
Abstract
Temperature stress such as heat, cold, or freezing is a principal cause for yield reduction in crops. In particular, heat stress is very common and dangerous for plants since this stress can impact several plant and cellular functions. In spite of their role in sensing local stress and in controlling fundamental processes including PCD, the responses of cellular structures and organelles to heat stress are poorly investigated. In this work, we investigated the possible changes induced by mild heat stress, medium heat stress, and heat shock (HS; 5 min at 35°C, 45°C, or 50°C, respectively) on actin cytoskeleton and endoplasmic reticulum (ER) of tobacco BY-2 cultured cells. While mild and medium heat stresses are ineffective, HS induces depolymerization of actin microfilaments and changes in ER morphology accompanied by accumulation of the HSP70 binding protein (BiP). These effects of HS are prevented by the inhibitor of ethylene production Co2+. While the analyzed cell structures do not seem to be involved in the establishment of mild and medium heat stresses at least in this experimental system, the strong modifications induced by the treatment at 50°C suggest that actin cytoskeleton and ER may be involved in the responses to HS. Besides, the inhibiting effect of Co2+ suggests a role for ethylene as a regulative molecule in the responses to HS here observed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Temperature stress such as heat, cold, or freezing is a principal cause for yield reduction in crops (Mittler 2006). In particular, heat stress is very common and dangerous for plants as they need light energy which can easily cause temperature increase in the exposed tissues. Heat stress can impact several plant and cellular functions. For example, high temperatures are known to alter membrane fluidity and impair enzyme function via protein denaturation (Kampinga et al. 1995). Both these effects can result in elevated reactive oxygen species production that causes oxidative stress possibly leading to programmed cell death (PCD) (Dat et al. 1998; Vacca et al. 2004). At the plant level, heat stress can lead to reduced photosynthesis and assimilate translocation, altered growth and reproduction, and premature and enhanced senescence (Hall 2001). Plants can face heat stress by the activation of proper defense mechanisms in order to minimize damage and ensure protection of cellular homeostasis. A heat stress of sufficient magnitude elicits a characteristic heat shock (HS) response, the elements of which are highly conserved. HS response is characterized by a rapid activation of HS genes and the synthesis and accumulation of HS proteins. There is now extensive evidence in literature that many of these proteins are molecular chaperones which play important roles in the tolerance of plants to a variety of biotic and abiotic stresses (for a review, see Kotak et al. 2007). Molecular chaperones are key components contributing to cellular homeostasis under both optimal and adverse growth conditions. They are responsible for protein folding, assembly, translocation, and degradation in a broad array of normal cellular processes. Molecular chaperones also function in the stabilization of proteins and membranes, and can assist in protein refolding under stress conditions. Different members of this family of proteins are targeted to the nucleus, cytosol, chloroplasts, mitochondria, endoplasmic reticulum (ER), and peroxisomes, implicating these proteins in the protection of practically all cellular compartments (Wang et al. 2004).
Many of the plant responses to heat stress, including downward bending and abscission of leaves and flowers, induction of adventitious root formation, root hair formation, and inhibition of root elongation, have been associated with enhanced ethylene production (Klassen and Bugbee 2004). Ethylene is an important plant hormone and its biosynthesis is stimulated by a variety of abiotic and biotic stresses, including heat stress (Hays et al. 2007; Suzuki et al. 2008). Many ethylene responses involve changes in gene expression and a large family of plant-specific transcription factors referred to as ethylene-response-element binding proteins (EREBPs) have been found in the Arabidopsis and other plant species genomes (Riechmann and Meyerowitz 1998). It has been shown that ethylene induces the synthesis of enzymes such as chitinases, β-1,3-glucanases, peroxidases, phenylalanine-ammonia-lyase and other defense-related proteins as well as the formation of phytoalexins, lignin, and other phenolic compounds (Broekaert et al. 2006 and reference therein) and the Arabidopsis gene encoding the HS protein APX1 (defined as such because of the presence of a heat shock transcription factor binding site in its promoter; Storozhenko et al. 1998) is induced by ethephon, an ethylene-releasing compound (Wu 1995).
In spite of their role in sensing local stress and in controlling fundamental processes including PCD (Ferri and Kroemer 2001), the responses of cellular structures and organelles to heat stress are poorly investigated. A cell-type-specific disruption and recovery of the cytoskeleton upon HS has been observed in Arabidopsis thaliana epidermal root cells (Müller et al. 2007) and changes in mitochondrial morphology have been observed during the onset of cell death induced by HS in A. thaliana leaf tissue or isolated protoplasts (Scott and Logan 2008). Cell cultures represent an optimal system to study the responses of cellular structures and organelles to stress conditions as they are formed by more homogeneous cells than those present in complex tissues. In addition, the administration and the reproducibility of the stress conditions are easy in this more controlled system. Tobacco Bright-Yellow 2 (BY-2) cultured cells appear very useful for these investigations since their biochemical responses to heat stress have been extensively investigated (Vacca et al. 2004; Locato et al. 2008, 2009). Mild heat stress do not affect cell viability but induces modulation of the antioxidant network, while HS induces a strong modification in the antioxidant network accompanied by PCD showing apoptotic features, i.e., cytoplasmic shrinkage, specific DNA fragmentation (laddering) and release of cytochrome c from mitochondria (Vacca et al. 2004; Locato et al. 2008). In this work, we investigated the effect of mild heat stress, medium heat stress, and HS (5 min at 35°C, 45°C, or 50°C, respectively) on actin cytoskeleton and endoplasmic reticulum (ER) of tobacco BY-2 cultured cells and its sensitivity to the inhibitor of ethylene production Co2+.
Materials and methods
Cell culture growth and heat treatment
The suspension of tobacco BY-2 (Nicotiana tabacum L. cv Bright-Yellow 2) cells was routinely cultured in a growth chamber at 25°C on a gyratory shaker (120 rpm) according to Nagata et al. (1992). A stationary (7-day-old) culture was diluted 4:100 (v/v) with fresh growth medium and cultured for 4 days before heat treatment. For heat treatments, 20 ml of cell culture in 100-ml culture flasks were maintained into a water bath with shaking at 35°C, 45°C, or 50°C for a heating time of 5 min in the absence or presence of 100 μM CoCl2 added 10 min before heat treatment. After heat treatments, the flasks were returned to the growth chamber at 25°C and at the indicated times after this return aliquots of cells were withdrawn and used for microscopic analyses or frozen in liquid nitrogen and stored at −80°C until use for western blot analyses (see below).
Cell viability assay and determination of cells undergoing PCD
For the cell viability assay, the cells were collected and stained for 5 min with the Evans Blue dye at a final concentration of 150 μg/ml. Dead cells stained blue, while living cells remained unstained. The cells were examined with a light microscope and the percentage of dead (Evans Blue-stained) cells was calculated from the observation of at least 1,000 cells. For PCD determination, we utilized cytoplasmic shrinkage, a morphological modification usually observed in cultured cells undergoing PCD with apoptotic features (McCabe and Leaver 2000; Lam 2004). The cells were examined with a light microscope and the percentage of cells showing cytoplasmic shrinkage was calculated from the observation of at least 1,000 cells.
Confocal microscopy
The actin microfilaments were visualized by confocal laser scanner microscopy after staining of the fixed cells with phalloidin-TRITC as described (Malerba et al. 2008). Briefly, about 100,000 cells were collected by gentle centrifugation and fixed for 10 min in 1.8% (w/v) paraformaldehyde in standard buffer (50 mM piperazine-N,N′-bis(2-ethanesulfonic acid), pH 7.0, 5 mM MgCl2, and 10 mM EGTA). After a subsequent 10-min fixation in standard buffer containing 10% (v/v) glycerol, the cells were rinsed twice (for 10 min) and resuspended in 0.5 ml of standard buffer. Then, 0.5 ml of 0.66 μM phalloidin-TRITC, prepared freshly from a 66-μM stock solution in absolute ethanol by dilution in phosphate-buffered saline (PBS), was added to the resuspended cells. After 35 min of incubation, the cells were washed three times for 10 min in PBS and observed immediately. The ER was analyzed utilizing the vital dye 3,3′-dihexyloxacarbocyanine iodide (DiOC6) as described (Malerba et al. 2004). Briefly, about 100,000 cells were washed, resuspended in fresh growth medium diluted 1:10 (v/v) with deionized water, and stained for 5 min with 4 μg of the dye/ml. The analysis of actin cytoskeleton and ER was performed on at least 100 cells for each temperature and experimental time with a Leica DMIRE2 confocal laser scanner microscope equipped with a ×63 oil immersion objective and Leica TCS-NT software. The data acquisition settings (laser power, pinhole size, scan conditions, detector settings, and so on) were identical for all observations. The images were edited by Photoshop 6.0 (Adobe, San Jose, CA, USA).
Cell fraction preparation
Cells were collected by gentle centrifugation, frozen in liquid nitrogen and homogenized for 5 min at maximum speed with a Ultra-Turrax T25 device (International PBI, Milan, Italy) at a density of 1 g of fresh weight per 2 ml of homogenizing buffer (25 mM 2-(N-morpholino)ethanesulfonic acid-2-(bis(2-hydroxymethyl)amino)-2-(hydroxymethyl)-1,3-propanediol, pH 7.8, 250 mM sucrose, 5 mM EDTA, 0.2% bovine serum albumin, and 0.2% casein) freshly supplied with 2 mM dithiothreitol and 10 μl of plant protease inhibitor cocktail (Sigma cat. nr. P 9599) per ml of cell homogenate. The homogenate was centrifuged at 1,000×g for 10 min and the supernatant was centrifuged again at 10,000×g for 15 min. The supernatant was centrifuged at 48,000×g for 60 min and the resultant pellet, representing the microsomal fraction, was resuspended in 10 mM Tris–HCl, pH 6.5, 1 mM EDTA, 1 mM dithiothreitol and 20% (v/v) glycerol supplemented with the protease inhibitor cocktail, and stored at −80°C until use. The supernatant was centrifuged at 200,000×g for 3 h and the resultant supernatant, representing the cytosolic (soluble) fraction, was stored at −80°C until use. All the above-reported procedures were performed at 4°C.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and western blot experiments
Equal amounts of proteins, determined with the Bio-Rad microassay using bovine serum albumin as standard, were separated by discontinuous sodium dodecyl sulfate–polyacrylamide gel electrophoresis (4% stacking, 10% resolving gel), performed as described previously (Laemmli 1970) in a Mini Protean II apparatus (Bio-Rad). The proteins were electrotransferred onto polyvinylidene difluoride membranes (Immobilon-P; Millipore Corporation, Billerica, MA, USA) using a Bio-Rad Mini Gel Trans Blot cell and immunodecorated. Immunodecoration of actin was performed on cytosolic fraction utilizing a polyclonal antibody against A. thaliana protein (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Immunodecoration of binding protein (BiP) was performed on the microsomal fraction with an antibody against tobacco BiP, a generous gift from Dr. A. Vitale, Istituto di Biologia e Biotecnologia Agraria, CNR, Milano, Italy. The relative abundance of immunodecorated proteins was quantified using ImageJ 1.32 J program (Wayne Rasband, National Institutes of Health, USA, http://rsb.info.nih.gov/ij/).
Results
Effect of heat treatments and CoCl2 on the actin cytoskeleton
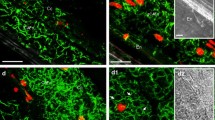
During the past decades, the plant cytoskeleton has been identified as an important target for a manifold of signal chains that are triggered either by environmental cues such as light, temperature, gravity, or touch or by internal signals including hormones or developmental inputs. More recently, it has emerged that the cytoskeleton is more than a mere target of signaling; in fact, it participates very actively in signal transduction itself (Nick 2007). In particular, the actin cytoskeleton has been identified as major target and effector of several signaling cascades, including those leading to PCD, in yeast, animals, and plants (Gourlay and Ayscough 2005; Hussey et al. 2006). Besides cytoplasmic shrinkage, a typical morphological hallmark often observed in cultured cells undergoing PCD with apoptotic features, including tobacco BY-2 cells, is presumably caused by breakdown of the cytoskeleton (Vacca et al. 2004; Hussey et al. 2006). Thus, we investigated whether heat treatment can induce changes in the actin cytoskeleton. Representative confocal extended focus images of tobacco BY-2 cells stained with phalloidin-TRITC for actin visualization are presented in Fig. 1. Long filaments of actin were present in control cells (a) as well as in cells treated at 35°C (b) or 45°C (c). The treatment at 50°C induced large fragmentation of the filaments which was already evident immediately after heat treatment (d) and progressively increased after 4 h and 16 h (e and f). Western blot experiments show that this fragmentation is accompanied by a decrease in the protein level only at the last experimental time (16 h after 50°C treatment; Fig. 2). Interestingly, both the actin depolymerization (Fig. 1g) and the decrease in the protein level (Fig. 2) induced by HS were prevented by the ethylene production inhibitor Co2+.

Confocal microscopy analysis of the effect of HS and Co2+ on actin cytoskeleton. a Control cells; b cells treated for 5 min at 35°C; c cells treated for 5 min at 45°C; d, e, f cells treated for 5 min at 50°C and observed 0, 4, 16 h after the return to the normal growth conditions, respectively; g cells fed with 100 μM CoCl2, treated for 5 min at 50°C and observed 16 h after the return to the normal growth conditions

Western blot analysis of the level of actin. Lane C control cells; lanes 0, 4, 16 cells treated for 5 min at 50°C and collected 0, 4, 16 h after the return to the normal growth conditions, respectively; 16 h + CoCl 2 cells fed with 100 μM CoCl2, treated for 5 min at 50°C and collected 16 h after the return to the normal growth conditions. The results of a typical experiment (n = 4) are shown. An arbitrary value of 100 was assigned to the amount of immunodecorated actin of the control cells. Fifty micrograms of proteins were run in each lane
Effect of heat treatments and CoCl2 on the ER
The ER is a point of integration of cell stress signals. In fact, the accumulation of unfolded proteins or the inhibition of transport between ER and Golgi apparatus results in the so-called ER stress response (Oyadomari et al. 2002). ER is also an important intracellular store of Ca2+, one of the most versatile and ubiquitous second messengers, and, thus, it may regulate the responses to stress conditions (Scorrano et al. 2003). So, we investigated the effect of heat treatments on the ER of tobacco BY-2 cells. The ER was examined at the morphological level by confocal microscopy utilizing the lipophilic dye DiOC6. Although this dye stains all organelles (Watanabe et al. 1998), it is possible to focus on the ER (Sabnis et al. 1997) and, thus, DiOC6 is one of the most commonly employed dyes for delineating the ER architecture in live plant cells (Hepler and Gunning 1998). Representative confocal extended focus images of tobacco BY-2 cells stained with this dye are shown in Fig. 3. The ER of control cells (a) is composed of tubular elements and, to a lesser extent, cisternae and this aspect was unaffected by the treatment at 35°C (b) and 45°C (c). On the contrary, marked changes in the ER architecture were observed after the treatment at 50°C. In fact, already after 4 h, the cisternae were prevalent (d) and after 16 h the tubular elements were almost completely absent and the ER appeared to be composed only of perforated cisternae (e). As demonstrated by western blot experiments (Fig. 4), these morphological modifications were accompanied by the accumulation of BiP, a widely distributed and highly conserved ER-resident molecular chaperone member of the HSP70 family of proteins the synthesis of which in plants is stimulated by a variety of abiotic and biotic stresses (see “Discussion” section). Again, the administration of Co2+ prevented the changes in the ER morphology (Fig. 3f) as well as the increase in BiP level induced by HS (Fig. 4).

Confocal microscopy analysis of the effect of HS and Co2+ on ER architecture. a Control cells; b cells treated for 5 min at 35°C; c cells treated for 5 min at 45°C; d, e cells treated for 5 min at 50°C and observed 4 and 16 h after the return to the normal growth conditions, respectively; f cells fed with 100 μM CoCl2, treated for 5 min at 50°C and observed 16 h after the return to the normal growth conditions

Western blot analysis of the level of BiP. Lane C control cells; lanes 4 and 16 cells treated for 5 min at 50°C and collected 4 and 16 h after the return to the normal growth conditions, respectively; 16 h + CoCl 2 cells fed with 100 μM CoCl2, treated for 5 min at 50°C and collected 16 h after the return to the normal growth conditions. The results of a typical experiment (n = 4) are shown. An arbitrary value of 100 was assigned to the amount of immunodecorated BiP of the control cells. Fifty micrograms of proteins were run in each lane
Effect of CoCl2 on HS-induced cell death and cytoplasmic shrinkage
The importance of the cellular structures and organelles in the regulation of PCD processes and the above-reported inhibition of the HS-induced changes by Co2+ prompted us to test the effect of the inhibitor of ethylene production on HS-induced cell death and cytoplasmic shrinkage. Interestingly, while Co2+ had little or no effect on control cells as well as on cells treated at 35°C or 45°C, both cell death and cytoplasmic shrinkage induced by treatment at 50°C were inhibited by Co2+ (Fig. 5a and b).

Effect of HS and Co2+ on cell viability (a) and cytoplasmic shrinkage (b). At the indicated times, the cells were collected and stained with Evans Blue (a) or observed for cytoplasmic shrinkage (b). Means ± SD (n ≥ 6) are presented
Discussion
In order to test the effects of heat stress on actin cytoskeleton and ER and the possible involvement of ethylene, in this work, tobacco BY-2 cultured cells were subjected to mild heat stress, medium heat stress, and HS (5 min at 35°C, 45°C or 50°C, respectively) in the absence or in the presence of Co2+, an inhibitor of ethylene biosynthesis.
Actin cytoskeleton
HS induces a large fragmentation of the actin filaments which is already evident immediately after the 5 min of treatment at 50°C and progressively increases over time (Fig. 1). Western blot experiments show that this fragmentation is accompanied by a decrease in the protein level at the last experimental time (16 h after 50°C treatment; Fig. 2). Both the actin depolymerization and the decrease in the protein level are prevented by the administration of CoCl2 (Figs. 1 and 2). The actinic component of cytoskeleton has been identified as a major target and effector of signaling cascades in both animals and plants (Gourlay and Ayscough 2005; Hussey et al. 2006). In particular, alteration of actin filaments dynamics can lead to PCD, in yeast, animals, and plants. Actually, reorganization of actin filaments occurs during developmental PCD in embryos of Picea abies and actin depolymerization is sufficient to induce PCD in self-incompatible pollen of Papaver rhoeas (Smertenko et al. 2003; Thomas et al. 2006). Finally, an actin-depolymerizing drug induces the accumulation of dead cells in sycamore cultures, while an actin-stabilizing drug reduces the cell death induced in these cultures by the phytotoxin fusicoccin (Malerba et al. 2008). These reports and the data presented in this paper suggest that the actin cytoskeleton may be involved in the pathway leading to PCD in response to different stimuli. The effect of CoCl2 strongly suggests a role for ethylene in the signaling transduction pathway(s) responsible for actin depolymerization.
ER architecture and BiP accumulation
HS remarkably affects ER architecture, which is mainly composed by tubular elements in control cells and by cisternae in treated cells (Fig. 3). This modification in ER architecture is accompanied by the accumulation of the ER-resident molecular chaperone BiP (Fig. 4). Both changes in ER architecture and BiP accumulation are prevented by CoCl2 (Figs. 3 and 4).
Changes in ER architecture in plant cells can be brought about by different factors or conditions including cell expansion, low temperatures, and treatment with different cell death inducers (Malerba et al. 2004 and reference therein). BiP is a widely distributed and highly conserved member of the HSP70 family of molecular chaperones. BiP is normally expressed during cell growth but its synthesis is induced in plants by a variety of biotic and abiotic stresses such as fungal and insect attack, cold and drought stress, and treatment with different cell death inducers (Malerba et al. 2004 and reference therein). Many of these stresses induce the accumulation of unfolded proteins in the ER that irreversibly bind BiP; this is thought to reduce the number of free BiP molecules leading to the induction of BiP transcription. The accumulation of the aggregates formed by unfolded proteins and BiP can cause modification in the ER architecture (Sparvoli et al. 2000) and overexpression of BiP in transgenic plants of tobacco relieves ER stress (Leborgne-Castel et al. 1999). Interestingly, a recent report shows that heat-shock-induced accumulation of hsp70 transcripts and HSP70 proteins in tobacco cells requires actin cytoskeleton reorganization. In particular, the actin microfilaments stabilizer jasplakinolide inhibits HS-induced accumulation of HSP70 and the destabilizer of microfilaments latrunculin B induces HSP70 accumulation in control cells (Suri and Dhindsa 2008). The data presented here support the view that changes in the ER architecture and BiP accumulation may be considered as a response of the ER to different stress conditions and suggest a role for ethylene in the signaling transduction pathway(s) responsible for the establishment of the response to HS.
Cell viability and cytoplasmic shrinkage
In addition to confirming the results obtained in this same material by Vacca et al. (2004) and in other materials, i.e., cucumber plants, carrot and Arabidopsis cell cultures (Balk et al. 1999; McCabe and Leaver 2000) our data show that HS-induced PCD can be prevented by the inhibitor of ethylene production Co2+ (Fig. 5). Ethylene is an important plant hormone that controls plant growth and developmental processes such as seed germination, root development, nodulation, flower and leaf senescence, and fruit development and ripening (Bleecker and Kende 2000). Enhanced ethylene production has been associated with reduced growth, accelerated senescence, and responses to several environmental stresses. In particular, heat-stress-induced ethylene production induces kernel abortion and increased maturation in developing wheat grains (Hays et al. 2007) and the perturbation of ethylene responses signal transduction pathway enhances tolerance to heat stress in Arabidopsis plants (Suzuki et al. 2005). Vice versa, ethylene is required for elicitin-induced oxidative burst but not for cell death induction in tobacco Bel W3 cell suspension cultures (Koehl et al. 2007). Our data strongly suggest the involvement of ethylene in the HS-induced PCD.
In conclusion, the data presented here indicate that HS induces depolymerization of actin microfilaments and changes in ER morphology accompanied by accumulation of the HSP70 BiP. The lack of effect of treatment at 35°C and 45°C suggests that the analyzed cell structures are not involved in the establishment of mild and medium heat stress conditions, at least in this experimental system. On the contrary, the strong modifications induced by the treatment at 50°C suggest that actin cytoskeleton and ER may be involved in the responses to HS. The effect of CoCl2 suggests a role for ethylene as regulative molecule in the responses to HS here observed.
Abbreviations
- BiP:
-
Binding protein
- DiOC6 :
-
3,3’-Dihexyloxacarbocyanine iodide
- ER:
-
Endoplasmic reticulum
- HS:
-
Heat shock
- PCD:
-
Programmed cell death
References
Balk J, Leaver CJ, McCabe PF (1999) Translocation of cytochrome c from the mitochondria to the cytosol occurs during heat-induced programmed cell death in cucumber plants. FEBS Lett 463:151–154
Bleecker AB, Kende H (2000) Ethylene: a gaseous signal molecule in plants. Annu Rev Cell Dev Biol 16:1–18
Broekaert WF, Delauré SL, De Bolle MFC, Cammue BPA (2006) The role of ethylene in host-pathogen interactions. Annu Rev Phytopathol 44:393–416
Dat JF, Lopez-Delgado H, Foyer CH, Scott IM (1998) Parallel changes in H2O2 and catalase during thermotolerance induced by salicylic acid or heat acclimation in mustard seedlings. Plant Physiol 116:1351–1357
Ferri KF, Kroemer G (2001) Organelle-specific initiation of cell death pathways. Nat Cell Biol 3:E255–E263
Gourlay CW, Ayscough KR (2005) The actin cytoskeleton in ageing and apoptosis. FEMS Yeast Res 5:1193–1198
Hall AE (2001) Crop responses to the environment. CRC, Boca Raton, FL
Hays DB, Do JH, Mason RE, Morgan G, Finlayson SA (2007) Heat stress induced ethylene production in developing wheat grains induces kernel abortion and increased maturation in a susceptible cultivar. Plant Sci 172:1113–1123
Hepler PK, Gunning BES (1998) Confocal fluorescence microscopy of plant cell. Protoplasma 201:121–157
Hussey PJ, Ketelaar T, Deeks MJ (2006) Control of the actin cytoskeleton in plant cell growth. Annu Rev Plant Biol 57:109–125
Kampinga HH, Brunsting JF, Stege GJ, Burgman PW, Konings AW (1995) Thermal protein denaturation and protein aggregation in cells made thermotolerant by various chemicals: role of heat shock proteins. Exp Cell Res 219:536–546
Klassen S, Bugbee B (2004) Ethylene synthesis and sensitivity in crop plants. HortScience 39:1546–1552
Koehl J, Djulic A, Kirner V, Nguyen TT, Heiser I (2007) Ethylene is required for elicitin-induced oxidative burst but not for cell death induction in tobacco cell suspension cultures. J Plant Physiol 164:1555–1563
Kotak S, Larkindale J, Lee U, von Koskull-Döring P, Vierling E, Scharf K-D (2007) Complexity of the heat stress response in plants. Curr Opin Plant Biol 10:310–316
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lam E (2004) Controlled cell death, plant survival and development. Nat Rev Mol Cell Biol 5:305–315
Leborgne-Castel N, Jelitto-Van Dooren EPWM, Crofts AJ, Denecke J (1999) Overexpression of BiP in tobacco alleviates endoplasmic reticulum stress. Plant Cell 11:459–469
Locato V, Gadaleta C, Vacca RA, De Gara L, de Pinto MC (2008) Production of reactive species and modulation of antioxidant network in response to heat shock: a critical balance for cell fate. Plant Cell Environ 31:1606–1619
Locato V, de Pinto MC, De Gara L (2009) Different involvement of mitochondrial, plastidial and cytosolic ascorbate-glutathione redox enzymes in heat shock responses. Physiol Plant 135:296–306
Malerba M, Cerana R, Crosti P (2004) Comparison between the effects of fusicoccin, Tunicamycin, and Brefeldin A on programmed cell death of cultured sycamore (Acer pseudoplatanus L.) cells. Protoplasma 224:61–70
Malerba M, Contran N, Tonelli M, Crosti P, Cerana R (2008) Role of nitric oxide in actin depolymerization and programmed cell death induced by fusicoccin in sycamore (Acer pseudoplatanus L.) cultured cells. Physiol Plant 133:449–457
McCabe PF, Leaver CJ (2000) Programmed cell death in cell cultures. Plant Mol Biol 44:359–368
Mittler R (2006) Abiotic stress, the field environment and stress combination. Trends Plant Sci 11:15–19
Müller J, Menzel D, Šamaj J (2007) Cell-type-specific disruption and recovery of the cytoskeleton in Arabidopsis thaliana epidermal root cells upon heat shock stress. Protoplasma 230:231–242
Nagata T, Nemoto Y, Hasezawa S (1992) Tobacco BY-2 cell line as the ‘HeLa’ cell in the cell biology of higher plants. Int Rev Cytol 132:1–30
Nick P (2007) The plant cytoskeleton—new jobs for a versatile network. Protoplasma 230:125–127
Oyadomari S, Araki E, Mori M (2002) Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis 7:335–345
Riechmann JL, Meyerowitz EM (1998) The AP2/EREBP family of plant transcription factors. Biol Chem 379:633–646
Sabnis RW, Deligeorgiev TG, Jachak MN, Dalvi TS (1997) DiOC6: a useful dye for staining the endoplasmic reticulum. Biotech Histochem 72:253–258
Scorrano L, Oakes SC, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300:135–139
Scott I, Logan DC (2008) Mitochondrial morphology transition is an early indicator of subsequent cell death in Arabidopsis. New Phytol 177:90–101
Smertenko AP, Bozhkov PV, Filonova LH, von Arnold S, Hussey PJ (2003) Re-organisation of the cytoskeleton during developmental programmed cell death in Picea abies embryos. Plant J 33:813–824
Sparvoli F, Faoro F, Daminati MG, Ceriotti A, Bollini R (2000) Misfolding and aggregation of vacuolar glycoproteins in plant cells. Plant J 24:825–836
Storozhenko S, Pauw PD, Van Montagu M, Inze D, Kushnir S (1998) The heat-shock element is a functional component of the Arabidopsis APX1 gene promoter. Plant Physiol 118:1005–1014
Suri SS, Dhindsa RS (2008) A heat-activated MAP kinase (HMAK) as activator of heat shock response in tobacco cells. Plant Cell Environ 31:218–226
Suzuki N, Rizhsky L, Bajad S, Liang H, Shuman J, Shulaev V, Mittler R (2005) Enhanced tolerance to environmental stress in transgenic plants expressing the transcriptional coactivator multiprotein bridging factor 1c. Plant Physiol 139:1313–1322
Suzuki N, Bajad S, Shuman J, Shulaev S, Mittler R (2008) The transcriptional co-activator MBF1c is a key regulator of thermotolerance in Arabidopsis thaliana. J Biol Chem 283:9269–9275
Thomas SG, Huang S, Li S, Staiger CJ, Franklin-Tong VE (2006) Actin depolymerization is sufficient to induce programmed cell death in self-incompatible pollen. J Cell Biol 174:221–229
Vacca RA, de Pinto MC, Valenti D, Passarella S, Marra E, De Gara L (2004) Production of reactive oxygen species, alteration of cytosolic ascorbate peroxidase, and impairment of mitochondrial metabolism are early events in heat shock-induced programmed cell death in tobacco Bright-Yellow 2 cells. Plant Physiol 134:1100–1112
Wang W, Vinocur B, Shoseyov O, Altman A (2004) Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci 9:244–252
Watanabe N, Che FS, Iwano M, Takayama S, Nakano T, Yoshida S, Isogai A (1998) Molecular characterization of photomixotrophic tobacco cells resistant to protoporphyrinogen oxidase-inhibiting herbicides. Plant Physiol 118:751–758
Wu C (1995) Heat shock transcription factors: structure and regulation. Annu Rev Cell Dev Biol 11:441–469
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Malerba, M., Crosti, P. & Cerana, R. Effect of heat stress on actin cytoskeleton and endoplasmic reticulum of tobacco BY-2 cultured cells and its inhibition by Co2+ . Protoplasma 239, 23–30 (2010). https://doi.org/10.1007/s00709-009-0078-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00709-009-0078-z