated its active role in detoxification process against Cd stress (Figs. 3 and 4b). Invertase activity reached its maximal level at 300-μM Cd exposure. Its activity in acidic, neutral, and alkaline medium was increased by 10.12-, 31.98-, and 14.26-folds than control, respectively (Fig. 4b). As invertase activity increased, total sugar and total reducing sugar increased, which states that plants need more availability of carbon source under heavy metal stress (Singh and Luthra 1988). This is particularly important as the tissue under Cd treatment have shown impairment in photosynthetic activities. It is inferred from the results that in vitro shoots of W. somnifera could induce sugar metabolism for protection against Cd stress. Possibly, the demand of reducing power as NADPH is being met by the substantially enhanced levels of sugar mobilization by upregulated invertases, yielding glucose to enter into pentose phosphate pathway (Couee et al. 2006). Increased GSH, NPSH, and AsA and total reducing sugar help in maintaining reduced conditions in cell for normal growth of the plant as shown in W. somnifera. These results suggest that upon Cd exposure, W. somnifera shoots elicit its ability to tolerate ROS by increasing their antioxidants and/or reducing power activity (Fig. 7).
The levels of antioxidative enzymes were found to be varying considerably under Cd treatment (Figs. 4a and 5). The enhancements for APX (15-folds), POD (12.11-folds), and G-POD (10.60-folds) were maximum at 200 μM, while CAT activity was maximum at 100 μM (1.69-folds); GPX and GST activities were maximum at 300 μM by 5.96- and 3.14-folds higher than control, respectively (Fig. 4a). Native PAGE analysis also showed that Cd significantly altered the isozymes of antioxidative enzymes at mild to moderate Cd concentrations (Fig. 5). Changes in the profiles of antioxidative enzymes indicate their involvement in detoxification mechanism in W. somnifera. Three SOD (SOD 1, SOD 2, and SOD 3) isozymes were found at mild Cd concentration, whereas SOD 1 and SOD 3 were observed at moderate treatment (Fig. 5). Decrease in SOD activity indicated that SOD fails to be sufficient to detoxify O2 ·− radical to protect plant from cellular damages which resulted in generation of more O2 ·− under Cd stress. H2O2 is highly toxic, which must be scavenged by various peroxidases. The activities of CAT, APX, G-POD, POD, and GPX were increased by 1.7-, 15.0-, 11-, 2-, and 6-folds, respectively, in comparison to control (Fig. 4a). The increased activities upon Cd exposure suggested their involvement in H2O2 detoxification mechanism. APX is also an essential regulator of ROS scavenging through ascorbate–glutathione cycle (Figs. 4a and 5). Three isozymes of POX and GPX each were observed in treated shoots. One isozyme (POX 1) appeared in treated shoots only. Intensity and number of bands in POX and GPX increased, suggesting their involvement in adapting under Cd stress (Khatun et al. 2008). However, number of isozymic forms of GR decreased under Cd treatment, reflecting its decreased activity in W. somnifera under Cd stress. Three GR (GR1, GR2, and GR3) isozymes were found in control, while in treated shoots, only GR2 and GR 3 were found and also their intensity was decreased from mild to toxic Cd concentrations in comparison to control (Fig. 5). It is reported that heavy metals might cleave the highly conserved disulphide bridge between CYS76 and CYS 81 present in GR and decreased the level of GR activity (Khatun et al. 2008). Decrease in activity of SOD and GR suggested that both these enzymes may be of focal point to further expand the detoxification mechanism of Cd oxidative stress. The GST activity also increased with increasing Cd concentration, indicating its role in detoxification process in W. somnifera. It is reported that GST acts as metal chelators which detoxify the toxic compounds via GSH conjugation (Beltagi et al. 2010).

Isozymic pattern of metabolic enzymes in in vitro shoot cultures of W. somnifera after 30 days of treatment. Lane 1 control, lane 2 5 μM, lane 3 10 μM, lane 4 20 μM, lane 5 50 μM, lane 6 100 μM, lane 7 150 μM, lane 8 200 μM, and lane 9 300 μM
Newer isozymic forms of housekeeping enzyme such as GOT, EST, and DIA increased, indicating their involvement in Cd detoxification and tolerance mechanism. One of the most active enzymes, GOT, in the cell possesses an important role in N metabolism and in the flow of carbon between the organic acid and amino acid pools (Siregar et al. 2010). A total of four GOT isozymes were found in W. somnifera under Cd supplementation. Isozymes GOT1, GOT2, and GOT 3 were found at moderate to higher Cd concentrations, while in control, only GOT1 and GOT 4 were noticed. Whereas GOT 2 and GOT 3 were found as newer isozymes at moderate to toxic Cd concentrations in comparison with control (Fig. 5). Diaphoreses is one of the flavin reductase enzymes in plants, and it uses two electron transport mechanisms. A total of four isozymes, viz, DIA 1, DIA 2, DIA 3, and DIA 4 were found in W. somnifera under Cd stress. Three isozymes DIA 1, DIA3, and DIA 4 were detected in treated shoots, suggesting their involvement in Cd detoxification and ROS-scavenging mechanism. Diaphoreses detoxifies the redox active compounds like quinines and circumvents the adverse effect of oxidative stress in cells generated by metal stress. Five EST isozymes were observed at 150 μM Cd concentration, and only two isozymes of EST (1 and 3) at 300 μM (Fig. 5) were noticed. Increase in band intensity and isozyme number of EST at mild to moderate Cd concentrations in comparison to control suggests its active role in Cd tolerance mechanism as reported earlier (Sabir et al. 2012).
Total ROS was maximum at 50 μM (2.39-folds) and minimum at 300 μM (0.25-folds). H2O2 content decreased upon Cd exposure due to high-H2O2 scavenging activity. The total content of H2O2 was minimum at 300 μM by 0.17-folds lower and its percentage scavenging activity was increased by 2.31-folds than control (Figs. 6 and 7). O2 ·− and ·OH radicals were increased upon Cd exposure due to the decrease in their scavenging activities. O2 ·− and ·OH radicals were maximum at 300 μM by 2.4- and 5.21-folds higher, and their percentage scavenging activity decreased by 0.15- and 0.10-folds lower in comparison with control, respectively (Figs. 6 and 7). H2O2 scavenging activity increased, and free radicals (O2 ·– and ·OH) scavenging activity decreased due to enhanced levels of various peroxidase activities and inhibition of SOD activity during Cd stress in W. somnifera.

Effect of Cd supplementation on total ROS, H2O2, and superoxide and hydroxyl radicals in in vitro shoot cultures of W. somnifera after 30 days of treatment. Average values are given. Error bar represents the standard deviation (n = 3)

Effect of Cd supplementation on the ROS-scavenging (hydrogen peroxide, hydroxyl, and superoxide radicals) activity, total reducing power, and antioxidative activity in in vitro shoots of W. somnifera after 30 days of treatment. Average values are given. Error bar represents the standard deviation (n = 3)
Conclusions
In conclusion, our study reveals for the first time that in vitro-regenerated shoots of W. somnifera have the ability to accumulate (more than 2,000-fold higher as compared to control) and tolerate Cd. Exposure to toxic and lethal Cd concentrations (150–1,000 μM) exerted the N, P, and K deficiency in shoots, which are very essential for plant growth and development. Due to N, P, and K deficiency, shoots showed severe toxic symptoms like stunted growth, chlorosis and necrosis and decreased photosynthetic pigment contents. Toxic Cd concentration also caused oxidative damage in W. somnifera shoots as confirmed by enhanced MDA and ROS (O2 ·– and ·OH) and decreased FW and DW of multiple shoot cultures. Various enzymatic and nonenzymatic antioxidants are involved in Cd detoxification mechanism. Enhanced sugar metabolism also suggests its active role in Cd detoxification and maintaining carbon source to shoots under stress. The toxicity of Cd was decreased by boosting of nonenzymatic responses such as increment in level of total sugars, phenolics, ASA, DHA, tocopherol, flavonoid, GSH, NPSH, and proline. The results clearly showed that interplay of enzymatic and nonenzymatic antioxidative responses, sugar metabolism, and phenolics and proline metabolism together count to tolerance of the Cd-induced oxidative stress and as well as for the mechanism of efficient scavenging of ROS. The plant resorts to modulate its metabolism in such a way to avoid the destructive effect of oxidative stress and evolves a biochemical machinery to detoxify Cd-generated toxicity. More detailed studies are needed to elucidate the mechanisms of Cd-induced responses related to secondary metabolites such as withanolides. Further, understanding the detoxification mechanism and strategies in W. somnifera against the heavy metal stress at plant level will provide insights into manipulating heavy metal tolerance in medicinally important plant.
Abbreviations
- ASC:
-
Ascorbate
- APX:
-
Ascorbate peroxidase
- BSA:
-
Bovine serum albumin
- CAD:
-
Cinnamyl alcohol dehydrogenase
- CAT:
-
Catalase
- Cd:
-
Cadmium
- DAB:
-
3, 3′-Diaminobenzidine
- DCPIP:
-
2,6-Dichlorophenolindophenol
- DCFH-DA:
-
2′,7′-Dichlorodihydrofluorescein diacetate
- DHAR:
-
Dehydroascorbate reductase
- DHA:
-
Dehydroascorbate
- DIA:
-
Diaphorase
- DTNB:
-
5-5'-Dithiobis-(2-ntrobenzoic acid)
- DTT:
-
Dithiothreitol
- DNPH:
-
2,4-Dinitrophenylhydrazine
- EDTA:
-
Ethylenediaminetetraacetic acid
- EST:
-
Amyl esterase
- FRAP:
-
Ferric-reducing antioxidant power property
- GOT:
-
Glutamate oxaloacetate transaminase
- G-POD:
-
Guaiacol peroxidase
- GPX:
-
Glutathione peroxidase
- GR:
-
Glutathione reductase
- GSH:
-
Reduced glutathione
- GSSG:
-
Oxidized glutathione
- GST:
-
Glutathione-S-transferase
- G6PDH:
-
Glucose-6-phosphate dehydrogenase
- ICP:
-
Inductively coupled plasma
- KPB:
-
Potassium phosphate buffer
- MDA:
-
Malondialdehyde
- MDHAR:
-
Monodehydroascorbate reductase
- MS:
-
Murashige and Skoog
- MTT:
-
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
- NBT:
-
Nitroblue tetrazolium
- NEM:
-
N-Ethylmaleimide
- NPSH:
-
Nonprotein thiol
- OAT:
-
Ornithine aminotransferase
- PAGE:
-
Polyacrylamide gel electrophoresis
- PAL:
-
Phenylalanine ammonia lyase
- P5C:
-
Pyrroline-5-corboxylate reductase
- POD:
-
Peroxidase
- PO:
-
Proline oxidase
- PPO:
-
Polyphenol oxidase
- PVPP:
-
Polyvinylpolypyrrolidone
- ROS:
-
Reactive oxygen species
- SKDH:
-
Shikimate dehydrogenase
- SOD:
-
Superoxide dismutase
- TBA:
-
2-Thiobarbituric acid
- TDW:
-
Triple distilled water
- TBARS:
-
2-Thiobarbituric acid reactive substances
- TCA:
-
Trichloroacetic acid
- TPTZ:
-
2,4,4-Tripyridyl-2-triazine
- TEMED:
-
N,N,N′,N′-Tetramethylethylenediamine
References
Ali MB, Hahn EJ, Paek KY (2007) Methyl jasmonate and salicylic acid induced oxidative stress and accumulation of phenolics in Panax ginseng bioreactor root suspension cultures. Molecules 12:607–621
Bates LS, Waldran RP, Teare ID (1973) Rapid determination of free proline for water stress srudies. Plant Soil 39:205–208
Beltagi HS, Mohamed AA, Rashed MM (2010) Response of antioxidative enzymes to Cd stress in leaves and roots of radish (Raphanus sativus L.). Not Sci Biol 4:76–82
Bernstein N, Shoresh M, Xu Y, Huang B (2010) Involvement of the plant antioxidative response in the differential growth sensitivity to salinity of leaves vs roots during cell development. Free Radic Biol Med 49:1161–1171
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem 72:248–254
Chandrashekar S, Umesha S (2012) Induction of antioxidant enzymes associated with bacterial spot pathogenesis in tomato. Int J Food Agric Vet Sci 2:22–34
Chang CC, Yang MH, Wen HM, Chern JC (2002) Estimation of total flavonoid content in Propolis by two complementary colorimetric methods. J Food Drug Anal 10:178–182
Couee I, Sulmon CC, Gouesbet G, Amrani AE (2006) Involvement of soluble sugars in reactive oxygen species balance and responses to oxidative stress in plants. J Exp Bot 57:449–459
Gill SS, Tuteja N (2011) Cd stress tolerance in crop plants probing the role of sulfur. Plant Signal Behav 6:215–222
Hart JJ, Welch RM, Norvell WA, Sullivan LA (1998) Characterization of cadmium binding, uptake, and translocation in intact seedlings of bread and durum wheat cultivars. Plant Physiol 116:1413–1420
Heath R, Packer L (1968) Photoperoxidation in isolated chloroplasts: I. Kinetics and stoichiometry of fatty acid peroxidation. Arch Biochem Biophys 196:385–395
Hiscox JD, Israelstam GF (1979) A method for the extraction of chlorophyll from leaf tissue without maceration. Can J Bot 57:1332–1334
Jaleel CA, Riadh K, Gopi R, Manivannan P, Ines J, Al-Juburi HJ, Xing ZC, Bo SH, Panneerselvam R (2009) Antioxidant defense responses: physiological plasticity in higher plants under abiotic constraints. Acta Physiol Plant 31:427–436
Kaur G, Singh HP, Batish DR, Kohil RK (2012) A time course assessment of changes in reactive oxygen species generation and antioxidant defense in hydroponically grown wheat in response to lead ions (Pb+2). Protoplasma 249:1091–1100
Khan MA, Ahmad I, Rahman IU (2007) Effect of environmental pollution on heavy metals content of Withania somnifera. J Chin Chem Soc 54:339–343
Khatun S, Ali MB, Hahn EJ, Paek KY (2008) Copper toxicity in Withania somnifera: growth and antioxidant enzymes response of in vitro grown plants. Env Exp Bot 64:279–285
Kivack B, Akay S (2005) Quantitative determination of a-tocopherol in Pistacia lentiscus, Pistacia lentiscus var. chia and Pistacia terebinthus by TLC-densitometry and colorimetry. Fitoterapia 76:62–66
Kramer U (2010) Metal hyperaccumulation in plants. Annu Rev Plant Biol 61:517–534
Kováčik J, Klejdus B, Hedbavny J, Štork F, Bačkor M (2009) Comparison of cadmium and copper effect on phenolic metabolism, mineral nutrients and stress-related parameters in Matricaria chamomilla plants. Plant Soil 320:231–242
Lombardi L, Sebastiani L (2005) Copper toxicity in Prunus cerasifera: growth and antioxidant enzymes responses of in vitro grown plants. Plant Sci 168:797–802
Madan S, Nainawatee HS, Jain RK, Chowdhury JB (1995) Proline and proline metabolizing enzymes in in-vitro selected NaCl-tolerant Brassica juncea L. under salt stress. Ann Bot 76:51–57
Mahmood T, Gupta KJ, Kaiser WM (2009) Cd stress stimulates nitric oxide production by wheat roots. Pak J Bot 41:1285–1290
Metwally A, Safronova VI, Belimov AA, Dietz KJ (2005) Genotypic variation of the response to cadmium toxicity in Pisum sativum L. J Exp Bot 56:167–178
Mishra MK, Prakash V (2010) Response of non-enzymatic antioxidants to zinc induced stress at different pH in Glycine max L. cv. Merrill. Acad J Plant Sci 1:1–10
Mittler R, Vanderauwera S, Gollery M, Breusegem FV (2004) Reactive oxygen gene network of plants. Plant Sci 9:491–498
Molan AL, Faraj AM, Mahdy AS (2012) Antioxidant activity and phenolic content of some medicinal plants traditionally used in northern Iraq. Phytopharmaco 2:224–233
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco cultures. Physiol Plant 15:473–497
Namdjoyan S, Namdjoyan S, Kermanian H (2012) Induction of phytochelatin and responses of antioxidants under cadmium stress in safflower (Carthamus tinctorius) seedlings. Turk J Bot 36:495–502
Nazar R, Iqbal N, Masood A, Iqbal M, Khan R, Syeed S, Khan NA (2012) Cadmium toxicity in plants and role of mineral nutrients in its alleviation. Am J Plant Sci 3:1476–1489
Nedjimi B, Daoud Y (2009) Cadmium accumulation in Atriplex halimus subsp. schweinfurthii and its influence on growth, proline, root hydraulic conductivity and nutrient uptake. Flora 204:316–324
Nelson N (1944) A photometric adaptation of the Somogyi method for the determination of glucose. J Biol Chem 163:375–380
Sabir F, Sangwan RS, Kumar R, Sangwan NS (2012) Salt stress-induced responses in growth and metabolism in callus cultures and differentiating in vitro shoots of Indian ginseng (Withania somnifera Dunal). J Plant Growth Regul 31:537–548
Sabir F, Mishra S, Sangwan RS, Jadaun JS, Sangwan NS (2013) Qualitative and quantitative variations in withanolides and expression of some pathway genes during different stages of morphogenesis in Withania somnifera Dunal. Protoplasma. doi:10.1007/s00709-012-0438-y
Saha P, Chatterijee P, Biswas AK (2010) NaCl pretreatment alleviates salt stress by enhancement of antioxidative defense system and osmolyte accumulation in Mungbean (Vigna radiate L. Wilczek). Indian J Exp Biol 48:593–600
Sangwan NS, Yadav U, Sangwan RS (2003) Genetic diversity among elite varieties of the aromatic grasses, Cymbopogon martini. Euphytica 130:117–130
Sangwan RS, Chaurasiya ND, Mishra LN, Lal P, Uniyal GC, Sharma R, Sangwan NS, Suri KA, Qazi GN, Tuli R (2004) Phytochemical variability in commercial herbal products and preparations of Withania somnifera (ashwagandha). Curr Sci 86:461–465
Sangwan RS, Chaurasiya ND, Lal P, Misra L, Uniyal GC, Tuli R, Sangwan NS (2007) Withanolide A biogeneration in in vitro shoot cultures of ashwagandha (Withania somnifera Dunal), a main medicinal plant in Ayurveda. Chem Pharm Bull 55:1371–1375
Sangwan RS, Chaurasiya ND, Lal P, Mishra LN, Tuli R, Sangwan NS (2008) Root contained withanolide A is inherently de novo synthesized within roots in ashwagandha (Withania somnifera). Physiol Plant 133:278–287
Schopfer P, Plachy C, Frahry G (2001) Release of reactive oxygen intermediates (superoxide radicals, hydrogen peroxide, and hydroxyl radicals) and peroxidase in germinating radish seeds controlled by light, gibberellin, and abscisic acid. Plant Physiol 125:1591–1602
Singh N, Luthra R (1988) Sucrose metabolism and essential oil accumulation during lemongrass (Cymbopogon flexuosus Stapf) leaf development. Plant Sci 57:127–133
Siregar IZ, Yunanto T (2010) The genetics of glutamate oxaloacetate transaminase (GOT) in Pinus merkusii Jungh. et de Vriese. Biodiversitas 11:5–8
Tuli, R, Sangwan RS, Kumar S, Bhattacharya S, Misra LN, Mandal C, Raghubir R, Trivedi PK, Tewari SK, Mishra P, Chaturvedi P, Sangwan NS, Nair KN, Rawat AKS, Srivastava V, Srivastava RK, Ojha SK, Mehrotra S, Khajuria A, Suri KA (2009) Ashwagandha (Withania somnifera) A model Indian medicinal plant. Eds R Tuli and RS Sangwan, Publisher NMITLI-CSIR, New Delhi. ISBN No.978-93-80235-29-5
Tzure-Meng WU, Yi-Ting HSU, Tse-Min LEE (2009) Effects of Cd on the regulation of antioxidant enzyme activity, gene expression, and antioxidant defenses in the marine macroalga Ulva fasciata. Bot Studies 50:25–34
Uchida R (2000) Essential nutrients for plant growth: nutrient functions and deficiency symptoms. College of Tropical Agriculture and Human Resources, University of Hawaii at Manoa 31:55
Vajanathappa J, Badami S (2009) Antiedematogenic and free radical scavenging activity of swertiamarin isolated from Enicostemma axillare. Planta Med 75:12–17
Vogeli LR, Wagner GJ (1990) Subcellular localization of cadmium and cadmium-binding peptides in tobacco leaves: implication of a transport function for cadmium-binding peptides. Plant Physiol 92:1086–1093
Yannarelli GG, Alvarez AJF, Santa-Cruz DM, Tomaro ML (2007) Glutathione reductase activity and isoforms in leaves and roots of wheat plants subjected to Cd stress. Phytochemistry 68:505–512
Yen Duh PD (1993) Antioxidative properties of methanolic extracts from peanut hulls. J Amer Oil Chem Society 70:383–386
Zeng X, Ma LQ, Oiu R, Tang Y (2009) Responses of nonprotein thiols to Cd exposure in Cd hyperaccumulator Arabis paniculata Franch. Env Exp Bot 66:242–248
Zheng G, Lv HP, Gao S, Wang SR (2010) Effect of Cd on growth and antioxidant response in Glycyrrhiza uralensis seedlings. Plant Soil Environ 56:508–515
Acknowledgments
Authors are thankful to the NMITLI, CSIR, New Delhi for the financial assistance. The help by Soil Science department (Drs. Sukhmal Chand, P. Khare, A. Kalra and Mr. Kundan) in ICP analysis is gratefully acknowledged. BM and NSS thank Academy of Scientific and Innovative Research (AcSIR), New Delhi. Authors are thankful to Director, CSIR-CIMAP for constant encouragement and support.
Conflict of interest
The authors declare they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling Editor: Peter Nick
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
(DOC 40 kb)
Supplementary Fig. 1A
Effect of Cd supplementation on growth of in vitro shoots of W. somnifera after 30 days of treatment (A control; B 5 μM; C 10 μM; D 20 μM; E 50 μM; F 100 μM; G 150 μM; H 200 μM; I 300 μM). (PDF 140 kb)
Supplementary Fig. 1B
Inhibition of growth and branching in in vitro grown shoots of W. somnifera after 30 days of treatment. a control. b 300 μM (PDF 115 kb)
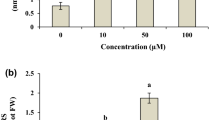
Supplementary Fig. 2
Effect of Cd supplementation on FW and DW of multiple shoot cultures of W. somnifera after 30 days of treatment. Average values are given. Error bar represents the standard deviation (n = 3). (PDF 26 kb)
Rights and permissions
About this article
Cite this article
Mishra, B., Sangwan, R.S., Mishra, S. et al. Effect of cadmium stress on inductive enzymatic and nonenzymatic responses of ROS and sugar metabolism in multiple shoot cultures of Ashwagandha (Withania somnifera Dunal). Protoplasma 251, 1031–1045 (2014). https://doi.org/10.1007/s00709-014-0613-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00709-014-0613-4