
Abstract
The Aurora kinases play a key role in mitosis and are overexpressed in multiple human tumor types; there has been considerable interest in developing Aurora kinase inhibitors as antitumor agents, particularly Aurora A and Aurora B kinases. A series of novel hydrazide hydrochlorides of pyrazolo[1,5-a]pyrimidine carboxamides were designed and synthesized and their inhibitory activities against Aurora kinases were evaluated. Some of the tested compounds exhibited low micromolar to nanomolar activity with respect to the inhibition of Aurora A kinase. The most potent compound in this series was found to be a potent inhibitor of Aurora A in an HTRF enzymatic assay with an IC50 as low as 23 nM. A structure–activity relationship study indicated that halogen substitution in the benzene ring of amide plays an important role in kinase inhibitory potency.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aurora kinases are the members of serine/threonine kinases and they are strongly associated with human cancer [1, 2]. Humans feature a family of Aurora kinases with three members: isoforms A, B, and C. Both Aurora A and Aurora B are significantly overexpressed in a wide range of human cancer cell lines. The overexpression of Aurora A causes aberrant phosphorylation of normal cell cycle targets and cytoplasmic targets, leading to chromosomal instability, oncogenic transformation, tumor progression, and development of chemoresistance [3]. Similarly, the overexpression of Aurora B increases the phosphorylation of histone H3, forming more aggressive tumors in transgenic mouse models [4, 5]. Mechanistically, Aurora kinases (A, B, and C) are regulatory proteins and play key roles in the mitotic events of cell division [6, 7].
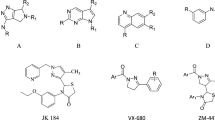
Over the past decade, extensive research has been carried out towards the discovery of Aurora-selective small-molecule inhibitors. As a result, a handful of small-molecule Aurora inhibitors have been identified. Among them, VX-680 [8] and SNS-314 [9, 10] have entered human clinical trials as pan-Aurora kinase inhibitors; MLN8237 [11] and MK-5108 [12] are undergoing clinical assessment as Aurora A-specific inhibitors as shown in Fig. 1. Recently, selective Aurora B inhibitor barasertib (AZD1152) [13] and the pan-aurora kinase inhibitor danusertib (PHA-739358) [14] have been utilized in clinical trials for the treatment of leukemia, myeloma, and other solid tumors [15, 16].

The structures of Aurora kinase inhibitors
In addition, a number of several heterocyclic amide derivatives have been detailed as potent Aurora kinase inhibitors. We envisioned that there are less reports for amides from pyrazolo[1,5-a]pyrimidine pharmacophore in this category and it was thought it would be worthwhile to synthesize some new pyrazolo[1,5-a]pyrimidine-bearing amides. In this present article, we describe the design, synthesis, and biochemical evaluation of a novel series of pyrazolo[1,5-a]pyrimidine analogs with representative example as shown in Fig. 2 showing good antiproliferative activity against both Aurora A and Aurora B.

Chemical structure of compound 11f which showed good aurora kinase activity
Results and discussion
Chemistry
The novel hydrazide hydrochloride derivatives of 6-amino-pyrazolo[1,5-a]pyrimidine-3-carboxamides described herein were synthesized over eight consecutive steps. The first four steps involved the synthesis of novel compound 6-aminopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester 5 as shown in Scheme 1. Previously described methods were used for the synthesis of both 5-amino-1H-pyrazole-4-carboxylic acid methyl ester (2) [17–19] and 6-bromopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester 3 [20]. The treatment of ester 3 with benzophenone imine in presence of cesium carbonate, xantphos, and Pd2(dba)3 in 1,4-dioxane at 110 °C for 10 h gave methyl 6-[(diphenylmethylidine)amino] pyrazolo[1,5-a]pyrimidine-3-carboxylate 4 [21–23]. The treatment of imine with HCl in MTBE gave 6-aminopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester 5.

The further design involved synthesis of substituted benzoylamino-pyrazolo[1,5-a]pyrimidine-3-carboxylic acids 8a–8h in two steps, as shown in Scheme 2. Step 5 involved the synthesis of carboxylic acid methyl esters 7a–7h which were obtained by the treatment of 6-aminopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester 5 with the corresponding acid chlorides 6a–6h in the presence of DIPEA at room temperature. The above carboxylic acid methyl esters 7a–7h were subsequently treated with lithium hydroxide monohydrate at 50 °C in THF, MeOH, and H2O for 2 h to give the corresponding benzoylaminopyrazolo[1,5-a]pyrimidine-3-carboxylic acids 8a–8h via step 6.

The final step involved the synthesis of the target compounds which involved the synthesis of corresponding hydrochloride salts of hydrazides 11a–11h in two steps, as shown in Scheme 3. Step 7 involved the synthesis of respective hydrazine carboxylic acid tert-butyl esters 10a–10h from 8a–8h using tert-butyl carbazate (9) in the presence of DIPEA, HOAT, EDC.HCl in DMF at room temperature. The above hydrazine carboxylic acid tert-butyl esters 10a–10h were reacted with HCl in 1,4-dioxane to obtain the target molecules 11a–11h via step 8.

Biological screening
An in vitro kinase assay was performed at a single-dose concentration of 10 µM over Aurora A and Aurora B to evaluate the Aurora kinase inhibitory activity of the synthesized pyrazolo[1,5-a]pyrimidine carboxamide compounds 10a–10h and 11a–11h. Table 1 shows the percentage of inhibition of kinase activity as an average of duplicate assay compared to the known non-selective kinase inhibitor, staurosporine, as a positive control. Out of these 16 derivatives, 4 analogs (11b, 11e, 11f, and 11g) showed high potency over 90% inhibitions against Aurora A, while the mean inhibitions were moderate in the rest of the compounds. Interestingly, most of these analogs were relatively less inhibitive against Aurora B (20.3–80.1% inhibition at 10 µM). Meanwhile, it is observed that analog 11f showed good highest inhibition against both Aurora A and Aurora B, while, analog 11a showed almost a lack of the mean inhibition against both Aurora A and Aurora B.
After a single-dose preliminary screening, compounds 11a–11h were tested for further activity study (Table 2). These compounds were tested against Aurora A and Aurora B by measuring the ability to inhibit phosphorylation of a biotinylated-polypeptide substrate (p-GAT, CIS Bio International) in a homogeneous time-resolved fluorescence (HTRF) assay at an ATP concentration of 30 µM. The results were reported as a 50% inhibition concentration value (IC50). The IC50 values for these selected compounds were determined in a 10-dose IC50 mode with threefold serial dilution starting at 30 µM as listed in Table 2. Most of the compounds inhibited Aurora A with IC50 values in the nanomolar to low micromolar range. Especially, compounds 11e, 11f, and 11g revealed nanomolar IC50 values. These results were encouraging with respect to the internal standard staurosporine which showed a potency of IC50 = 1.5 nM in the internal enzymatic Aurora A assay. Once again, these analogs were relatively less active against Aurora B. Only the most potent compound 11f showed the highest Aurora B activity with IC50 value reaching 9.63 µM in the enzymatic assay, whereas the internal standard staurosporine which showed a potency of IC50 = 9 nM in the internal enzymatic Aurora assay.
Structure–activity relationship
As can be seen from Table 1, a number of compounds exhibited a robust inhibitory activity against isolated Aurora A enzyme. With varied halogen, methoxy, and trihalomethyl substituents in benzamide position, it was observed that 3,5-difluoro, o-fluoro, m-fluoro, and p-fluoro substituted benzamide hydrazine hydrochlorides (10c, 10f, 10i, and 10m) gave better enzymatic activity. Amongst these derivatives 11f gave the highest activity (IC50 = 23 nM) against isolated Aurora A enzyme, which contains 3,5-difluoro in the benzamide group as shown in Table 2. The activity order came in the following sequence 3,5-difluoro > o-F > m-F > p-F > m-CF3. More or less the same activity sequence was observed against Aurora B enzyme. Compound 11f showed highest Aurora B activity (IC50 = 9.63 µM) in the enzymatic assay.
In general, the structure–activity relationship study indicated that halogen substitution plays important roles in kinase inhibitory potency. It is worth noting that compound 11a showed almost a lack of the mean inhibition against both Aurora A and Aurora B, suggesting that the—CF3 group of the benzamide played a key role in the binding of inhibitors to Aurora kinase. Interestingly, compound 11f showed good inhibition against both Aurora A and Aurora B, due to the presence of 3,5-difluoro group in benzamide.
Conclusion
A new 6-aminopyrazolo[1,5-a]pyrimidine-3-carboxylic acid scaffold, useful for kinase inhibition, has been designed and synthesized. We have disclosed the synthesis of a novel series of carboxamide containing hydrazide hydrochloride derivatives of the above scaffold as potent inhibitors of Aurora kinase. The compounds also provide an opportunity of laying the foundation for promising molecules of anticancer potency.
Experimental
Chemicals were of analytical grade and obtained from Sigma-Aldrich Co. The purification of all intermediates and final compounds was carried out by column chromatography using Merck silica gel with 230–400 mesh size. The purity of synthesized compounds was determined by thin layer chromatography experiments, performed on alumina-backed silica gel 40 F254 plates (Merck, Darmstadt, Germany). TLC observation of these plates were carried out by illuminating under UV (254 nm) lamp and KMnO4 stain solution. Melting points were determined using Büchi B-540 instrument. All 1H NMR and 13C NMR spectra were recorded on Bruker AM-300 (300 MHz for 1H NMR and 75 MHz for 13C NMR) and Bruker AM-400 (400 MHz for 1H NMR and 100 MHz for 13C NMR), Bruker BioSpin Corp., Germany. The chemical shifts are reported in ppm (δ) with reference to TMS as an internal standard. The signals are designed as follows: s, singlet; d, doublet; t, triplet; m, multiplet; brs, broad singlet. IR spectra for all the compounds were recorded using a Bruker Alpha FTIR spectrometer using a diamond ATR single reflectance module (24 scans). Molecular weights for all the synthesized compounds were checked by LC–MS 6200 series Agilent Technology. Elemental analysis was carried out with a Perkin-Elmer model 240-C apparatus. The results of elemental analysis (C, H, and N) were within ±0.4% of the calculated amounts.
5-Amino-1H-pyrazole-4-carboxylic acid methyl ester (2) [17–19] and 6-bromopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester 3 [20] were synthesized according to previously described methods.
Methyl 6-[(Diphenylmethylidene)amino]pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (4, C21H16N4O2)
To a degassed suspension of 12.00 g of 6-bromopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (3, 46.86 mmol) and 21.37 g of cesium carbonate (65.60 mmol) in 180 cm3 of 1,4-dioxane was added 2.44 g of 4,5-bis(diphenylphosphino)-9,9-dimethylanthene (4.21 mmol) and 1.40 g of tris(dibenzylideneacetone)dipalladium(0) dichloromethane complex (1.40 mmol), followed by the drop wise addition of 12.73 g of benzophenone imine (70.29 mmol). The resultant reaction mixture was heated to 110 °C for 10 h under argon atmosphere. The completion of reaction was monitored by TLC. The reaction mixture was filtered through Celite bed and was washed with 50 cm3 of ethyl acetate; the filtrate was concentrated to dryness under reduced pressure to afford the crude product. The crude product was purified by column chromatography over silica gel with chloroform/methanol (100:2, v/v) to afford methyl 6-[(diphenylmethylidene)amino]pyrazolo[1,5-a]pyrimidine-3-carboxylate (4) as a pale yellow solid (12.52 g, 75%). M.p.: 211.6–213.1 °C; TLC: R f = 0.22 (EtOAc-hexane 2:8); IR (ATR): \(\bar{\nu }\) = 1240 (C–N stretch), 1704 (C=N stretch), 1751 (C=O stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 3.77 (3H, s, ester CH3), 7.30–7.32 (3H, m, ArH), 7.37–7.47 (2H, m, ArH), 7.52 (2H, dd, J 1 = 7.6 Hz, J 2 = 1.6 Hz, ArH), 7.59–7.63 (1H, m, ArH), 7.73 (2H, dd, J 1 = 7.2 Hz, J 2 = 1.6 Hz, ArH), 8.38 (1H, d, J = 2.4 Hz, ArH), 8.51 (1H, s, ArH), 8.84 (1H, d, J = 2.0 Hz, ArH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 51.5, 101.8, 127.4,129.0 (3 peaks), 129.4 (2 peaks),129.6 (2 peaks), 129.8 (2 peaks), 132.4, 135.2, 136.4, 138.3, 144.2, 147.5, 149.5, 162.4, 173.4 ppm; LC–MS (ESI): m/z = 357.2 ([M+H]+).
6-Aminopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (5, C8H8N4O2)
To a suspension of 12.00 g of methyl 6-[(diphenylmethylidene)amino]pyrazolo[1,5-a]pyrimidine-3-carboxylate (4, 133.67 mmol) in 240 cm3 of methyl tert-butyl ether was added 30 cm3 of 4.5 M hydrochloric acid in methyl tert-butyl ether maintaining the temperature between 10 and 15 °C over a period of 20 min under nitrogen atmosphere. The resultant reaction mixture was stirred at room temperature (28 °C) for 8 h. The completion of reaction was monitored by TLC. The precipitate from the reaction mixture was filtered, washed with 40 cm3 of methyl tert-butyl ether and dried under vacuum at room temperature (28 °C) for 6 h to afford hydrochloric salt of 6-aminopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester. The suspension of hydrochloric salt of amine in 60 cm3 of water was basified (pH ~10–12) with 10% aqueous potassium carbonate and the product was extracted to 2 × 60 cm3 of n-butanol. The combined n-butanol layer was dried over anhydrous sodium sulfate and concentrated to dryness under reduced pressure to afford 6-aminopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (5) as a pale yellow solid (5.95 g, 93%). M.p.: 197.2–198.6 °C; TLC: R f = 0.31 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1255 (C–N stretch), 1550 (N–H bend), 3232 and 3399 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 3.76 (3H, s, ester CH3), 5.52 (2H, s, amine NH2), 8.28 (1H, d, J = 2.4 Hz, ArH), 8.33 (1H s, ArH), 8.48 (1H, d, J = 2.4 Hz, ArH) ppm; 13C NMR (400 MHz, DMSO-d 6): δ = 51.2, 101.0, 117.0, 135.5, 141.7, 145.6, 147.2, 162.7 ppm; LC–MS (ESI): m/z = 192.9 ([M+H]+).
General procedure for the synthesis of substituted carboxylic acid methyl esters 7a – 7h
6-Aminopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (5, 0.55 g, 2.86 mmol) was dissolved in 5.50 cm3 of N,N-dimethylformamide and 1.10 g of N,N-diisopropylethylamine (8.58 mmol) and corresponding acid chlorides 6a–6h (3.14 mmol) were added drop wise under nitrogen atmosphere. The reaction mixture was gradually allowed to attain room temperature (28 °C) and stirred for 2–3 h. The completion of reaction was monitored by TLC. The reaction mixture was poured to 60 cm3 of ice cold water, stirred for 1 h and the precipitate formed was filtered, washed with 5 cm3 of water and dried under vacuum at room temperature (28 °C) for 12 h to afford the crude product. The crude product was crystallized by digesting in 5 cm3 of ethyl acetate and 25 cm3 of n-heptane at room temperature (28 °C), stirred for 1 h, filtered, washed with 5 cm3 of n-heptane and dried under vacuum at room temperature (28 °C) to afford the corresponding substituted carboxylic acid methyl esters 7a–7h.
6-[3-(Trifluoromethyl)benzoylamino]pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (7a, C16H11F3N4O3)
This compound was prepared by the reaction of 6-aminopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (5) with 0.65 g of 3-(trifluoromethyl)benzoyl chloride (6a, 3.14 mmol) in the presence of N,N-diisopropylethylamine in N,N-dimethylformamide. The crude product was obtained as a yellow solid, which on crystallization afforded 7a as a pale yellow solid (0.88 g, 85%). M.p.: 223.1–224.9 °C; TLC: R f = 0.41 (EtOAc-hexane 4:6); IR (ATR): \(\bar{\nu }\) = 1572 (N–H bend), 1660 (amidic C=O), 1701 (ester C=O), 3311 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 3.82 (3H, s, ester CH3), 7.84 (1H, t, J = 7.0 Hz, ArH), 8.03 (1H, d, J = 7.6 Hz, ArH), 8.31–8.36 (2H, m, ArH), 8.61 (1H, s, ArH), 9.04 (1H, d, J = 2.4 Hz, ArH), 9.72 (1H, d, J = 2.0 Hz, ArH), 11.07 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 51.6, 102.2, 120.3, 123.0, 125.7, 128.4 (q, 1 J C-F = 271.0 Hz), 124.7, 124.8, 125.2, 127.4, 129.3, 129.3 (d, 3 J C-F = 3.0 Hz), 129.6, 129.9, 130.3 (q, 2 J C-F = 31.0 Hz), 130.4, 132.4, 134.7, 144.4, 147.7, 149.0, 162.5, 165.1 ppm; LC–MS (ESI): m/z = 363.0 ([M−H]−).
6-(4-Fluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (7b, C15H11FN4O3)
This compound was prepared by the reaction of 6-aminopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (5) with 0.50 g of 4-fluorobenzoyl chloride (6b, 3.14 mmol) in the presence of N,N-diisopropylethylamine in N,N-dimethylformamide. The crude product was obtained as a yellow solid, which on crystallization afforded 7b as a pale yellow solid (0.77 g, 86%). M.p.: 232.5–234.4 °C; TLC: R f = 0.41 (EtOAc-hexane 4:6); IR (ATR): \(\bar{\nu }\) = 1560 (N–H bend), 1659 (amidic C=O), 1708 (ester C=O), 3306 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 3.82 (3H, s, ester CH3), 7.44 (2H, t, J = 8.8 Hz, ArH), 8.11 (2H, dd, J 1 = 5.4 Hz, J 2 = 9.2 Hz, ArH), 8.60 (1H, s, ArH), 9.05 (1H, d, J = 2.4 Hz, ArH), 9.72 (1H, d, J = 2.4 Hz, ArH), 10.89 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 51.6, 102.1, 116.0, 116.2 (d, 2 J C-F = 22.0 Hz), 125.5, 127.1, 130.3, 130.3 (d, 4 J C-F = 2.0 Hz), 131.0, 131.1 (d, 3 J C-F = 10.0 Hz), 144.4, 147.7, 149.1, 162.5, 163.7, 166.2 (d, 1 J C-F = 249.0 Hz), 165.5 ppm; LC–MS (ESI): m/z = 315.2 ([M+H]+).
6-(4-Chlorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (7c, C15H11ClN4O3)
This compound was prepared by the reaction of 6-aminopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester 5 with 0.55 g of 4-chlorobenzoyl chloride (6c, 3.14 mmol) in the presence of N,N-diisopropylethylamine in N,N-dimethylformamide. The crude product was obtained as a yellow solid, which on crystallization afforded 7c as a pale yellow solid (0.78 g, 83%). M.p.: 231.4–233.1 °C; TLC: R f = 0.41 (EtOAc-hexane 4:6); IR (ATR): \(\bar{\nu }\) = 1563 (N–H bend), 1661 (amidic C=O), 1710 (ester C=O), 3307 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 3.82 (3H, s, ester CH3), 7.68 (2H,d, J = 8.4 Hz, ArH), 8.05 (2H, d, J = 8.8 Hz, ArH), 8.61 (1H, s, ArH), 9.04 (1H, d, J = 2.4 Hz, ArH), 9.73 (1H, d, J = 2.4 Hz, ArH), 10.93 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 51.6, 102.1, 125.4, 127.2, 129.2 (2 peaks), 130.2 (2 peaks), 132.5, 137.7, 144.4, 147.7, 149.1, 162.5, 165.5 ppm; LC–MS (ESI): m/z = 329.6 ([M−H]−).
6-(4-Bromobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (7d, C15H11BrN4O3 )
This compound was prepared by the reaction of 6-aminopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (5) with 0.69 g of 4-bromobenzoyl chloride (6d, 3.14 mmol) in the presence of N,N-diisopropylethylamine in N,N-dimethylformamide. The crude product was obtained as a yellow solid, which on crystallization afforded 7d as a pale yellow solid (0.85 g, 80%). M.p.: 229.4–232.0 °C; TLC: R f = 0.41 (EtOAc-hexane 4:6); IR (ATR): \(\bar{\nu }\) = 1560 (N–H bend), 1657 (amidic C=O), 1709 (ester C=O), 3305 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 3.82 (3H, s, ester CH3), 7.82 (2H, dd, J 1 = 2.0 Hz, J 2 = 6.8 Hz, ArH), 7.97 (2H, dd, J 1 = 1.8 Hz, J 2 = 6.6 Hz, ArH), 8.62 (1H, s, ArH), 9.04 (1H, d, J = 2.4 Hz, ArH), 9.72 (1H, d, J = 2.4 Hz, ArH), 10.93 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 51.6, 102.1, 125.4, 126.7, 127.2, 130.3 (2 peaks), 132.1 (2 peaks), 132.9, 144.4, 147.7, 149.1, 162.5, 165.6 ppm; LC–MS (ESI): m/z = 376.7 ([M+H]+).
6-(3-Fluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (7e, C15H11FN4O3)
This compound was prepared by the reaction of 6-aminopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester 5 with 0.50 g of 3-fluorobenzoyl chloride (6e, 3.14 mmol) in the presence of N,N-diisopropylethylamine in N,N-dimethylformamide. The crude product was obtained as a yellow solid, which on crystallization afforded 7e as a pale yellow solid (0.77 g, 86%). M.p.: 243.4–244.9 °C; TLC: R f = 0.41 (EtOAc-hexane 4:6); IR (ATR): \(\bar{\nu }\) = 1560 (N–H bend), 1658 (amidic C=O), 1709 (ester C=O), 3305 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6); δ = 3.82 (3H, s, ester CH3), 7.52 (1H, dt, J 1 = 6.4 Hz, J 2 = 10.8 Hz, ArH), 7.64 (1H, td, J 1 = 5.6 Hz, J 2 = 8.0 Hz, ArH), 7.81–7.88 (2H, m, ArH), 8.60 (1H, s, ArH), 9.04 (1H, d, J = 2.4 Hz, ArH), 9.71 (1H, d, J = 2.4 Hz, ArH), 10.92 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 51.6, 102.1, 114.9, 115.2 (d, 2 J C-F = 23.0 Hz), 119.6, 119.8 (d, 2 J C-F = 21.0 Hz), 124.5, 124.5 (d, 4 J C-F = 2.0 Hz), 125.3, 127.2, 131.3, 131.4 (d, 3 J C-F = 8.0 Hz), 136.0, 136.1 (d, 3 J C-F = 7.0 Hz), 144.4, 147.7, 149.0, 161.2, 163.6 (d, 1 J C-F = 243.0 Hz), 162.5, 165.2, 165.2 ppm; LC–MS (ESI): m/z = 313.1 ([M−H]−).
6-(3,5-Difluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (7f, C15H10F2N4O3)
This compound was prepared by the reaction of 6-aminopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester 5 with 0.55 g of 3,5-difluorobenzoyl chloride (6f, 3.14 mmol) in the presence of N,N-diisopropylethylamine in N,N-dimethylformamide. The crude product was obtained as a yellow solid, which on crystallization afforded 7f as a pale yellow solid (0.79 g, 83%). M.p.: 228.9–230.8 °C; TLC: R f = 0.41 (EtOAc-hexane 4:6); IR (ATR): \(\bar{\nu }\) = 1562 (N–H bend), 1680 (amidic C=O), 1697 (ester C=O), 3360 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 3.82 (3H, s, ester CH3), 7.61 (1H, dt, J 1 = 6.4 Hz, J 2 = 11.6 Hz, ArH), 7.74 (2H, dd, J 1 = 2.0 Hz, J 2 = 8.0 Hz, ArH), 8.63 (1H, s, ArH), 9.03 (1H, d, J = 2.0 Hz, ArH), 9.71 (1H, d, J = 2.4 Hz, ArH), 10.99 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 51.6, 102.2, 108.0, 108.3, 108.5 (t, 2 J C-F = 26.0 Hz), 111.6, 111.8 (d, 2 J C-F = 19.0 Hz), 111.7, 111.9 (d, 2 J C-F = 20.0 Hz), 125.1, 127.5, 137.1, 137.2, 137.3 (t, 3 J C-F = 9.0 Hz), 144.5, 147.8, 149.0, 161.4, 163.9 (d, 1 J C-F = 246.0 Hz), 161.6, 164.0 (d, 1 J C-F = 246.0 Hz), 162.5, 164.2 ppm; LC–MS (ESI): m/z = 331.1 ([M−H]−).
6-(2-Fluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (7g, C15H11FN4O3)
This compound was prepared by the reaction of 6-aminopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester 5 with 0.50 g of 2-fluorobenzoyl chloride (6g, 3.14 mmol) in the presence of N,N-diisopropylethylamine in N,N-dimethylformamide. The crude product was obtained as a yellow solid, which on crystallization afforded 7g as a pale yellow solid (0.78 g, 87%). M.p.: 236.7–238.5 °C; TLC: R f = 0.41 (EtOAc-hexane 4:6); IR (ATR): \(\bar{\nu }\) = 1560 (N–H bend), 1658 (amidic C=O), 1709 (ester C=O), 3305 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 3.82 (3H, s, ester CH3), 7.37–7.44 (2H, m, ArH), 7.62–7.68 (1H, m, ArH), 7.77 (1H, dt, J 1 = 6.0 Hz, J 2 = 13.6 Hz, ArH), 8.61 (1H, s, ArH), 8.94 (1H, d, J = 2.0 Hz, ArH), 9.71 (1H, d, J = 2.4 Hz, ArH), 11.05 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 51.6, 102.2, 116.7, 117.0 (d, 2 J C-F = 22.0 Hz), 123.6, 123.8 (d, 2 J C-F = 14.0 Hz), 125.2, 125.2 (d, 3 J C-F = 7.0 Hz), 125.2, 126.7, 130.6, 130.6 (d, 4 J C-F = 3.0 Hz), 133.9, 134.0 (d, 3 J C-F = 9.0 Hz), 144.4, 147.7, 148.5, 158.3, 160.8 (d, 1 J C-F = 249.0 Hz), 162.5, 163.9 ppm; LC–MS (ESI): m/z = 313.0 ([M−H]−).
6-(4-Methoxybenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (7h, C16H14N4O4)
This compound was prepared by the reaction of 6-aminopyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester 5 with 0.53 g of 4-methoxybenzoyl chloride (6h, 3.14 mmol) in the presence of N,N-diisopropylethylamine in N,N-dimethylformamide. The crude product was obtained as a yellow solid, which on crystallization afforded 7h as a pale yellow solid (0.82 g, 88%). M.p.: 241.6–243.5 °C; TLC: R f = 0.41 (EtOAc-hexane 4:6); IR (ATR): \(\bar{\nu }\) = 1560 (N–H bend), 1658 (amidic C=O), 1709 (ester C=O), 3305 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 3.82 (3H, s, ester CH3), 3.85 (3H, s, methoxy CH3), 7.12 (2H, d, J = 8.8 Hz, ArH), 8.03 (2H, d, J = 8.8 Hz, ArH), 8.59 (1H, s, ArH), 9.06 (1H, d, J = 2.4 Hz, ArH), 9.73 (1H, d, J = 2.4 Hz, ArH), 10.70 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 51.5, 53.9, 102.0, 114.3 (2 peaks), 125.8, 125.8, 126.8, 130.3 (2 peaks), 144.3, 147.59, 149.2, 162.5, 162.9, 165.9 ppm; LC–MS (ESI): m/z = 325.2 ([M−H]−).
General procedure for the synthesis of substituted carboxylic acids 8a–8h
To a suspension of substituted carboxylic acid methyl ester 7a–7h (1.00 mmol) in 2.5 cm3 of tetrahydrofuran, 2.5 cm3 of methanol, and 2.2 cm3 of water was added lithium hydroxide monohydrate (3.00 mmol) and heated to 72 °C under nitrogen atmosphere for 3–4 h. The completion of reaction was monitored by TLC. The reaction mixture was gradually allowed to attain room temperature (28 °C) and concentrated to dryness under reduced pressure. The residue was diluted with 10 cm3 of ice cold water, acidified (pH ~5–6) with concentrated hydrochloric acid and stirred for 1 h. The precipitate formed was filtered, washed with 2 cm3 of water and dried at room temperature (28 °C) for 12 h to afford the crude product. The crude product was crystallized by digesting in 10 cm3 of methanol for 1 h at room temperature (28 °C), filtered, washed with 2 cm3 of methanol, and dried under vacuum at room temperature (28 °C) for 4 h to afford the corresponding substituted carboxylic acids 8a–8h.
6-[3-(Trifluoromethyl)benzoylamino]pyrazolo[1,5-a]pyrimidine-3-carboxylic acid (8a, C15H9F3N4O3)
This compound was prepared by the reaction of 0.75 g of 6-[3-(trifluoromethyl)benzoylamino]pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (7a, 2.04 mmol) with 0.26 g of lithium hydroxide monohydrate (6.17 mmol) in tetrahydrofuran, methanol, and water. The crude product was obtained as a yellow solid, which on crystallization afforded 8a as a pale yellow solid (0.54 g, 76%). M.p.: 262.3–264.1 °C; TLC: R f = 0.10 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1561 (N–H bend), 1633 (amidic C=O), 1671 (acid C=O), 3105 (N–H stretch), 3435 (O–H) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 7.85 (1H, t, J = 8.0 Hz, ArH), 8.04 (1H, d, J = 7.6 Hz, ArH), 8.26–8.37 (1H, m, ArH), 8.45 (1H, d, J = 2.8 Hz, ArH), 8.56 (1H, s, ArH), 9.02 (1H, d, J = 2.4 Hz, ArH), 9.71 (1H, d, J = 2.0 Hz, ArH), 11.07 (1H, s, amide NH), 12.44 (1H, s, acid-OH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 103.3, 120., 123.0, 125.7, 128.4 (q, 1 J C-F = 271.0 Hz), 124.7, 124.8, 125.0, 127.3, 129.2, 129.3 (d, 3 J C-F = 4.0 Hz), 129.3, 129.6, 129.9, 130.3 (q, 2 J C-F = 32.0 Hz), 130.5, 132.4, 134.8, 144.6, 147.9, 148.7, 163.5, 163.7, 165.1 ppm; LC–MS (ESI): m/z = 348.9 ([M−H]−).
6-(4-Fluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid (8b, C14H9FN4O3)
This compound was prepared by the reaction of 0.75 g of 6-(4-fluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (7b, 2.38 mmol) with 0.30 g of lithium hydroxide monohydrate (7.16 mmol) in tetrahydrofuran, methanol, and water. The crude product was obtained as a yellow solid, which on crystallization afforded 8b as a pale yellow solid (0.56 g, 78%). M.p.: 274.8–276.9 °C; TLC: R f = 0.10 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1562 (N–H bend), 1633 (amidic C=O), 1674 (acid C=O), 3104 (N–H stretch), 3436 (O–H) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 7.44 (2H, t, J = 8.6 Hz, ArH), 8.11 (2H, dd, J 1 = 5.4 Hz, J 2 = 8.6 Hz, ArH), 8.51 (1H, s, ArH), 9.01 (1H, d, J = 2.0 Hz, ArH), 9.70 (1H, d, J = 2.4 Hz, ArH), 10.85 (1H, s, amide NH), 12.43 (1H, s, acid-OH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 103.1, 116.0, 116.2 (d, 2 J C-F = 22.0 Hz), 125.3, 126.9, 130.3, 130.3 (d, 4 J C-F = 2.0 Hz), 131.0, 131.1 (d, 3 J C-F = 9.0 Hz), 144.5, 147.8, 148.7, 163.6, 166.1 (d, 1 J C-F = 249.0 Hz), 163.5, 165.4 ppm; LC–MS (ESI): m/z = 300.9 ([M+H]+).
6-(4-Chlorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid (8c, C14H9ClN4O3)
This compound was prepared by the reaction of 0.75 g of 6-(4-chlorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (7c, 2.26 mmol) with 0.28 g of lithium hydroxide monohydrate (6.80 mmol) in tetrahydrofuran, methanol, and water. The crude product was obtained as a yellow solid, which on crystallization afforded 8c as a pale yellow solid (0.55 g, 77%). M.p.: 270.3–272.5 °C; TLC: R f = 0.10 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1632 (amidic C=O), 1677 (acid C=O), 3120 (N–H stretch), 3322 (O–H) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 7.64 (2H, d, J = 6.0 Hz, ArH), 8.08 (2H, d, J = 8.4 Hz, ArH), 8.45 (1H, s, ArH), 8.99 (1H, s, ArH), 9.69 (1H, s, amide NH), 11.13 (1H, s, acid-OH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 105.9, 124.8, 127.0, 129.1 (2 peaks), 130.2 (2 peaks), 132.6, 137.5, 144.2, 147.6, 148.1, 164.4, 165.4 ppm; LC–MS (ESI): m/z = 316.0 ([M−H]−).
6-(4-Bromobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid (8d, C14H9BrN4O3)
This compound was prepared by the reaction of 0.75 g of 6-(4-bromobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (7d, 2.00 mmol) with 0.25 g of lithium hydroxide monohydrate (6.00 mmol) in tetrahydrofuran, methanol, and water. The crude product was obtained as a yellow solid, which on crystallization afforded 8d as a pale yellow solid (0.57 g, 79%). M.p.: 268.2–269.9 °C; TLC: R f = 0.10 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1566 (N–H bend), 1633 (amidic C=O), 1678 (acid C=O), 3119 (N–H stretch), 3323 (O–H) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 7.81 (2H, d, J = 8.4 Hz, ArH), 8.00 (2H, d, J = 8.4 Hz, ArH), 8.47 (1H, s, A–H), 9.00 (1H, d, J = 2.0 Hz, ArH), 9.69 (1H, s, amide NH), 11.07 (1H, s, acid-OH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 105.4, 124.9, 126.5, 127.0, 130.4 (2 peaks), 132.1 (2 peaks), 133.0, 144.3, 147.7, 148.2, 164.1, 165.6 ppm; LC–MS (ESI): m/z = 359.0 ([M−2H]−).
6-(3-Fluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid (8e, C14H9FN4O3)
This compound was prepared by the reaction of 0.75 g of 6-(3-fluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (7e, 2.38 mmol) with 0.30 g of lithium hydroxide monohydrate (7.15 mmol) in tetrahydrofuran, methanol, and water. The crude product was obtained as a yellow solid, which on crystallization afforded 8e as a pale yellow solid (0.54 g, 76%). M.p.: 268.3–270.1 °C; TLC: R f = 0.10 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1562 (N–H bend), 1635 (amidic C=O), 1673 (acid C=O), 3104 (N–H stretch), 3436 (O–H) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 7.51 (1H, dt, J 1 = 5.6 Hz, J 2 = 13.6 Hz, ArH), 7.65 (1H, td, J 1 = 6.0 Hz, J 2 = 8.0 Hz, ArH), 7.82-7.89 (2H, m, ArH), 8.55 (1H, s, ArH), 9.02 (1H, d, J = 2.4 Hz, ArH), 9.71 (1H, d, J = 2.4 Hz, ArH), 10.89 (1H, s, amide NH), 12.44 (1H, s, acid-OH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 103.2, 114.9, 115.2 (d, 2 J C-F = 23.0 Hz), 119.5, 119.8 (d, 2 J C-F = 21.0 Hz), 124.4, 124.4 (d, 4 J C-F = 3.0 Hz), 125.1, 127.0, 131.3, 131.3 (d, 3 J C-F = 7.0 Hz), 136.0, 136.1 (d, 3 J C-F = 7.0 Hz), 144.5, 147.8, 148.6, 161.2, 163.6 (d, 1 J C-F = 243.0 Hz), 163.5, 165.1, 165.1 ppm; LC–MS (ESI): m/z = 299.1 ([M−H]−).
6-(3,5-Difluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid (8f, C14H8F2N4O3)
This compound was prepared by the reaction of 0.75 g of 6-(3,5-difluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (7f, 72.25 mmol) with 0.28 g lithium hydroxide monohydrate (6.77 mmol) in tetrahydrofuran, methanol, and water. The crude product was obtained as a yellow solid, which on crystallization afforded 8f as a pale yellow solid (0.54 g, 76%). M.p.: 259.6–261.4 °C; TLC: R f = 0.10 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1556 (N–H bend), 1631 (amidic C=O), 1668 (acid C=O), 3128 (N–H stretch), 3363 (O–H) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 7.60 (1H, dt, J 1 = 7.2 Hz, J 2 = 16.0 Hz, ArH), 7.74 (2H, t, J = 6.4 Hz, ArH), 8.55 (1H, s, ArH), 8.99 (1H, d, J = 2.4 Hz, ArH), 9.68 (1H, d, J = 2.4 Hz, ArH), 10.93 (1H, s, amide NH), 12.45 (1H, s, acid-OH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 103.3, 108.0, 108.2, 108.5 (t, 2 J C-F = 26.0 Hz), 116.6, 111.8 (d, 2 J C-F = 19.0 Hz), 111.6, 111.8 (d, 2 J C-F = 20.0 Hz), 124.8, 127.2, 137.1, 137.2, 137.3 (t, 3 J C-F = 8.0 Hz), 144.6, 147.9, 148.6, 161.4, 163.9 (d, 1 J C-F = 248.0 Hz), 161.5, 164.0 (1 J C-F = 246.0 Hz), 163.4, 163.9 ppm; LC–MS (ESI): m/z = 317.1 ([M−H]−).
6-(2-Fluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid (8g, C14H9FN4O3)
This compound was prepared by the reaction of 0.75 g of 6-(2-fluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (7g, 2.38 mmol) with 0.30 g of lithium hydroxide monohydrate (7.16 mmol) in tetrahydrofuran, methanol, and water. The crude product was obtained as a yellow solid, which on crystallization afforded 8g as a pale yellow solid (0.55 g, 77%). M.p.: 269.9–271.6 °C; TLC: R f = 0.10 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1563 (N–H bend), 1635 (amidic C=O), 1673 (acid C=O), 3104 (N–H stretch), 3436 (O–H) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 7.37–7.44 (2H, m, ArH), 7.62–7.68 (1H, m, ArH), 7.78 (1H, dt, J 1 = 6.0, J 2 = 13.2 Hz, ArH), 8.55 (1H, s, ArH), 8.92 (1H, d, J = 2.4 Hz, ArH), 9.69 (1H, d, J = 2.4 Hz, ArH), 11.02 (1H, s, amide NH), 12.43 (1H, s, acid-OH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 103.2, 116.7, 117.0 (d, 2 J C-F = 21.0 Hz), 123.7, 123.8 (d, 3 J C-F = 14.0 Hz), 125.0, 125.2 (d, 2 J C-F = 21.0 Hz), 126.6, 130.6, 130.64 (d, 4 J C-F = 2.0 Hz), 133.9, 134.0 (d, 3 J C-F = 9.0 Hz), 144.6, 147.9, 148.2, 158.3, 160.8 (d, 1 J C-F = 249.0 Hz), 163.52, 163.9 ppm; LC–MS (ESI): m/z = 299.2 ([M−H]−).
6-(4-Methoxybenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid (8h, C15H12N4O3)
This compound was prepared by the reaction of 0.75 g of 6-(4-methoxybenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid methyl ester (7h, 2.29 mmol) with 0.29 g of lithium hydroxide monohydrate (6.89 mmol) in tetrahydrofuran, methanol, and water. The crude product was obtained as a yellow solid, which on crystallization afforded 8h as a pale yellow solid (0.56 g, 78%). M.p.: 263.4–265.3 °C; TLC: R f = 0.10 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1563 (N–H bend), 1635 (amidic C=O), 1673 (acid C=O), 3104 (N–H stretch), 3436 (O–H) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 3.85 (3H, s, methoxy CH3), 7.11 (2H, d, J = 8.8 Hz, ArH), 8.03 (2H, d, J = 9.2 Hz, ArH), 8.52 (1H, s, Ar–H), 9.03 (1H, d, J = 2.4 Hz, ArH), 9.70 (1H, d, J = 2.0 Hz, ArH), 10.65 (1H, s, amide NH), 12.41 (1H, s, acid-OH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 55.9, 103.0, 114.3, 125.6, 125.8, 126.6, 130.2, 144.4, 147.7, 148.7, 162.9, 163.5, 165.9 ppm; LC–MS (ESI): m/z = 311.1 ([M−H]−).
General procedure for the synthesis of compounds 10a–10h
To a suspension of substituted carboxylic acid 8a–8h (1.00 mmol) in 2.5 cm3 of N,N-dimethylformamide was added tert-butyl carbazate (9, 1.10 mmol), ethyl carbodiimide hydrochloride (1.50 mmol), 1-hydroxy-7-azabenzotriazole (1.20 mmol), followed by the drop wise addition of N,N-diisopropylethylamine (3.00 mmol) and stirred at room temperature (28 °C) under nitrogen atmosphere for 12 h. The completion of reaction was monitored by TLC. The reaction mixture was poured into 25 cm3 of ice cold water, stirred for 1 h, filtered, washed with 5 cm3 of water, and dried under vacuum at room temperature (28 °C) for 8 h to afford the crude product. The crude product was purified by column chromatography over silica gel with chloroform/methanol (100:2 v/v) to afford the compounds 10a–10h.
N′-[6-[3-(Trifluoromethyl)benzoylamino]pyrazolo[1,5-a]pyrimidine-3-carbonyl]hydrazinecarboxylic acid tert-butyl ester (10a, C20H19F3N6O4)
This compound was prepared by the reaction of 0.50 g of 6-[3-(trifluoromethyl)benzoylamino]pyrazolo[1,5-a]pyrimidine-3-carboxylic acid (8a, 1.42 mmol) with 0.20 g of tert-butyl carbazate (9, 1.57 mmol) in presence of 0.33 g of ethyl carbodiimide hydrochloride (2.14 mmol), 0.40 g of 1-hydroxy-7-azabenzotriazole (1.71 mmol), and 0.55 g of N,N-diisopropylethylamine (4.28 mmol) in N,N-dimethylformamide. The crude product was obtained as a brown solid, which on column chromatography purification afforded 10a as a beige solid (0.55 g, 83%). M.p.: 198.5–200.4 °C; TLC: R f = 0.71 (MeOH–CHCl3 0.5:9.5); IR (ATR): \(\bar{\nu }\) = 1161 (alkoxy C–O), 1535, 1556, 1618 (N–H bend), 1666, 1687, 1722 (C=O), 3097, 3215, 3284 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 1.43 (9H, s, boc CH3), 7.85 (1H, t, J = 8.0 Hz, ArH), 8.05 (1H, d, J = 7.6 Hz, ArH), 8.33 (1H, d, J = 7.6 Hz, ArH), 8.37 (1H, s, ArH), 8.61 (1H, s, ArH), 9.05 (1H, d, J = 5.6 Hz, amide NH), 9.04 (1H, s, ArH), 9.33 (1H, s, ArH), 9.75 (1H, d, J = 1.6 Hz, amide NH), 11.07 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 28.5 (3 peaks), 79.8, 104.1, 120.3, 123.0, 125.7, 128.4 (q, 1 J C-F = 271.0 Hz), 124.7, 124.8, 124.9 (t, 3 J C-F = 4.0 Hz), 127.7, 127.8, 127.9 (t, 3 J C-F = 4.0 Hz), 129.3, 129.6, 130.0, 130.3 (q, 2 J C-F = 32.0 Hz), 130.5, 132.4, 134.8, 143.3, 146.2, 148.5, 155.9, 161.4, 165.1 ppm; LC–MS (ESI): m/z = 463.2 ([M−H]−).
N’-[6-(4-Fluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carbonyl]hydrazinecarboxylic acid tert-butyl ester (10b, C19H19FN6O4)
This compound was prepared by the reaction of 0.5 g of 6-(4-fluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid (8b, 1.66 mmol) with 0.24 g of tert-butyl carbazate (9, 1.83 mmol) in presence of 0.38 g of ethyl carbodiimide hydrochloride (2.49 mmol), 0.27 g of 1-hydroxy-7-azabenzotriazole (2.00 mmol), and 0.64 g of N,N-diisopropylethylamine (5.00 mmol) in N,N-dimethylformamide. The crude product was obtained as a brown solid, which on column chromatography purification afforded 10b as a beige solid (0.58 g, 84%). M.p.: 208.5–210.2 °C; TLC: R f = 0.71 (MeOH–CHCl3 0.5:9.5); IR (ATR): \(\bar{\nu }\) = 1161 (alkoxy C–O), 1510, 1564, 1602 (N–H bend), 1666, 1687, 1708 (C=O), 3078, 3221, 3311 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 1.43 (9H, s, boc CH3), 7.44 (2H, t, J = 8.8 Hz, ArH), 8.11 (2H, dt, J 1 = 3.6 Hz, J 2 = 10.4 Hz, ArH), 8.60 (1H, s, ArH), 9.02 (1H, d, J = 6.4 Hz, amide NH), 9.04 (1H, s, ArH), 9.32 (1H, s, ArH), 9.75 (1H, d, J = 2.0 Hz, amide NH), 10.87 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 28.5 (3 peaks), 79.8, 104.0, 116.0, 116.2 (d, 2 J C-F = 22.0 Hz), 125.2, 127.4, 130.3, 130.3 (d, 4 J C-F = 3.0 Hz), 131.0, 131.1 (d, 3 J C-F = 9.0 Hz), 143.2, 146.1, 148.5, 155.9, 161.4, 163.7, 166.2 (d, 1 J C-F = 249.0 Hz), 165.5 ppm; LC–MS (ESI): m/z = 413.2 ([M−H]−).
N′-[6-(4-Chlorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carbonyl]hydrazinecarboxylic acid tert-butyl ester (10c, C19H19ClN6O4)
This compound was prepared by the reaction of 0.5 g of 6-(4-chlorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid (8c, 1.57 mmol) with 0.23 g of tert-butyl carbazate (9, 1.73 mmol) in presence of 0.36 g of ethyl carbodiimide hydrochloride (2.36 mmol), 0.25 g of 1-hydroxy-7-azabenzotriazole (1.89 mmol), and 0.61 g of N,N-diisopropylethylamine (4.73 mmol) in N,N-dimethylformamide. The crude product was obtained as a brown solid, which on column chromatography purification afforded 10c as a beige solid (0.60 g, 89%). M.p.: 212.8–214.1 °C; TLC: R f = 0.71 (MeOH–CHCl3 0.5:9.5); IR (ATR): \(\bar{\nu }\) = 1157 (alkoxy C–O), 1521, 1560, 1591 (N–H bend), 1666, 1687, 1708 (C=O), 3103, 3221, 3294 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 1.43 (9H, s, boc CH3), 7.68 (2H, d, J = 8.4 Hz, ArH), 8.06 (2H, d, J = 8.4 Hz, ArH), 8.60 (1H, s, ArH), 9.02 (1H, d, J = 6.4 Hz, amide NH), 9.03 (1H, s, ArH), 9.32 (1H, s, ArH), 9.75 (1H, d, J = 2.0 Hz, amide NH), 10.92 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 28.5 (3 peaks), 79.8, 104.0, 125.1, 127.4, 129.1 (2 peaks), 130.1 (2 peaks), 132.5, 137.6, 143.2, 146.1, 148.4, 155.9, 161.4, 165.4 ppm; LC–MS (ESI): m/z = 429.3 ([M−H]−).
N’-[6-(4-Bromobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carbonyl]hydrazinecarboxylic acid tert-butyl ester (10d, C19H19BrN6O4)
This compound was prepared by the reaction of 0.50 g of 6-(4-bromobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid (8d, 1.38 mmol) with 0.20 g of tert-butyl carbazate (9, 1.52 mmol) in presence of 0.32 g of ethyl carbodiimide hydrochloride (2.07 mmol), 0.22 g of 1-hydroxy-7-azabenzotriazole (1.66 mmol), and 0.53 g of N,N-diisopropylethylamine (4.14 mmol) in N,N-dimethylformamide. The crude product was obtained as a brown solid, which on column chromatography purification afforded 10d as a beige solid 0.56 g, 86%). M.p.: 215.6–217.7 °C; TLC: R f = 0.71 (MeOH–CHCl3 0.5:9.5); IR (ATR): \(\bar{\nu }\) = 1157 (alkoxy C–O), 1525, 1562, 1589 (N–H bend), 1666, 1687, 1708 (C=O), 3099, 3221, 3254 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 1.43 (9H, s, boc CH3), 7.82 (2H, d, J = 8.4 Hz, ArH), 7.98 (2H, d, J = 8.4 Hz, ArH), 8.60 (1H, s, ArH), 9.02 (1H, d, J = 6.8 Hz, amide NH), 9.03 (1H, s, ArH), 9.32 (1H, s, ArH), 9.74 (1H, d, J = 2.0 Hz, amide NH), 10.92 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 28.5 (3 peaks), 79.8, 104.0, 125.1, 126.6, 127.4, 130.3 (2 peaks), 132.1 (2 peaks), 132.8, 143.1, 146.1, 148.4, 155.9, 161.4, 165.6 ppm; LC–MS (ESI): m/z = 474.9 ([M−H]−).
N′-[3-(2-Fluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carbonyl]hydrazinecarboxylic acid tert-butyl ester (10e, C19H19FN6O4)
This compound was prepared by the reaction of 0.50 g of 6-(3-fluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid (8e, 1.66 mmol) with 0.24 g of tert-butyl carbazate (9, 1.83 mmol) in presence of 0.38 g of ethyl carbodiimide hydrochloride (2.49 mmol), 0.27 g of 1-hydroxy-7-azabenzotriazole (2.00 mmol), and 0.64 g of N,N-diisopropylethylamine (5.00 mmol) in N,N-dimethylformamide. The crude product was obtained as a brown solid, which on column chromatography purification afforded 10e as a beige solid (0.60 g, 87%). M.p.: 206.1–207.9 °C; TLC: R f = 0.71 (MeOH–CHCl3 0.5:9.5); IR (ATR): \(\bar{\nu }\) = 1160 (alkoxy C–O), 1512, 1563, 1601 (N–H bend), 1666, 1687, 1705 (C=O), 3078, 3221, 3311 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 1.43 (9H, s, boc CH3), 7.52 (1H, dt, J 1 = 6.4 Hz, J 2 = 15.2 Hz, ArH), 7.65 (1H, dt, J 1 = 6.0 Hz, J 2 = 10.0 Hz, ArH), 7.83-7.90 (2H, m, ArH), 8.61 (1H, s, ArH), 9.03 (1H, d, J = 6.8 Hz, amide NH), 9.04 (1H, s, ArH), 9.32 (1H, s, ArH), 9.75 (1H, d, J = 2.0 Hz, amide NH), 10.93 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 28.5 (3 peaks), 79.8, 104.1, 115.0, 115.2 (d, 2 J C-F = 22.0 Hz), 119.6, 119.8 (d, 2 J C-F = 21.0 Hz), 124.5, 124.5 (d, 4 J C-F = 2.0 Hz), 125.0, 127.5, 131.3, 131.4 (d, 3 J C-F = 8.0 Hz), 136.1, 136.1 (d, 3 J C-F = 7.0 Hz), 143.2, 146.1, 148.5, 155.9, 161.2, 161.4, 163.6, 165.2 (d, 1 J C-F = 258.0 Hz) ppm; LC–MS (ESI): m/z = 413.2 ([M−H]−).
N′-[6-(3,5-Difluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carbonyl]hydrazinecarboxylic acid tert-butyl ester (10f, C19H18F2N6O4)
This compound was prepared by the reaction of 0.50 g of 6-(3,5-difluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid (8f, 1.57 mmol) with 0.22 g of tert-butyl carbazate (9, 1.72 mmol) in presence of 0.36 g of ethyl carbodiimide hydrochloride (2.35 mmol), 0.25 g of 1-hydroxy-7-azabenzotriazole (1.88 mmol), and 0.60 g of N,N-diisopropylethylamine (4.71 mmol) in N,N-dimethylformamide. The crude product was obtained as a brown solid, which on column chromatography purification afforded 10f as a beige solid (0.58 g, 86%). M.p.: 203.8–205.7 °C; TLC: R f = 0.71 (MeOH–CHCl3 0.5:9.5); IR (ATR): \(\bar{\nu }\) = 1159 (alkoxy C–O), 1511, 1565, 1609 (N–H bend), 1667, 1667, 1709 (C=O), 3077, 3220, 3311 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 1.43 (9H, s, boc CH3), 7.62 (1H, dt, J 1 = 4.8 Hz, J 2 = 13.6 Hz, ArH), 7.75 (2H, dt, J 1 = 4.8 Hz, J 2 = 6.4 Hz, ArH), 8.61 (1H, s, ArH), 9.01 (2H, d, J = 2.4 Hz, amide NH, ArH), 9.33 (1H, s, ArH), 9.74 (1H, d, J = 2.0 Hz, amide NH), 10.98 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 28.5 (3 peaks), 79.8, 104.1, 108.0, 108.2, 108.5 (t, 2 J C-F = 26.0 Hz), 111.6, 111.8 (d, 2 J C-F = 19.0 Hz), 111.7, 111.8 (d, 2 J C-F = 19.0 Hz), 124.7, 127.6, 137.1, 137.2, 137.3 (t, 3 J C-F = 8.0 Hz), 143.2, 146.2, 148.3, 155.9, 161.4, 163.9 (d, 1 J C-F = 252.0 Hz) 161.5, 164.0 (d, 1 J C-F = 252.0 Hz), 161.5, 163.9, 164.0 ppm; LC–MS (ESI): m/z = 432.1 ([M−H]−).
N′-[6-(2-Fluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carbonyl]hydrazinecarboxylic acid tert-butyl ester (10g, C19H19FN6O4)
This compound was prepared by the reaction of 0.50 g of 6-(2-fluorobenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid (8g, 1.66 mmol) with 0.024 g of tert-butyl carbazate (9, 1.83 mmol) in presence of 0.38 g of ethyl carbodiimide hydrochloride (2.49 mmol), 0.27 g of 1-hydroxy-7-azabenzotriazole (2.00 mmol), and 0.64 g of N,N-diisopropylethylamine (5.00 mmol) in N,N-dimethylformamide. The crude product was obtained as a brown solid, which on column chromatography purification afforded 10g as a beige solid (0.61 g, 89%). M.p.: 200.5–202.6 °C; TLC: R f = 0.71 (MeOH–CHCl3 0.5:9.5); IR (ATR): \(\bar{\nu }\) = 1159 (alkoxy C–O), 1511, 1565, 1601 (N–H bend), 1666, 1687, 1708 (C=O), 3075, 3220, 3310 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 1.43 (9H, s, boc CH3), 7.37–7.44 (2H, m, ArH), 7.63–7.80 (1H, m, ArH), 7.78 (1H, t, J = 6.8 Hz, ArH), 8.61 (1H, s, ArH), 8.94 (1H, d, J = 2.0 Hz, amide NH), 9.01 (1H, s, ArH), 9.32 (1H, s, ArH), 9.74 (1H, d, J = 1.6 Hz, amide NH), 11.04 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 27.4 (3 peaks), 78.7, 103.1, 115.7, 115.9 (d, 2 J C-F = 22.0 Hz), 122.6, 122.7 (d, 3 J C-F = 14.0 Hz), 123.8, 124.1 (d, 2 J C-F = 26.0 Hz), 124.1, 125.9, 129.5, 129.5 (d, 4 J C-F = 2.0 Hz), 132.8, 132.9 (d, 3 J C-F = 8.0 Hz), 142.1, 145.1,146.9, 154.8, 157.2, 159.7 (d, 1 J C-F = 249.0 Hz), 160.3, 162.8 ppm; LC–MS (ESI): m/z = 413.2 ([M−H]−).
N′-[6-(4-Methoxybenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carbonyl]hydrazinecarboxylic acid tert-butyl ester (10h, C20H22N6O5)
This compound was prepared by the reaction of 0.50 g of 6-(4-methoxybenzoylamino)pyrazolo[1,5-a]pyrimidine-3-carboxylic acid (8h, 1.60 mmol) with 0.23 g of tert-butyl carbazate (9, 1.76 mmol) in presence of 0.37 g of ethyl carbodiimide hydrochloride (2.40 mmol), 0.26 g of 1-hydroxy-7-azabenzotriazole (1.92 mmol), and 0.62 g of N,N-diisopropylethylamine (4.80 mmol) in N,N-dimethylformamide. The crude product was obtained as a brown solid, which on column chromatography purification afforded 10h as a beige solid (0.56 g, 83%). M.p.: 213.8–215.5 °C; TLC: R f = 0.71 (MeOH–CHCl3 0.5:9.5); IR (ATR): \(\bar{\nu }\) = 1157 (alkoxy C–O), 1523, 1563, 1595 (N–H bend), 1666, 1687, 1708 (C=O), 3100, 3221, 3260 (N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 1.43 (9H, s, boc CH3), 3.85 (3H, s, methoxy CH3), 7.11 (2H, d, J = 8.88 Hz, ArH), 8.03 (2H, d, J = 8.8 Hz, ArH), 8.58 (1H, s, ArH), 9.01 (1H, s, ArH), 9.05 (1H, d, J = 2.4 Hz, amide NH), 9.31 (1H, s, ArH), 9.74 (1H, d, J = 2.0 Hz, amide NH), 10.68 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 27.4 (3 peaks), 54.9, 78.7, 102.9, 113.2 (2 peaks), 124.4, 124.7, 126.0, 129.2 (2 peaks), 142.0, 144.9, 147.45 154.9, 160.4, 161.8, 164.8 ppm; LC–MS (ESI): m/z = 425.2 ([M−H]−).
General procedure for the synthesis of compounds 11a – 11h
To a suspension of compounds 10a–10h (1.00 mmol) in 4 cm3 of dichloromethane was added 1 cm3 of 4.5 M hydrochloric acid in 1,4-dioxane at 0 °C. The temperature was gradually allowed to attain room temperature (28 °C) and stirred for 12 h under nitrogen atmosphere. The completion of reaction was monitored by TLC. The precipitate from the reaction mixture was filtered, washed with 1 cm3 of dichloromethane and dried under vacuum at room temperature (28 °C) for 4 h to afford the compounds 11a–11h.
N-[3-(Hydrazinocarbonyl)pyrazolo[1,5-a]pyrimidin-6-yl]-3-(trifluoromethyl)benzamide hydrochloride (11a, C15H12ClF3N6O2)
From the reaction of 0.40 g of 10a (0.86 mmol) with 4.5 M hydrochloric acid in 1,4-dioxane in dichloromethane medium was obtained 11a as a pale yellow solid (0.29 g, 85%). M.p.: 236.7–238.8 °C; TLC: R f = 0.10 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1521, 1598 (N–H bend), 1657, 1689 (C=O), 3050, 3147 (N–H stretch), 3235, 3412 (1° amine, N–H stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 7.82 (1H, t, J = 8.0 Hz, ArH), 8.01 (1H, d, J = 7.6 Hz, ArH), 8.45 (2H, d, J = 6.4 Hz, ArH), 8.72 (1H, s, ArH), 9.31 (1H, d, J = 2.4 Hz, ArH), 9.80 (1H, d, J = 2.0 Hz, ArH), 10.48 (2H, s, amide NH), 11.53 (1H, s, amide NH) ppm; 13C NMR (75 MHz, DMSO-d 6): δ = 102.2, 118.8, 122.5, 126.1, 129.7 (q, 1 J C-F = 270.9 Hz), 125.0, 125.1, 125.2 (t, 3 J C-F = 4.0 Hz), 125.5, 127.5, 127.6, 127.7 (t, 3 J C-F = 4.0 Hz), 129.2, 129.5, 129.9, 130.2 (q, 2 J C-F = 23.5 Hz), 132.5, 134.4, 143.4, 146.3, 149.0, 160.7, 165.0 ppm; LC–MS (ESI): m/z = 365.1 ([M+H−HCl]).
4-Fluoro-N-[3-(hydrazinocarbonyl)pyrazolo[1,5-a]pyrimidin-6-yl]benzamide hydrochloride (11b, C14H12ClFN6O2)
From the reaction of 0.40 g of 10b (0.96 mmol) with 4.5 M hydrochloric acid in 1,4-dioxane in dichloromethane medium was obtained 11b as a pale yellow solid (0.29 g, 87%). M.p.: 229.6–231.4 °C; TLC: R f = 0.10 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1522, 1596 (N–H bend), 1655, 1687 (C=O), 3053, 3154 (2° N–H stretch), 3238, 3417 (1° N–H2 stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 7.42 (2H, dt, J 1 = 6.8 Hz, J 2 = 15.6 Hz, ArH), 8.22 (2H, dt, J 1 = 3.2 Hz, J 2 = 10.0 Hz, ArH), 8.71 (1H, s, ArH), 9.30 (1H, d, J = 2.0 Hz, ArH), 9.80 (1H, d, J = 2.4 Hz, ArH), 10.48 (1H, s, amide NH), 11.28 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 102.2, 115.9, 116.1 (d, 2 J C-F = 22.0 Hz), 125.8, 127.4, 130.1, 130.1 (d, 4 J C-F = 3.0 Hz), 131.3, 131.3 (d, 3J C-F = 9.0 Hz), 143.4, 146.3, 149.1, 160.8, 163.7, 166.2 (d, 1 J C-F = 249.0 Hz), 165.4 ppm; LC–MS (ESI): m/z = 315.1 ([M+H−HCl]).
4-Chloro-N-[3-(hydrazinocarbonyl)pyrazolo[1,5-a]pyrimidin-6-yl]benzamide hydrochloride (11c, C14H12Cl2N6O2)
From the reaction of 0.40 g of 10c (0.92 mmol) with 4.5 M hydrochloric acid in 1,4-dioxane in dichloromethane medium was obtained 11c as a pale yellow solid (0.31 g, 91%). M.p.: 238.1–239.9 °C; TLC: R f = 0.10 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1523, 1597 (N–H bend), 1656, 1687 (C=O), 3053, 3155 (2° N–H stretch), 3238, 3417 (1° N–H2 stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 7.95 (2H, d, J = 8.4 Hz, ArH), 8.16 (2H, d, J = 8.4 Hz, ArH), 8.72 (1H, s, ArH), 9.30 (1H, d, J = 2.4 Hz, ArH), 9.80 (1H, d, J = 2.4 Hz, ArH), 10.48 (1H, s, amide NH), 11.36 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 102.2, 125.7, 127.4, 129.1 (2 peaks), 130.4 (2 peaks), 132.3, 137.6, 143.4, 146.3, 149.0, 160.8, 165.5 ppm; LC–MS (ESI): m/z = 329 ([M–H−HCl]).
4-Bromo-N-[3-(hydrazinocarbonyl)pyrazolo[1,5-a]pyrimidin-6-yl]benzamide hydrochloride (11d, C14H12ClBrN6O2)
From the reaction of 0.40 g of 10d (0.84 mmol) with 4.5 M hydrochloric acid in 1,4-dioxane in dichloromethane medium was obtained 11d as a pale yellow solid (0.30 g, 89%). M.p.: 239.7–242.0 °C; TLC: R f = 0.10 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1520, 1595 (N–H bend), 1656, 1687 (C=O), 3054, 3157 (2° N–H stretch), 3238, 3415 (1° N-H2 stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 7.78 (2H, d, J = 8.4 Hz, ArH), 8.08 (2H, d, J = 8.4 Hz, ArH), 8.71 (1H, s, ArH), 9.29 (1H, d, J = 2.4 Hz, ArH), 9.79 (1H, d, J = 2.0 Hz, ArH), 10.48 (1H, s, amide NH), 11.34 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 102.3, 125.7, 126.7, 127.5, 130.5 (2 peaks), 132.0 (2 peaks), 132.7, 143.4, 146.3, 149.0, 160.8, 165.6 ppm; LC–MS (ESI): m/z = 377.3 ([M+2H−HCl]).
3-Fluoro-N-[3-(hydrazinocarbonyl)pyrazolo[1,5-a]pyrimidin-6-yl]benzamide hydrochloride (11e, C14H12ClFN6O2)
From the reaction of 0.40 g of 10e (0.96 mmol) with 4.5 M hydrochloric acid in 1,4-dioxane in dichloromethane medium was obtained 11e as a pale yellow solid (0.29 g, 88%). M.p.: 234.5–236.4 °C; TLC: R f = 0.10 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1523, 1597 (N–H bend), 1656, 1687 (C=O), 3053, 3155 (2° N–H stretch), 3238, 3417 (1° N–H2 stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 7.48 (1H, dt, J 1 = 6.4 Hz, J 2 = 14.8 Hz, ArH), 7.61 (1H, dt, J 1 = 5.6 Hz, J 2 = 10.0 Hz, ArH), 7.97–8.01 (2H, m, ArH), 8.71 (1H, s, ArH), 9.34 (1H, d, J = 2.4 Hz, ArH), 9.80 (1H, d, J = 2.4 Hz, ArH), 10.49 (1H, s, amide NH), 11.44 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 102.3, 115.2, 115.4 (d, 2 J C-F = 23.0 Hz), 119.6, 119.8 (d, 2 J C-F = 21.0 Hz), 124.7, 124.7 (d, 4 J C-F = 2.0 Hz), 125.7, 127.6, 131.2, 131.3 (d, 3 J C-F = 8.0 Hz), 135.8, 135.9 (d, 3 J C-F = 7.0 Hz), 143.5, 146.3, 149.1, 160.8, 161.2, 163.6 (d, 1 J C-F = 243.0 Hz), 165.2, 165.2 ppm; LC–MS (ESI): m/z = 313.0 ([M−H−HCl]).
3,5-Difluoro-N-[3-(hydrazinocarbonyl)pyrazolo[1,5-a]pyrimidin-6-yl]benzamide hydrochloride (11f, C14H11ClF2N6O2)
From the reaction of 0.40 g of 10f (0.92 mmol) with 4.5 M hydrochloric acid in 1,4-dioxane in dichloromethane medium was obtained 11f as a pale yellow solid (0.31 g, 93%). M.p.: 228.8–230.5 °C; TLC: R f = 0.10 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1568, 1597 (N–H bend), 1622, 1678 (C=O), 3088, 3190 (2° N–H stretch), 3273, 3457 (1° N-H2 stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 7.60 (1H, d, J = 6.0 Hz, ArH), 7.87 (2H, s, ArH), 8.83 (1H, s, ArH), 9.29–9.33 (1H, m, ArH), 10.55 (1H, s, amide NH), 11.45 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 102.3, 108.0, 108.3, 108.5 (t, 2 J C-F = 26.0 Hz), 111.8, 112.0 (d, 2 J C-F = 20.0 Hz), 111.9, 112.1 (d, 2 J C-F = 19.0 Hz), 125.4, 127.7, 136.9, 137.0, 137.1 (t, 3 J C-F = 9.0 Hz), 143.5, 146.4, 149.0, 160.8, 161.4, 163.8 (d, 1 J C-F = 245.0 Hz), 161.5, 163.9, 164.0 ppm; LC–MS (ESI): m/z = 331.1 ([M−H−HCl]).
2-Fluoro-N-[3-(hydrazinocarbonyl)pyrazolo[1,5-a]pyrimidin-6-yl]benzamide hydrochloride (11g, C14H12ClFN6O2)
From the reaction of 0.40 g of 10g (0.96 mmol) with 4.5 M hydrochloric acid in 1,4-dioxane in dichloromethane medium was obtained 11g as a pale yellow solid (0.29 g, 87%). M.p.: 233.5–235.6 °C; TLC: R f = 0.10 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1556, 1579 (N–H bend), 1629, 1685 (C=O), 3050, 3152 (2° N–H stretch), 3235, 3414 (1° N-H2 stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 7.38-7.45 (2H, m, ArH), 7.64–7.69 (1H, m, ArH), 7.70 (1H, dt, J 1 = 6.0 Hz, J 2 = 13.2 Hz, ArH), 8.72 (1H, s, ArH), 9.03 (1H, d, J = 2.0 Hz, ArH), 9.75 (1H, d, J = 2.4 Hz, ArH), 10.37 (1H, s, amide NH), 11.09 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 102.4, 116.8, 117.0 (d, 2 J C-F = 21.0 Hz), 123.5, 123.7 (d, 3 J C-F = 13.0 Hz), 125.1, 125.2 (d, 2 J C-F = 21.0 Hz), 125.4, 127.1, 130.6, 130.6 (d, 4 J C-F = 2.0 Hz), 133.9, 134.0 (d, 3 J C-F = 9.0 Hz), 143.5, 146.3, 148.5, 158.3, 160.8 (d, 1 J C-F = 243.0 Hz), 160.8, 163.9 ppm; LC–MS (ESI): m/z = 313.0 ([M–H−HCl]).
N-[3-(Hydrazinocarbonyl)pyrazolo[1,5-a]pyrimidin-6-yl]-4-methoxybenzamide hydrochloride (11h, C15H15ClN6O3)
From the reaction of 0.40 g of 10h (0.93 mmol) with 4.5 M hydrochloric acid in 1,4-dioxane in dichloromethane medium was obtained 11h as a pale yellow solid (0.30 g, 90%). M.p.: 248.5–250.3 °C; TLC: R f = 0.10 (MeOH–CHCl3 1:9); IR (ATR): \(\bar{\nu }\) = 1120 (alkoxy C–O), 1558, 1606 (N–H bend), 1653, 1670 (C=O), 3202, 3304 (2° N–H stretch), 3335, 3400 (1° N–H2 stretch) cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 3.95 (3H, s, methoxy CH3), 7.13 (2H, d, J = 8.8 Hz, ArH), 8.8 (2H, d, J = 8.8 Hz, ArH), 8.70 (1H, d, J = 1.2 Hz, ArH), 9.17 (1H, s, ArH), 9.77 (1H, d, J = 2.0 Hz, ArH), 10.39 (1H, s, amide NH), 10.80 (1H, s, amide NH) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 55.9, 102.1, 114.2 (2 peaks), 125.7, 126.1, 127.2, 130.4 (2 peaks), 143.3, 146.2, 149.1, 160.8, 162.9, 166.0 ppm; LC–MS (ESI): m/z = 327.3 ([M+H−HCl]).
The kinase assay
The kinase assays were conducted by hotspot kinase assay platform. The following base reaction buffer was used for the assay: 20 mM Hepes (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.02 mg/cm3 BSA, 0.02% Brij35, 2 mM DTT, 0.1 mM Na3VO4, and 1% DMSO.
The reaction procedure is used as follows: the required cofactor for the enzymatic reaction was added to a freshly prepared buffer solution, followed by the addition of the Aurora kinase at a concentration of 20 µM. The contents were mixed gently and the compound dissolved in DMSO was added to the reaction mixture at 10 µM concentration. Compounds were evaluated in a 10-dose IC50 mode with threefold serial dilution starting at 30 µM for IC50 determination. 33P-ATP was added to the mixture to initiate the reaction, and the mixture was incubated at room temperature for 2–3 h. Staurosporine was used as the control compound in a five-dose IC50 mode with tenfold serial dilutions starting at 20 µM, and the reaction was carried out at 10 µM ATP concentration.
References
Fu J, Bian M, Jiang Q, Zhang C (2007) Mol Cancer Res 5:1
Pollard JR, Mortimore M (2009) J Med Chem 52:2629
Carvajal RD, Tse A, Schwartz GK (2006) Clin Cancer Res 12:6869
Katayama H, Brinkley WR, Sen S (2003) Cancer Metastasis Rev 22:451
Ota T, Suto S, Katayama H, Han ZB, Suzuki F, Maeda M, Tanino M, Terada Y, Tatsuka M (2002) Cancer Res 62:5168
Carmena M, Earnshaw WC (2003) Nat Rev Mol Cell Biol 4:842
Marumoto T, Zhang D, Saya H (2005) Nat Rev Cancer 5:42
Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, Su M, Golec JM, Miller KM (2004) Nat Med 10:262
Oslob JD, Yu CH (2007) Pyrazolopyrimidines useful as Aurora kinase inhibitors and their preparation, pharmaceutical compositions and use in the treatment of Aurora kinase mediated diseases. Patent WO2007013964, Feb 1, 2007
Lew W, Baskaran S, Oslob JD, Yoburn JC, Zhong M (2006) Thienopyrimidines useful as Aurora kinase inhibitors and their preparation, pharmaceutical compositions, and their use for treatment of Aurora kinase-mediated diseases. Patent WO2006036266, Apr 6, 2006
Sloane DA, Trikic MZ, Chu ML, Lamers MB, Mason CS, Mueller I, Savory WJ, Williams DH, Eyers PA (2010) ACS Chem Biol 5:563
Shimomura T, Hasako S, Nakatsuru Y, Mita T, Ichikawa K, Kodera T, Sakai T, Nambu T, Miyamoto M, Takahashi I, Miki S, Kawanishi N, Ohkubo M, Kotani H, Iwasawa Y (2010) Mol Cancer Ther 9:157
Mortlock AA, Foote KM, Heron NM, Jung FH, Pasquet G, Lohmann JJ, Warin N, Renaud F, De-Savi C, Roberts NJ, Johnson T, Dousson CB, Hill GB, Perkins D, Hatter G, Wilkinson RW, Wedge SR, Heatson SP, Odedra R, Keen NJ, Crafter C, Brown E, Thompson K, Brightwell S, Khatri L, Brady MC, Kearney S, McKillop D, Rhead S, Parry T, Green S (2007) J Med Chem 50:2213
Carpinelli P, Ceruti R, Giorgini ML, Cappella P, Gianellini L, Croci V, Degrassi A, Texido G, Rocchetti M, Vianello P, Rusconi L, Storici P, Zugnoni P, Arrigoni C, Soncini C, Alli C, Patton V, Marsiglio A, Ballinari D, Pesenti E, Fancelli D, Moll J (2007) Mol Cancer Ther 6:3158
Bavetsias V, Linardopoulos S (2015) Front Oncol 5:278
Falchook GS, Bastida CC, Kurzrock R (2015) Semin Oncol 42:832
McCall JM, Kelly RC, Romero DL (2012) Pyrazolopyrimidinone compounds for the inhibition of PASK and their preparation. Patent WO2012149157, Nov 1, 2012
Rajadhyaksha MN, Kolekar SL, Baviskar AY, Panandikar AM (2011) Process for the preparation of a pyrazole derivative. Patent WO2011064798, Jun 3, 2011
Kumar AKA, Nair KB, Bodke YD, Sambasivam G, Bhat KG (2016) Monatsh Chem 147:2221
Bremberg U, Linden A, Lundback T, Nilsson J, Wiik M, Bergner M, Brandt P, Hammer K, Ringom R (2008) Preparation of pyrazolo[1,5-a]pyrimidines as inhibitors of stearoyl-CoA desaturase. Patent WO 2008116898, Oct 2, 2008. Chem Abstr 149:425–961
Lees S, Jorgensen M, Hartwig JF (2001) Org Lett 3:2729
Huang X, Buchwald SL (2001) Org Lett 3:3417
Vo GD, Hartwig JF (2009) J Am Chem Soc 131:11049
Acknowledgements
We are thankful to Anthem Biosciences management, Anthem Biosciences, Bangalore, India, for their invaluable support and allocation of resources for this work. We would like to thank Analytical chemistry team, Department of Analytical Chemistry, Anthem Biosciences, Bangalore, India, for having carried out all the analytical work. Also, we would like to thank molecular biology team, Department of Molecular Biology, Anthem Biosciences, Bangalore, India, for executing the Aurora Kinase activity studies.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kumar, A.K.A., Bodke, Y.D., Sambasivam, G. et al. Design, synthesis, and evaluation of novel hydrazide hydrochlorides of 6-aminopyrazolo[1,5-a]pyrimidine-3-carboxamides as potent Aurora kinase inhibitors. Monatsh Chem 148, 1767–1780 (2017). https://doi.org/10.1007/s00706-017-1943-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-017-1943-7