
Abstract
The new anthraquinonylhydrazones were obtained by the interaction of 9,10-dioxoanthracene-1-diazonium sulfates with a number of α- and β-carbonyl-containing compounds under modified conditions of the Japp–Klingemann reaction, and a probable mechanism of the formation has been proposed. It was found that hydrazones, unsaturated in the second position of the anthraquinone ring, containing acetyl or ethoxycarbonyl moieties in the ylidene part of the molecule, are capable of eliminating these fragments. It has been experimentally established that hydrazones, free rotation around the N=C bond of which is possible, exist as one isomer due to the presence of an intramolecular hydrogen bond in the molecule. The anthraquinonylhydrazone of dimedone with action against the bacteria strains of Staphylococcus aureus 209-P, Mycobacterium luteum B-917, and fungus Candida tenuis VKM Y-70 was found. The hydrazones of dimedone and barbituric acid with a higher trolox equivalent antioxidant coefficients of antioxidant action were found using CUPRAC assay. In addition, the hydrazones of dimedone and barbituric acid exhibited better activity against catalase enzyme. Correlations between the structure of the synthesized hydrazones and their antioxidant activity have been defined.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hydrazones are valuable objects in the organic synthesis due to their ability to interact with electrophile and nucleophile reagents [1]. Compounds with anti-inflammatory, analgesic, anticonvulsant, antituberculosis, antitumor, anti-HIV, antimicrobial, and other activity have been found among them [2].
The synthesis of hydrazones based on anthrone or its derivatives is presented in works [3,4,5]. In addition, the arylation of phenylacetylene and 2-naphthol with diazonium salts of 9,10-anthracenedione was studied, and several representatives of hydrazones were obtained [6]. Until recently, the interaction of diazonium salts of 9,10-anthracenedione with methylene-active compounds was represented by the synthesis of only one derivative, an intermediate, anthraquinonylhydrazone of ethyl pyruvate [7], obtained by the reaction of the corresponding 2-diazonium chloride 9,10-anthracenedione with ethyl 2-methylacetoacetate under classical conditions of the Japp–Klingemann reaction. At the same time, the anthraquinonylhydrazones of ketones and aldehydes are not described, that could possibly due to the tendency of (anthraquinone-1-yl)hydrazine to an intramolecular cyclocondensation [8].
Previously [9], we proposed an effective way of obtaining of anthraquinonylhydrazones 4a–4e (Scheme 1) containing acyl and/or alkoxycarbonyl moieties in the ylidene part of the molecule, which are convenient for further chemical transformations [10].

Taking into account the limited number of publications on the synthesis and transformation of hydrazones that contain the 9,10-anthraquinone fragment in the hydrazone part, it seems promising to obtain new derivatives by the coupling reaction of 9,10-dioxoanthracene-1-diazonium sulfates with α- and β-carbonyl-containing compounds for further chemical conversions to obtain biologically active substances.
Results and discussion
Chemistry
Continuing research in a direction of the synthesis and modification of anthraquinonylhydrazones, in this paper, we reported a synthesis of new hydrazones by the interaction of freshly prepared diazonium salt 2 obtained by the method of [11] with acetone (3a), ethylmethyl ketone (3b), acetylacetone (3c), malonic esters 3d, and acetoacetate 3e esters under the modified conditions of the Japp–Klingemann reaction in water (Scheme 1).
The formation of hydrazones 4a–4e and 5a–5e probably occurs according to the following mechanism (Scheme 1). The initial step of the reaction is the azo coupling of diazonium sulfate 1 or 2 with the enol form of compound 3 in an aqueous medium, which leads to intermediate A, which then, as a result of the thermodynamically favorable 1,5-hydrogen migration, is converted to hydrazone 4 or 5.
The hydrazone structures of 4a–4e [9] and 5a–5e are confirmed by the presence of corresponding signals of the protons and carbon atoms of the carbonyl-containing groups in the 1H and 13C NMR spectra, as well as by the presence of corresponding molecular peaks in the spectra of liquid chromatography–mass spectrometry (LC–MS). In the 1H and 13C NMR spectra of compounds 4a, 4b, 4e, 5a, 5b, and 5e, for which rotation around a bond is possible, only one set of resonance signals is present, which indicates the existence of these derivatives in the form of one geometric isomer. This behavior is explained by the presence of an intramolecular hydrogen bond between the NH group of the hydrazone fragment and the proton acceptor in the ylidene part of the molecule (C=O of the ester group of the substituent [12, 13]), which is confirmed by the corresponding weak-polar placement of the proton signal of the NH group [14,15,16] in 1H NMR spectra (12.29–14.48 ppm).
In turn, the presence of an electron-withdrawing conjugated quinone substituent in the hydrazone fragment in combination with an intramolecular hydrogen bond causes the bifurcation of proton of the NH group in the hydrazone fragment (Fig. 1) [17], which lead to the complete predominance of only one geometrical isomer.

Bifurcated hydrogen in hydrazones
The signals of the ylidene proton in compounds 4a, 4b and 5a, 5b are superimposed with the protons of the anthraquinone fragment in the range from 7.71 to 7.99 ppm. The proton signal of the NH group of the anthraquinonylhydrazones 4a–4e and 5a–5e resonates as a wide singlet within the range of 12.29–14.48 ppm.
It was found that the formation of the 4d, 4e compounds is accompanied by the by-product 4f in an amount of 10 and 13%, respectively, which was separated from the main product by chromatography on silica gel (eluent benzene) (Scheme 2).

It should be noted that in the case of the presence of a carboxyl group in the second position of the anthraquinone ring in the compounds 5d, 5e, the formation of the by-product of elimination of the acetyl or ethoxyl fragment is not observed. It is probably explained by the stabilizing effect of the COOH group on an intramolecular hydrogen bond in the formed hydrazones 5d, 5e. The formation of hydrazone 4f was confirmed by the data of 1H NMR, 13C NMR, and LC–MS. In particular, in the 1H NMR spectrum of 4f, the proton signal of the methylidene group superimposes with the protons of the anthraquinone fragment. The ethoxyl fragment is represented by multiplets of two protons of the methylene group at 4.35 ppm and a triplet of three protons of the methyl group at 1.30 ppm. In the 13C NMR spectrum, in addition to the signals of the carbon in anthraquinone and methylidene fragments, is characterized by the presence of carbon signals of one ethoxycarbonyl group. The LC–MS spectrum contains the corresponding molecular peak with a mass of 322 [M+].
Functionalization of the ylidene part with carbocyclic and heterocyclic fragments has been carried out by coupling of the 9,10-dioxoanthracene-1-diazonium sulfates 1 and 2 with cyclic β-dicarbonyl compounds 6a–6c (5,5-dimethyl-1,3-cyclohexanedione, 2,4,6-pyrimidinetrione, and 2-thioxopyrimidine-4,6-dione) with obtaining the hydrazones 7a–7c and 8a–8c (Scheme 3).

Analysis of the data of 1H and 13C NMR spectra of the obtained hydrazones 7a–7c, 8a–8c showed that compounds 7a, 7b, 8a, 8b exist in the ketone form and hydrazones 7c, 8c in the thioketone form in DMSO-d6 solution. In the 1H NMR spectra of compounds 7b, 7c and 8b, 8c, the singlet signals of the proton of NH group of the hydrazone fragment are presented in the range from 13.45 to 15.98 ppm and amide NH protons of a heterocyclic fragment are observed in the range 11.61–11.90 ppm, respectively.
Antimicrobial activity
The synthesized compounds 4a–4e, 5a–5e, 7a–7c, 8a–8c were evaluated for their antibacterial and antifungal activity against strains of Escherichia coli B-906, Staphylococcus aureus 209-P, Mycobacterium luteumB-917, Candida tenuis VKM Y-70, and Aspergillus niger VKM F-1119 by the diffusion technique [18] and by the serial dilution technique (determination of minimal inhibition concentrations MIC) [19]. Their activities were compared with those of the known antibacterial agent vancomycin and antifungal agent nystatin (control C).
The strain of bacteria E. coli appeared not to be sensitive to the tested hydrazones 4a–4e, 5a–5e, 7a–7c, 8a–8c investigated by the diffusion technique at concentrations of 0.1 and 0.5% (Table 1). S. aureus was highly sensitive to compound 8a and moderately sensitive to derivative 8b (the diameter of the inhibition zone was 20.0 and 14.0 mm, respectively) at a concentration of 0.5%. The test culture of bacteria M. luteum was highly sensitive to hydrazone 8a at the same concentration (the diameter of the inhibition zone was 21.0 mm). Other compounds were not active against these strains of bacteria. Strains of fungi C. tenuis and A. niger were slightly sensitive to compounds 5c, 5e, 8b at a concentration of 0.5% (the diameter of the inhibition zone was 7.0–15.0 mm).
Evaluation of the antibacterial activity of synthesized compounds using the serial dilution technique (Table 2) showed that only compounds 7c, 8a, 8b have low antibacterial action at MIC 250.0 and 500.0 μg/cm3 against strain E. coli. The hydrazones 4a, 4b, 5c, 8a, 8b have MIC 125–500 μg/cm3 against test-culture bacteria S. aureus. Strain M. luteum was sensitive at MIC 62.5–500.0 μg/cm3 to the compounds. Derivatives 4a, 4b, 5e, 7c, 8a, 8b show antifungal action against test-culture C. tenuis at MIC 62.5–250.0 μg/cm3. Only compound 8b had inhibitory effect against A. niger (MIC 500.0 μg/cm3). The other hydrazones did not show any antifungal activity at the studied concentration against fungi C. tenuis and A. niger.
CUPRAC antioxidant capacity
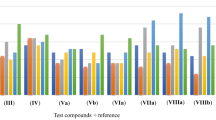
Antioxidant capacity of anthraquinonylhydrazones 4a–4e, 5a–5e, 7a–7c, 8a–8c was determined by comparing with trolox (TR) as the standard reference compound using CUPRAC assay (at room temperature) [20]. The CUPRAC molar absorption coefficient (ε) of the tested compound divided by that of TR under the same conditions gave the TEAC-CUPRAC values (Fig. 2). Amongst the compounds screened for antioxidant capacity, 8a and 8b exhibited the highest antioxidant capacities, and the trolox equivalent antioxidant coefficients (TEAC) of these compounds were 0.89 and 0.87, respectively (Fig. 2). Based on the experimental testing of hydrazone derivatives, the effect of substituents on the side of the ylidene fragment on the antioxidant activity of the compounds has been established. Antioxidant activity of 5d and 8a, 8b approximates to the level of the reference preparation. Replacement of the acetyl fragments in compound 5c with ethoxycarbonyl (compound 5d) increases the TEAC coefficient, and replacing one of ethoxycarbonyl substituents (5c) with acetyl fragment (5e) decreases this index. The presence of the carboxyl group in the second position of the anthraquinone ring in compounds 5a–5e and 8a–8c increases the TEAC coefficient in comparison with hydrazones 4a–4e and 7a–7c.

TEAC coefficients (N = 3) of the hydrazones with respect to the CUPRAC assay
All synthesized compounds were tested in vitro for their catalase activity [21], which is an integral part of antioxidant protection in cells, using the CUPRAC method. The inhibition values are given in Fig. 3.

Catalase enzyme activities (U/cm3) of the hydrazones
The results (Fig. 3) showed that the highest activity was characteristic for compounds 4d (0.77 U/cm3), 5e (0.75 U/cm3), 8a (0.79 U/cm3), 8b (0.78 U/cm3), and 8c (0.76 U/cm3). The presence of the carboxyl group in the second position of the anthraquinone ring in compounds 8a–8c increases the catalase enzyme activities in comparison with hydrazones 7a–7c. Replacement of one ethoxycarbonyl fragment in compound 5d with acetyl fragment (compound 5e) increases the TEAC coefficient. The results of catalase enzyme activity of hydrazones 8a, 8b are consistent with the data of TEAC investigation.
Conclusion
The synthesis and probable mechanism of formation of new anthraquinonylhydrazones by the coupling reaction of 9,10-dioxoanthracene-1-diazonium sulfates with an active α-ketones and β-carbonyl compounds in neutral aqueous medium under mild conditions are presented. It was determined that the obtained hydrazones exist in the form of a single geometric isomer due to the presence of an intramolecular hydrogen bond between the NH group of the hydrazone fragment and the proton acceptor in the ylidene part of the molecule. All the synthesized compounds were tested for their antibacterial, antifungal, and antioxidant activity. The anthraquinonylhydrazone 8a exhibited a promising antimicrobial activity against bacteria S. aureus, M. luteum, and antifungal effect against fungus C. tenuis. All compounds were tested for their antioxidant capacity using the CUPRAC method and for inhibitory activities against catalase enzyme. The synthesized compounds 8a, 8b exhibited better antioxidant capacity than the other compounds. The results revealed that 8a, 8b exhibited high catalase enzyme inhibition activity compared to the other hydrazones.
Experimental
Melting points were measured using a Buchi B-540 melting point apparatus. Elemental analysis was performed on a Perkin Elmer 2400 CHN-analyzer, and their results were found to be in good agreement with the calculated values. 1H NMR spectra (in DMSO-d6) and 13C NMR spectra (in CF3COOD) were recorded on a Varian Mercury-400 spectrometer with TMS as internal standard. Mass spectra were recorded on an Agilent 1100 Series G1956B LC/MSD SL LCMS system, using electrospray ionization at atmospheric pressure (70 eV). Thin-layer chromatography (TLC) was performed on Merck silica gel plates (60F254) in benzene, and detection was carried out with ultraviolet light (254 nm).
All chemicals were of reagent grade and used without further purification. Diazonium salts 1 and 2 were obtained according to a published method [9] from 1-aminoanthracene-9,10-dione and 1-amino-9,10-dioxo-9,10-dihydroanthracene-2-carboxylic acid (Sigma-Aldrich). Hydrazones 4a–4e were obtained by the method described in [9].
General method of obtaining of hydrazones 5a–5e, 7a–7c, 8a–8c
9,10-Dioxoanthracene-1-diazonium sulfate (1 or 2) (9 mmol) was added to a solution of appropriate compound containing active methylene group (3a–3e, 6a–6c) (30 mmol) in 150 cm3 water at 0–5 °C. The reaction mixture was stirred for 1 h at 0–5 °C. The precipitate was filtered off, washed with water (2 × 100 cm3) and dried. If necessary, the products can be purified by recrystallization from acetic acid.
Ethyl 2-[2-(9,10-dioxo-9,10-dihydroanthracene-1-yl)hydrazineylidene]acetate (4f, C18H14N2O4)
Compound 4f was separated from the main product (4d or 4e) by column chromatography on silica gel (Merck silica gel 60 H), eluent benzene. Yield 10% (from compound 4d), 13% (from compound 4e); m.p.: 238–239 °C; 1H NMR (400 MHz, DMSO-d6): δ = 1.31 (t, 3H, J = 7.2 Hz, CH3), 4.35 (m, 2H, CH2), 7.78–7.88 (m, 5H, 4HAr and CH=), 8.03–8.15 (m, 3H, HAr), 12.55 (br s, 1H, NH) ppm; 13C NMR (100 MHz, CF3COOD): δ = 12.31 (CH3), 63.60 (CH2), 114.63, 121.52, 122.75, 128.02, 128.66, 130.04, 130.57, 133.13, 133.32, 134.82, 135.48, 135.88, 147.14 (CAr), 163.30, 185.31, 185.82 (C=O) ppm; LC–MS (70 eV): m/z = 322 (M+).
9,10-Dioxo-1-[2-(2-oxopropylidene)hydrazinyl]-9,10-dihydroanthracene-2-carboxylic acid (5a, C18H12N2O5)
Yield 62%; m.p.: 260–261 °C; 1H NMR (400 MHz, DMSO-d6): δ = 2.07 (s, 3H, CH3), 7.92–7.99 (m, 5H, 4HAr and CH=), 8.16–8.21 (m, 2H, HAr), 12.98 (br s, 1H, OH), 14.01 (br s, 1H, NH) ppm; 13C NMR (100 MHz, CF3COOD): δ = 22.78 (CH3), 114.70, 121.86, 122.85, 127.19, 127.43, 127.64, 132.05, 133.45, 135.00, 135.62, 136.22, 138.33, 145.69 (CAr), 169.40, 185.41, 187.06, 204.79 (C=O) ppm; LC–MS (70 eV): m/z = 336 (M+).
9,10-Dioxo-1-[2-(2-oxobutylidene)hydrazinyl]-9,10-dihydroanthracene-2-carboxylic acid (5b, C19H14N2O5)
Yield 56%; m.p.: 278–280 °C; 1H NMR (400 MHz, DMSO-d6): δ = 2.14 (t, 3H, CH3, J = 7.2 Hz), 2.42 (m, 2H, CH2), 7.68–7.76 (m, 5H, 4HAr and CH=), 7.87–8.14 (m, 2H, HAr), 13.07 (br s, 1H, OH), 14.03 (br s, 1H, NH) ppm; 13C NMR (100 MHz, CF3COOD): δ = 7.34 (CH3), 22.27 (CH2), 114.51, 121.82, 122.39, 127.45, 127.71, 132.23, 133.36, 133.87, 134.75, 135.57, 136.15, 145.73, 146.55 (CAr), 170.09, 186.20, 187.48, 203.69 (C=O) ppm; LC–MS (70 eV): m/z = 350 (M+).
1-[2-(2,4-Dioxopentan-3-ylidene)hydrazinyl]-9,10-dioxo-9,10-dihydro-anthracene-2-carboxylic acid (5c, C20H14N2O6)
Yield 68%; m.p.: > 310 °C; 1H NMR (400 MHz, DMSO-d6): δ = 2.30 (s, 6H, CH3), 7.83–7.93 (m, 4H, HAr), 8.15–8.23 (m, 2H, HAr), 12.74 (br s, 1H, OH), 13.22 (br s, 1H, NH) ppm; 13C NMR (100 MHz, CF3COOD): δ = 24.68 (CH3), 30.24 (CH3), 121.01, 124.75, 127.49, 127.81, 127.89, 131.75, 133.78, 135.30, 135.76, 135.85, 136.09, 136.17, 141.92 (CAr), 172.90, 184.78, 185.29, 200.39, 203.04 (C=O) ppm; LC–MS (70 eV): m/z = 378 (M+).
1-[2-(1,3-Diethoxy-1,3-dioxopropan-2-ylidene)hydrazinyl]-9,10-dioxo-9,10-dihydroanthracene-2-carboxylic acid (5d, C22H18N2O8)
Yield 67%; m.p.: 295–297 °C; 1H NMR (400 MHz, DMSO-d6): δ = 1.36 (s, 6H, CH3), 4.28–4.41 (m, 4H, CH2), 7.82–7.85 (m, 4H, HAr), 7.89–8.13 (m, 2H, HAr), 13.11 (br s, 1H, OH), 14.52 (br s, 1H, NH) ppm; 13C NMR (100 MHz, CF3COOD): δ = 13.08, 13.13 (CH3), 63.46, 63.98 (CH2), 116.88, 122.64, 123.44, 124.30, 127.36, 132.11, 133.53, 133.49, 133.67, 134.84, 135.54, 135.86, 144.45 (CAr), 166.80, 167.91, 169.65, 186.04, 186.71 (C=O) ppm; LC–MS (70 eV): m/z = 438 (M+).
1-[2-(1-Ethoxy-1,3-dioxobutan-2-ylidene)hydrazinyl]-9,10-dioxo-9,10-dihydroanthracene-2-carboxylic acid (5e, C21H16N2O7)
Yield 60%; m.p.: 224–226 °C; 1H NMR (400 MHz, DMSO-d6): δ = 1.39 (t, 3H, J = 7.2 Hz, CH3), 2.37 (s, 3H, CH3), 4.45 (q, 2H, J = 7.2 Hz, CH2), 7.90–7.92 (m, 4H, HAr), 8.12–8.17 (m, 2H, HAr), 13.43 (br s, 1H, OH), 14.08 (br s, 1H, NH) ppm; 13C NMR (100 MHz, CF3COOD): δ = 12.25 (CH3), 21.83 (CH3), 64.17 (CH2), 124.15, 127.39, 127.57, 127.99, 128.05, 131.82, 133.15, 133.55, 133.57, 135.51, 135.58, 136.02, 141.00 (CAr), 172.20, 173.35, 184.60, 186.71, 201.80 (C=O) ppm; LC–MS (70 eV): m/z = 408 (M+).
1-[2-(4,4-Dimethyl-2,6-dioxocyclohexylidene)hydrazinyl]anthracene-9,10-dione (7a, C22H18N2O4)
Yield 55%; m.p.: 243–245 °C; 1H NMR (400 MHz, DMSO-d6): δ = 1.09 (s, 6H, CH3), 2.67 (s, 2H, CH2), 2.75 (s, 2H, CH2), 7.93–8.02 (m, 4H, HAr), 8.15–8.22 (m, 2H, HAr), 8.36–8.38 (m, 1H, HAr), 16.05 (br s, 1H, NH) ppm; 13C NMR (100 MHz, CF3COOD): δ = 26.54, 26.65 (CH3), 30.16 (C(CH3)2), 50.58, 50.71 (CH2), 120.07, 123.49, 123.95, 127.36, 127.62, 130.12, 131.09, 131.95, 133.48, 133.97, 134.18, 135.73, 142.33 (CAr), 185.39, 186.13, 198.64, 201.90 (C=O) ppm; LC–MS (70 eV): m/z = 374 (M+).
5-[2-(9,10-Dioxo-9,10-dihydroanthracene-1-yl)hydrazinylidene]pyrimidine-2,4,6-(1H,3H,5H)-trione (7b, C18H10N4O5)
Yield 66.5%; m.p.: > 300 °C; 1H NMR (400 MHz, DMSO-d6): δ = 7.89–7.99 (m, 4H, HAr), 8.05–8.19 (m, 3H, HAr), 11.66 (br s, 1H, NH), 11.90 (br s, 1H, NH), 15.90 (br s, 1H, NH) ppm; 13C NMR (100 MHz, CF3COOD): δ = 119.59, 120.72, 122.55, 124.65, 126.85, 131.56, 132.21, 133.64, 133.72, 134.09, 135.97, 137.02, 144.15 (CAr), 150.35, 159.14, 160.09, 184.52, 185.30 (C=O) ppm; LC–MS (70 eV): m/z = 362 (M+).
5-[2-(9,10-Dioxo-9,10-dihydroanthracene-1-yl)hydrazono]-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (7c, C18H10N4O4S)
Yield 67%; m.p.: > 300 °C; 1H NMR (400 MHz, DMSO-d6): δ = 7.88–7.92 (m, 4H, HAr), 8.15–8.21 (m, 3H, HAr), 11.41 (br s, 1H, NH), 11.61 (br s, 1H, NH), 15.78 (br s, 1H, NH) ppm; 13C NMR (100 MHz, CF3COOD): δ = 114.47, 119.67, 124.67, 125.72, 126.70, 132.56, 133.11, 133.23, 133.30, 134.43, 134.86, 137.48, 142.44 (CAr), 159.68, 160.21 (C=O), 174.58 (C=S), 185.07, 185.89 (C=O) ppm; LC–MS (70 eV): m/z = 378 (M+).
1-[2-(4,4-Dimethyl-2,6-dioxocyclohexylidene)hydrazinyl]-9,10-dioxo-9,10-dihydroanthracene-2-carboxylic acid (8a, C23H18N2O6)
Yield 56%; m.p.: 274–276 °C; 1H NMR (400 MHz, DMSO-d6): δ = 1.07 (s, 6H, CH3), 2.62 (s, 2H, CH2), 2.76 (s, 2H, CH2), 7.94–7.99 (m, 3H, HAr), 8.08 (m, 1H, HAr), 8.17–8.23 (m, 2H, HAr), 13.30 (br s, 1H, OH), 16.02 (br s, 1H, NH) ppm; 13C NMR (100 MHz, CF3COOD): δ = 26.74, 26.84 (CH3), 30.53 (C(CH3)2), 43.50, 43.63 (CH2), 121.05, 126.04, 127.50, 127.87, 128.72, 131.16, 131.75, 133.56, 135.12, 135.43, 135.69, 135.99, 140.71 (CAr), 173.14, 184.16, 185.20, 199.05, 200.33 (C=O) ppm; LC–MS (70 eV): m/z = 418 (M+).
9,10-Dioxo-1-[2-(2,4,6-trioxotetrahydropyrimidin-5(2H)-ylidene)hydrazinyl]-9,10-dihydroanthracene-2-carboxylic acid (8b, C19H10N4O7)
Yield 57%; m.p.: > 300 °C; 1H NMR (400 MHz, DMSO-d6): δ = 7.91–7.99 (m, 3H, HAr), 8.04–8.05 (m, 1H, HAr), 8.14–8.20 (m, 2H, HAr), 11.39 (br s, 1H, NH), 11.66 (br s, 1H, NH), 13.45 (br s, 1H, OH), 15.70 (1H, br s, NH) ppm; 13C NMR (100 MHz, CF3COOD): δ = 114.23, 119.31, 124.69, 125.89, 126.85, 127.10, 131.60, 133.80, 134.56, 135.36, 135.72, 137.31, 142.46 (CAr), 150.23, 159.82, 160.09, 170.20, 184.62, 185.07 (C=O) ppm; LC–MS (70 eV): m/z = 406 (M+).
1-[2-(4,6-Dioxo-2-thioxotetrahydropyrimidin-5(2H)-ylidene)hydrazinyl]-9,10-dioxo-9,10-dihydroanthracene-2-carboxylic acid (8c, C19H10N4O6S)
Yield 59%; m.p.: > 300 °C; 1H NMR (400 MHz, DMSO-d6): δ = 7.90–7.93 (m, 4H, HAr), 8.17–8.23 (m, 2H, HAr), 11.66 (br s, 1H, NH), 11.88 (br s, 1H, NH), 13.49 (br s, 1H, OH), 15.98 (br s, 1H, NH) ppm; 13C NMR (100 MHz, CF3COOD): δ = 117.58, 119.86, 123.27, 124.92, 126.11, 131.82, 132.46, 133.14, 133.47, 136.31, 137.40, 137.86, 143.49 (CAr), 159.31, 160.06, 169.49 (C=O), 175.22 (C=S), 184.58, 185.15 (C=O) ppm; LC–MS (70 eV): m/z = 422 (M+).
Antimicrobial activity
Diffusion method
Antibacterial activity of compounds was evaluated by diffusion in peptone on nutrient medium (meat-extract agar for bacteria; wort agar for fungi). The microbial loading was 109 cells (spores)/1 cm3. The required incubation periods were as follows: 24 h at 35 °C for bacteria and 48–72 h at 28–30 °C for fungi. The results were recorded by measuring the zones surrounding the disk. Control disk contained vancomycin (for bacteria) or nystatin (for fungi) as a standard.
Serial dilution method
Testing was performed in a flat-bottomed 96-well tissue culture plate. The tested compounds were dissolved in dimethyl sulfoxide (DMSO) to the necessary concentration. The exact volume of solution of compounds was brought in nutrient medium. The inoculum of bacteria and fungi was in nutrient medium (meat-extract agar for bacteria; wort agar for fungi). The duration of incubation was 24–72 h at 37 °C for bacteria and 30 °C for fungi. The results were estimated according to the presence or absence of microorganism growth.
Antioxidant capacity
All reagents and solvents were of analytical reagent grade. Neocuproine (2,9-dimethyl-1,10-phenanthroline) was purchased from Sigma Chemical Co. (Steinheim, Germany). Copper(II) chloride dihydrate, ammonium acetate (NH4Ac), absolute ethanol (EtOH), methanol (MeOH), and dimethyl sulfoxide (DMSO) were purchased from Merck (Darmstadt, Germany). The spectra and absorption measurements were recorded in matched Helma quartz cuvettes of 1 cm thickness using a Perkin Elmer Lambda 35 UV–Vis spectrophotometer.
Preparation of solutions
The standard solutions of synthesized compounds were prepared in DMSO at a concentration of 1.0 mM. All standard solutions were stored at + 4 °C prior to analysis. The CuCl2 solution (10.0 mM) and ammonium acetate buffer solution (1 M, pH 7.0) were prepared in distilled water and neocuproine (Nc) solution (7.5 mM) in absolute ethanol.
CUPRAC antioxidant capacity
The CUPRAC method, as described by Apak et al. [20], is based on the reduction of a cupric neocuproine complex (Cu(II)-Nc) by compounds having antioxidant capacity to the yellow–orange coloured cuprous chelate (Cu(I)-Nc). The CUPRAC reaction mixture consisted of 1 cm3 CuCl2·2H2O (10 mM), 1 cm3 Nc (7.5 mM), 1 cm3 pH 7 NH4Ac buffer solution (1.0 M), × cm3 synthesized compound, and (1.1-×) cm3 DMSO. The mixture (4.1 cm3) was then incubated at 25 °C for 30 min, and afterwards, the absorbance at 450 nm was recorded against a reagent blank. Under the described experimental conditions, the calibration curves (absorbance versus concentration graphs) of each compound were constructed, and their TEAC coefficients were calculated.
Catalase enzyme activity
The method described by Bekdeşer et al. was employed for the determination of catalase enzyme activity [21]. The reaction mixture consisted of 0.5 cm3 H2O2 (1.0 mM), 1.8 cm3 H2O, 0.1 cm3 catalase solution (3.691 U/cm3), and 0.2 cm3 synthesized compound (1.0 mM). This mixture (total volume 2.6 cm3) was then incubated at 25 °C. After 30 min incubation period, the optical CUPRAC sensor [Cu(II)-Nc-impregnated membrane] was taken out and immersed in a test tube consisted of 2.0 cm3 of the incubation reaction mixture + 6.2 cm3 of EtOH. After 30 min agitation, the coloured membrane was taken out and its absorbance was recorded at 450 nm against that of a blank membrane without tested sample.
References
Belskaya N, Dehaen W, Bakulev V (2010) Arkivoc 2010:275
Ali R, Marella A, Alam T, Naz R, Akhter M, Shaquiquzzaman M, Saha R, Tanwar O, Alam M, Hooda J (2012) Indones J Pharm 23:193
Antonini I, Polucci P, Cola D, Palmieri G, Martelli S, Bontemps-Gracz M (1993) Farmaco 48:1641
Loskutov VA (2000) Russ J Org Chem 36:1478
Vaisburg AF, Etzlstorfer C, Falk H (1994) Monatsh Chem 125:1121
Bulhakova NA (2002) Synthesis, structure and properties of some 9,10-anthraquinone derivatives containing a nitrogen–nitrogen bond. Ph.D. thesis, Krasnoyarsk State Pedagogical University named after V.P. Astafieva, Krasnoyarsk ([in Russian])
Vorob’eva SL, Buyanov VN, Levina II, Suvorov NN (1989) Chem Heterocycl Compd 25:58
Kim MK, Wiemer DF (2004) Tetrahedron Lett 45:4977
Stasevych MV, Zvarych VI, Lunin VV, Vovk MV, Novikov VP (2017) Russ J Org Chem 53:468
Stasevych MV, Zvarych VI, Lunin VV, Khomyak SV, Vovk MV, Novikov VP (2017) Chem Heterocycl Compd 53:927
Zvarych V, Stasevych M, Lunin V, Deniz NG, Sayil C, Ozyurek M, Guclu K, Vovk M, Novikov V (2016) Monatsh Chem 147:2093
Butler RN, Quinn KF, Welke B (1992) J Chem Soc Chem Commun 1992:1481
Allen FH, Groom CR, Liebeschuetz JW, Bardwell DA, Olsson TSG, Wood JPA (2012) Chem Inf Model 52:857
Landge SM, Aprahamian I (2009) J Am Chem Soc 131:18269
Su X, Aprahamian I (2011) Org Lett 13:30
Landge SM, Tkatchouk E, Benítez D, Lanfranchi DA, Elhabiri M, Goddard WA, Aprahamian I (2011) J Am Chem Soc 133:9812
He L, Lu L, Zhang S, Freeman HS (2010) Color Technol 126:92
NCCLS (1990) Performance Standards for antimicrobial disk susceptibility tests, 4th edn: Approved standards. Document M2-A4, Villanova, PA
NCCLS (1998) Reference Method for broth dilution antifungal susceptibility testing of conidium forming filamentous fungi: proposed standard. Document M38-P, Wayne, PA
Apak R, Güçlü K, Özyürek M, Karademir SE (2004) J Agric Food Chem 52:7970
Bekdeşer B, Özyürek M, Güçlü K, Alkan FÜ, Apak R (2014) Spectrochim Acta Part A 132:485
Acknowledgements
The authors would like to express their gratitude to the Ministry of Education and Science of Ukraine, Scientific Research Project (Project number: 0116U004138) for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Stasevych, M., Zvarych, V., Lunin, V. et al. Synthesis and investigation of antimicrobial and antioxidant activity of anthraquinonylhydrazones. Monatsh Chem 149, 1111–1119 (2018). https://doi.org/10.1007/s00706-018-2157-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-018-2157-3