
Abstract
Electrochemical properties of base-modified cytosine or 7-deazaadenine nucleoside triphosphates (dNTPs) bearing electrochemically active anthraquinone or 3-nitrophenyl moieties were studied using cyclic voltammetry with the hanging mercury drop electrode. The anthraquinone moiety in the dNTPs gives well-pronounced reversible quinone/hydroquinone redox signals around −0.40 V (against Ag|AgCl|3M KCl reference electrode), while the nitro group in 3-nitrophenyl exhibits irreversible reduction to hydroxylamine around −0.45 V that can be reversibly oxidized to corresponding nitroso compound close to 0.0 V. Both anthraquinone and hydroxylamine redox groups can be selectively switched off by further electrochemical transformation, depending on negative potential applied and composition of the background electrolyte. Results of this study suggest that both nucleobase and the conjugate label moiety influence remarkably the adsorbability and/or intermolecular interactions taking part at the electrode surface. The potential analytical utilization of these phenomena is discussed.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Labeling of nucleic acids (NA) by various electroactive tags is of broad interest to scientists in connection with the development of electrochemical methods and biosensors for NA analysis, such as analysis of nucleotide sequences or sensing of DNA damage (reviewed in [1–3]). Redox labels can be introduced into NA via chemical phosphoramidite-based synthesis of oligonucleotides, by chemical modification of natural NA components (such as thymine residues in DNA by osmium tetroxide reagents [4] or 3′-terminal ribose in RNA by six-valent osmium complexes [5]), or enzymatically using polymerases and labeled (deoxy)nucleoside triphosphates [(d)NTPs] as monomer substrates. The latter approach has proved especially efficient and versatile for the preparation of not only the redox-labeled DNA, but also DNA bearing fluorophores [6, 7] or chemically reactive groups for further chemical transformations on DNA [8, 9] or for bioconjugation with proteins [10]. A number of modified dNTP bearing diverse electrochemically active moieties, such as ferrocene [11], organic nitrocompounds [12], [Ru/Os-(bpy)3] complexes [13], anthraquinone [14], benzofurazane [15], methoxyphenol [16], phenylazide [8], and others (reviewed in [2, 17]), have been developed and applied. A combination of these labels (to encode different nucleotide sequences, or even each nucleobase with different tags being electrochemically reduced or oxidized at different potentials) allows parallel analysis of multiple nucleotide sequences [18], typing of sequence polymorphisms [12, 13], or a simple monitoring of the conversion of one redox tag to another, e.g., to probe interactions of the modified DNA with proteins [8].
Anthraquinone (AQ), as a moiety exhibiting well-pronounced reversible electrochemistry [19–21], has been utilized for redox labeling of biomolecules [22–24]. Derivatives of AQ linked via various tethers to nucleosides were used, for example, to study DNA-mediated charge transfer [25–27]. Base-modified cytosine and 7-deazaadenine dNTPs bearing the AQ labels have recently been developed and used for polymerase synthesis of AQ-modified oligodeoxynucleotides, and utilization of the AQ tags for dual redox labeling of DNA in combination with earlier introduced nitrophenyl (PhNO2) labels in simple model applications have been tested [14]. However, a more detailed study of electrochemical properties of oligodeoxyribonucleotides (ODNs) bearing AQ or PhNO2 groups, or their combination, is to date missing. In this paper, we present a comparative study of base-modified AQ or PhNO2 dNTP conjugates using cyclic voltammetry with the hanging mercury drop electrode.
Results and discussion
In our previous study [14], anthraquinone-labeled dCTP and 7-deaza-dATP were synthesized and used for DNA labeling via incorporation of corresponding nucleotides into ODNs by DNA polymerases (for general methodologies of this strategy of modified nucleic acids construction, see reviews [2, 17, 28]). Electrochemical measurements revealed the modified ODNs to give well-developed signals due to reversible redox electrochemistry of the AQ moiety. Experiments focused on simultaneous detection of the AQ tags with another type of organic electrochemically active moieties attached to nucleobases in modified ODNs, 3-nitrophenyl [12], indicated possibilities of using the two labels for convenient dual DNA labeling when optimum conditions for their distinction are applied. Namely, conversion of the PhNO2 group into the corresponding hydroxylamine derivative (PhNHOH) via irreversible four-electron reduction of the nitro group facilitated resolution of AQ and PhNO2 signals when anodic responses were measured [14].
Cyclic voltammetry of AQ-dNTP conjugates
Here, we studied in more detail the electrochemical behavior of AQ and PhNO2-labeled dNTPs, dC PAQ TP, dA PAQ TP, \({\mathbf{dC}}^{{{\mathbf{PhNO}}_{{\mathbf{2}}} }} {\mathbf{TP}}\), and \({\mathbf{dA}}^{{{\mathbf{PhNO}}_{{\mathbf{2}}} }} {\mathbf{TP}}\). Cyclic voltammograms (CVs) of these four compounds at concentrations of 40 μM, measured in 0.3 M ammonium formate, 0.05 M sodium phosphate, pH 6.9 (a medium optimized for electrochemical analysis of DNA at mercury electrodes, suitable for simultaneous detection of natural electroactive DNA components [1, 3]) without pre-accumulation of the analytes, are presented in Fig. 1. Red curves correspond to CVs measured with initial potential E i = 0.0 V and switching potential E sw = −0.85 V, while black dotted curves were obtained for E sw = −1.85 V. Both AQ-dNTP conjugates (Fig. 1a, b), when measured with E sw = −0.85 V, gave a pair of peaks around −0.4 V corresponding to the reversible anthraquinone/anthrahydroquinone redox system:

Cyclic voltammograms of dC PAQ TP (a), dA PAQ TP (b), \({\mathbf{dC}}^{{{\mathbf{PhNO}}_{{\mathbf{2}}} }} {\mathbf{TP}}\) (c), and \({\mathbf{dA}}^{{{\mathbf{PhNO}}_{{\mathbf{2}}} }} {\mathbf{TP}}\) (d). Cyclic voltammetry (CV) at HMDE: E i +0.0 V (a, b), or +0.1 (c, d), E sw −1.85 or −0.85 V, scan rate 1 V/s, background electrolyte: 0.3 M ammonium formate, 0.05 M sodium phosphate, pH 6.9, and concentration of all substances was 40 μM
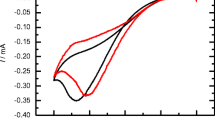
The behavior of dC PAQ TP (Fig. 1a) was nevertheless in some respects different from that of dA PAQ TP (Fig. 1b). First, a considerable difference in the heights of cathodic peak AQred and anodic peak AQH ox2 (the latter being 3 times higher than the former for 40 μM dC PAQ TP; see concentration dependences for further discussion) was observed for dC PAQ TP, while for dA PAQ TP the intensity of the cathodic peak AQred was about 3.5-times higher compared to the analogous signal of dC PAQ TP, and the anodic peak AQH ox2 of dA PAQ TP was higher by only 33 % as compared to the peak AQred of the same conjugate (Table 1; compare also solid curves in Fig. 2a, where details of the voltammograms are shown). Second, peak-to-peak separation for the AQ/AQH2 redox process was 19 mV in dC PAQ TP and 48 mV in dA PAQ TP, suggesting more facile electron transfer in the first instance. Third, for dA PAQ TP, clearly developed second pair of peaks at potentials more negative by 27 mV were observed. Differences in the relative intensities of the anodic and cathodic peaks of dC PAQ TP and dA PAQ TP can be attributed to different adsorbabilities of the two conjugates, with the dA PAQ TP adsorbing at the mercury surface more efficiently. The experiment, the results of which is shown in Fig. 2, supports such explanation: when dC PAQ TP was allowed to accumulate at the electrode surface with open current circuit for 60 s before the CVs were measured, the height of the peak AQred increased by about three times. Peak AQH ox2 became significantly higher after pre-accumulation and its height was practically the same as the height of the peak AQred. The dA PAQ TP exhibited similar effects upon the accumulation, giving wide cathodic and anodic peaks in which the two reversible pairs (distinguishable in CV measured without accumulation, Fig. 2) were clearly merged.

a Effects of adsorptive pre-accumulation (0 or 60 s at open current circuit) on CV responses of dC AQ TP and dA PAQ TP. b Repeated CV scans of dC PAQ TP (without pre-accumulation, E i +0.1 V, E sw −0.6 V, scan rate 1 V/s, and other conditions as in Fig. 1)
When the CVs were measured with the E sw = −1.85 V (i.e., with a setup previously used in DNA analysis to reduce guanine residues and obtain an anodic peak G of guanine at the mercury electrode [1, 3, 14]), the anodic peak AQH ox2 disappeared in both dC PAQ TP and dA PAQ TP, suggesting a deeper reduction of the AQ moiety upon applying the highly negative potentials. Blocking of the electrode surface by the reduction products then probably prevents fresh dNPAQTP from the bulk of solution to give peak AQH ox2 in the anodic scan. The dC PAQ TP conjugate (Fig. 1a) exhibited rather complicated behavior in a potential region between −0.9 and −1.7 V, producing several, under the given conditions, irreversible peaks in the cathodic scan that could be ascribed to reduction processes of ethynyl and carbamoyl groups in the linker, the above proposed reduction of the anthrahydroquinone (see the dependence of peak AQH ox2 height on E sw in Fig. 3, showing a steep decrease of the peak current between −1.1 V and −1.6 V), reduction of cytosine as well as tensammetric processes of the negatively charged dNTPs on the negatively charged surface (note the sharp “spike” at −1.45 V, Fig. 1a). On the contrary, dA PAQ TP yielded only one well-developed cathodic peak under the same conditions (peak Ared, Fig. 1b), which can be ascribed to reduction of the 7-deazaadenine nucleobase. Differences between the two dNTPs may be due to different adsorption modes (see above), influencing availability of different electroreducible groups for electronic communication with the electrode. Taken together, the shapes of CVs of individual PAQ conjugates, as well as differences observed between dC PAQ TP and dA PAQ TP both in the region of the AQ reversible electrochemistry and in the more negative potential region, suggest rather complicated processes undergone by these complex compounds on the mercury electrode surface (see also discussion of concentration dependences below).

Dependence of the intensity of peaks AQH ox2 and NHOHox on switching potential (dC PAQ TP and \({\mathbf{dC}}^{{{\mathbf{PhNO}}_{{\mathbf{2}}} }} {\mathbf{TP}}\), CV at HMDE, other conditions as in Fig. 1)
Cyclic voltammetry of PhNO2-dNTP conjugates
Both nitrophenyl-labeled dNTPs, \({\mathbf{dC}}^{{{\mathbf{PhNO}}_{{\mathbf{2}}} }} {\mathbf{TP}}\) (Fig. 1c) and \({\mathbf{dA}}^{{{\mathbf{PhNO}}_{{\mathbf{2}}} }} {\mathbf{TP}}\) (Fig. 1d), gave an irreversible cathodic signal, peak NO red2 , around −0.45 V (Table 1). Nitro group is known to be electrochemically reduced at various types of electrodes [29, 30] to hydroxylamine:
Due to the involvement of four electrons, the latter electrode reaction gives rise to a strong reduction signal allowing sensitive polarographic or voltammetric determination of various nitro compounds [31–39], including environmental pollutants [40–44]. In several proof-of-concept applications it has been utilized for convenient DNA labeling as well [8, 9, 12, 14]. The hydroxylamime moiety resulting from (2) is reversibly oxidizable by two electrons to the nitroso group [30]:
Hydroxylamine reduction is reflected in anodic signals (peak NHOHox) yielded by both PhNO2 dNTP conjugates close to 0.0 V (Fig. 1c, d; Table 1). In contrast to the anodic peak AQH ox2 , the peak NHOHox was observed on the CVs even when the measurements were performed with E sw = −1.85 V, displaying only partial decrease of its intensity. Dependence of the peak NHOHox height on E sw, measured for \({\mathbf{dC}}^{{{\mathbf{PhNO}}_{{\mathbf{2}}} }} {\mathbf{TP}}\) (Fig. 3), shows the peak current to be practically unchanged between E sw = −0.6 V and −1.4, and to decrease gradually with E sw being shifted to more negative potentials; for E sw = −1.85 V the peak NHOHox height corresponded to 55 % of value measured with E sw = −0.6 V. Such behavior suggests that either the corresponding redox moiety is not destroyed upon the electrode polarization to highly negative potentials, or the electrode surface does not get fully blocked by reduction products and the anodic peak NHOHox is produced by fresh analyte from the bulk of solution. Since the peak NHOHox measured with PhNO2-labeled ODN using ex situ voltammetric procedure (i.e., with adsorbed layer onto the electrode and no analyte present in the bulk of background electrolyte) completely disappeared for E sw = −1.2 V (not shown preliminary data; a complex study with labeled ODNs will be published elsewhere), the explanation based on involvement of fresh \({\text{dN}}^{{{\text{PhNO}}_{ 2} }} {\text{TP}}\) from solution appears to be more likely. Notably, significant decrease of the NHOHox height in \({\mathbf{dC}}^{{{\mathbf{PhNO}}_{{\mathbf{2}}} }} {\mathbf{TP}}\) was observed at E sw values coinciding with potential of reduction of the cytosine nucleobase (Fig. 1c), and partial electrode blocking with products of the latter reaction could cause the observed decrease of the peak NHOHox intensity. Similarly as in the case of corresponding AQ conjugates (Fig. 1a, b), \({\mathbf{dA}}^{{{\mathbf{PhNO}}_{{\mathbf{2}}} }} {\mathbf{TP}}\) differed from \({\mathbf{dC}}^{{{\mathbf{PhNO}}_{{\mathbf{2}}} }} {\mathbf{TP}}\) by absence of any distinct signals in the potential region between −0.6 and −1.85 V (Fig. 1d). The \({\mathbf{dA}}^{{{\mathbf{PhNO}}_{{\mathbf{2}}} }} {\mathbf{TP}}\) did not yield even the signal of 7-deazaadenine reduction in the ammonium formate medium (it was nevertheless observed in Britton–Robinson buffer at pH ≤ 6, see below). On the other hand, the behavior of dC PhNO2 TP in the same potential region was similar to that of dC PAQ TP, suggesting the nucleobase to be a critical component of the dNTP conjugate that dictates its behavior on the negatively charged mercury surface.
Effects of dNTP concentration, pH of background electrolyte, and scan rate
Dependences of intensities of peaks AQred/AQH ox2 measured without pre-accumulation for dC PAQ TP and dA PAQ TP, and of peaks NO red2 and NHOHox measured under the same conditions with \({\mathbf{dA}}^{{{\mathbf{PhNO}}_{{\mathbf{2}}} }} {\mathbf{TP}}\), are shown in Fig. 4. The height of the cathodic peak AQred of dC PAQ TP increased more or less linearly within the concentration region between 0 and 80 μM (Fig. 4a). A strikingly different concentration dependence was observed for the anodic peak AQH ox2 of the same dNTP conjugate. At low concentrations up to 25 μM dC PAQ TP, both peaks AQred and AQH ox2 followed an identical trend. However, between 25 and 40 μM dC PAQ TP, the height of peak AQH ox2 increased steeply, reached its maximum at 50 μM dC PAQ TP and then gradually decreased. The sigmoidal shape of the concentration dependence suggests intermolecular interactions at the electrode surface: the steep increase of the signal around 30 μM dC PAQ TP can be explained by positive cooperative effects of the already adsorbed (and electrochemically reduced) molecules of μM dC PAQ TP on adsorption of more molecules from the solution taking place from a critical surface coverage (i.e., distances between molecules at the surface), which under the given conditions is dictated by solution concentration of the dC PAQ TP. Alternatively, the reduction of dC PAQ TP may be accompanied by the formation of an ordered structure of the adsorbed layer and orientation of the AQH2 in a way facilitating the oxidation process. Since the cooperative effect was reflected in peak AQH ox2 , but not AQred heights, the presumptive intermolecular interactions were specific, in the case of dC PAQ TP, for its reduced form. Moreover, differences in the peak AQred and AQH ox2 intensities measured for 40 μM dC PAQ TP were retained in repeated CV scans (Fig. 2b), suggesting the presumptive interaction to be reversibly on/off switchable via changing the AQ redox state. In contrast to dC PAQ TP, for dA PAQ TP the S-shaped dependences of signal intensity were obtained for both cathodic and anodic peaks, indicating that analogous intermolecular interactions may have occurred in both reduced and oxidized forms of the latter conjugate, facilitating accumulation of the oxidized form at the electrode surface (see above). Concentration dependence of the dA PhNO2 TP peak NO red2 (Fig. 4c) involved a linear region between 0 and 10 μM, followed by a less steeply increasing part at higher concentration. Dependence of the height of peak NHOHox exhibited certain sign of transition around 20 μM; nevertheless, compared to signals of the AQ conjugates this effect was poorly pronounced.

Dependence of the intensity of peaks AQred, AQH ox2 , NO red2 , and NHOHox on dNTP concentration: a dC PAQ TP, b dA PAQ TP, and c \({\mathbf{dA}}^{{{\mathbf{PhNO}}_{{\mathbf{2}}} }} {\mathbf{TP}}\) (E sw −0.85 V and other conditions as in Fig. 1)
Dependences of heights and potentials of signals yielded by dA PAQ TP and dA PhNO2 TP on pH of the background electrolyte, measured in Britton–Robinson buffer, are shown in Fig. 5. The heights of peaks AQred and AQH ox2 were almost pH-independent in a wide range between pH 3 and 9 (Fig. 5a), indicating that the availability of protons were not limiting for reaction (1) to take place under the given conditions. A similar behavior was observed for peak NO red2 of dA PhNO2 TP (Fig. 5c). By contrast, peak NHOHox was detectable only in pH > 5 (Fig. 5c), most probably due to reduction of the hydroxylamine to amine that took place in the acidic media (indeed, an additional pH-dependent cathodic peak was detected in pH ≤ 5, but not in pH above 5; not shown). In both conjugates, peak Ared due to reduction of the nucleobase was observed only in pH ≤ 6, in agreement with earlier data showing that protonation was a prerequisite for the nucleobase reduction at the mercury electrode (reviewed in [3]). The potentials of all measured signals shifted to more negative potentials with increasing pH, exhibiting almost parallel trends (Fig. 5b, d).

Dependence of the intensity (a) and potential (b) of peaks AQred, AQH ox2 , and Ared for dA PAQ TP and intensity (c) and potential (d) of peaks NO red2 , NHOHox, and Ared of \({\mathbf{dA}}^{{{\mathbf{PhNO}}_{{\mathbf{2}}} }} {\mathbf{TP}}\) on pH of background electrolyte (E sw −0.85 V, scan rate 1 V/s, background electrolyte: Britton–Robinson buffers of the given pH and other conditions as in Fig. 1)
The effects of scan rate were studied in the ammonium formate medium and the results obtained for dC PAQ TP and dC PhNO2 TP are displayed in Fig. 6. As could be expected for rather complex molecules of the dNTP conjugates, involving hydrophobic, hydrophilic, and/or negatively charged parts, dependences of the measured signals on scan rate mostly did not fit into simple models valid for electrode processes driven by either diffusion or adsorption. Peak NO red2 of dC PhNO2 TP followed a slightly concave dependence on the scan rate (growing less steeply than a linear function, Fig. 6b); when the peak heights were plotted against square root of the scan rate (Fig. 6c), the resulting dependence was significantly convex (increasing more steeply than a line), together indicating the combined effects of diffusion and strong adsorption. The other peaks, AQred and AQH ox2 of dC PAQ TP and NHOHox of dC PhNO2 TP, exhibited strongly supralinear dependences of their heights on scan rate, suggesting more complex process possibly involving time-dependent desorption/reorientation of the dNTP molecules at the negatively charged surface or the above discussed lateral interactions of the AQ conjugates at the electrode surface.

Dependence of intensity of peaks AQred, AQH ox2 NO red2 , and NHOHox on scan rate for dC PAQ TP (a) and dC PhNO2 TP (b); dependence of peak NO red2 on square root of scan rate for dC PhNO2 TP (c) (E sw −0.85 V, other conditions as in Fig. 1)
Mixtures of AQ- and PhNO2-labeled dNTPs
Finally, we have been interested in the possibility of simultaneous voltammetric detection of the AQ- and PhNO2-labeled dNTPs in mixtures (Fig. 7). For this purpose, dC PAQ TP and C PhNO2 TP were mixed at ratios 1:1, 1:2, and 1:3 (keeping the dC PAQ TP concentration constant at 20 μM), and CVs were measured without pre-accumulation (dashed lines in Fig. 7) or after a 60-s pre-accumulation (solid lines). The results of these experiments demonstrated the above discussed preferential accumulation of the AQ conjugate at the electrode surface. When the CVs were measured without the pre-accumulation, peak NHOHox was detectable at the dNTPs ratio 1:1, but not the peak NO2, the potential of which (about −0.45 V) was close to the potential of AQ reduction. Upon increasing the C PhNO2 TP/dC PAQ TP ratio to 2:1 and 3:1, the peak NO2 was unmasked and peaks of the AQred/AQH ox2 depressed. However, after the pre-accumulation, both signals of C PhNO2 TP, peak NO red2 , and peak NHOHox, were strongly depressed even in threefold excess of the latter dNTP, suggesting that the more strongly adsorbing dC PAQ TP displaced the C PhNO2 TP from the electrode surface. Interestingly, in the presence of both conjugates, a new cathodic peak appeared around −0.65 V (well developed, at the C PhNO2 TP/dC PAQ TP ratio of 1:1 when the measurement was performed without pre-accumulation (Fig. 7a), and in measurements with pre-accumulation its intensity exhibited increasing trend with increasing concentration of C PhNO2 TP). This signal may indicate a chemical reaction between products or intermediate of electrochemical reduction of AQ and the nitro group at the electrode surface.

Voltammetric responses of mixtures of dC PAQ TP and dC PhNO2 TP at various ratios: a dC PAQ TP:dC PhNO2 TP = 1:1, b dC PAQ TP:dC PhNO2 TP = 1:2, c dC PAQ TP:dC PhNO2 TP = 1:3, accumulation time 0 s (dashed) or 60 s (solid; E sw −0.85 V, scan rate 1 V/s, and other conditions as in Fig. 1)
Conclusions
Cytosine or 7-deazaadenine dNTPs modified at the base residue with electrochemically active anthraquinone or 3-nitrophenyl moieties were studied using cyclic voltammetry with the hanging mercury drop electrode. AQ moiety in the dNTP conjugates is shown to retain its well-pronounced reversible electrochemistry around −0.40 V. The nitro group in PhNO2 exhibits characteristic irreversible reduction around −0.45 V. The product of this reduction, phenylhydroxylamine (NHOH), gives a well-developed signal close to 0.0 V due its reversible oxidation to the corresponding nitroso compound. Further electrochemical reduction of the AQH2 and NHOH redox groups can be used for their selective switching off, depending on the potential applied and composition of the background electrolyte (namely, in acidic media the signal of NHOH oxidation disappeared, suggesting irreversible reduction of the hydroxylamine to amine). The modified dNTPs studied differed in their adsorbability at the mercury electrode surface. In general, the tendency to being accumulated at the electrode was higher in AQ-modified dNTPs than in the PhNO2 derivatives, and among the former dA AQ TP was more efficiently adsorbed in its oxidized (AQ) form than dC AQ TP. The shapes of concentration dependences indicate intermolecular interactions of the AQ conjugates at the electrode surface that appear to be redox sensitive (specific for the reduced AQH2 form) in dC AQ TP. In our follow-up study (research in progress), the electrochemical properties of oligonucleotides modified with AQ- and PhNO2 conjugates are investigated and the possibilities of the analytical utilization of specific properties of the two types of DNA labels are tested (results will be published elsewhere).
Experimental
Synthesis of anthraquinone- and 3-nitrophenyl-labeled deoxynucleoside triphosphates
Anthraquinone-modified nucleoside triphosphates (dNTPs) bearing anthraquinone attached through a propargylcarbamoyl linker at the 5-position of cytosine (dC PAQ TP) or at the 7-position of 7-deazaadenine (dA PAQ TP) were prepared by Sonogashira cross-coupling of corresponding halogenated dNTPs with 2-(2-propynylcarbamoyl)anthraquinone according to [14]. Analogous 3-nitrophenyl-modified dNTPs were prepared by the Suzuki–Miyaura reaction of 7-iodo-7-deaza-2′-dATP (to obtain dA PhNO2 TP) or 5-iodo-2′-deoxycytidine 5′-dCTP (to obtain dC PhNO2 TP) with 3-nitrophenylboronic acid according to [12]. Both modified dNTPs were kindly donated by Prof. Michal Hocek.
Electrochemical analysis
Nucleoside triphosphates were analyzed by conventional in situ CV with a hanging mercury drop electrode. CV settings: scan rate 1 V/s, initial potential 0.0 V or +0.1 V, switching potentials −0.85 or −1.85 V. Background electrolyte: 0.3 M ammonium formate, 0.05 mM sodium phosphate, pH 6.9, if not stated otherwise. All measurements were performed at room temperature by using an Autolab analyzer (Eco Chemie, The Netherlands) in connection with VA-stand 663 (Metrohm, Herisau, Switzerland). The three-electrode system was used with an Ag|AgCl|3 M KCl electrode as a reference and platinum wire as an auxiliary electrode. Measurements were performed after deaeration of the solution by argon purging.
References
Fojta M, Jelen F, Havran L, Palecek E (2008) Curr Anal Chem 4:250
Hocek M, Fojta M (2011) Chem Soc Rev 40:5802
Palecek E, Bartosik M (2012) Chem Rev 112:3427
Fojta M, Kostecka P, Pivonkova H, Horakova P, Havran L (2011) Curr Anal Chem 7:35
Bartosik M, Trefulka M, Hrstka R, Vojtesek B, Palecek E (2013) Electrochem Commun 33:55
Dziuba D, Pohl R, Hocek M (2014) Bioconjugate Chem 1984
Riedl J, Pohl R, Ernsting NP, Orsag P, Fojta M, Hocek M (2012) Chem Sci 3:2797
Balintova J, Spacek J, Pohl R, Brazdova M, Havran L, Fojta M, Hocek M (2014) Chem Sci 6:575
Raindlova V, Pohl R, Klepetarova B, Havran L, Simkova E, Horakova P, Pivonkova H, Fojta M, Hocek M (2012) ChemPlusChem 77:652
Dadova J, Orsag P, Pohl R, Brazdova M, Fojta M, Hocek M (2013) Angew Chem Int Ed 52:10515
Brazdilova P, Vrabel M, Pohl R, Pivonkova H, Havran L, Hocek M, Fojta M (2007) Chem Eur J 13:9527
Cahova H, Havran L, Brazdilova P, Pivonkova H, Pohl R, Fojta M, Hocek M (2008) Angew Chem Int Ed 47:2059
Vrabel M, Horakova P, Pivonkova H, Kalachova L, Cernocka H, Cahova H, Pohl R, Sebest P, Havran L, Hocek M, Fojta M (2009) Chem Eur J 15:1144
Balintova J, Pohl R, Horakova P, Vidlakova P, Havran L, Fojta M, Hocek M (2011) Chem Eur J 17:14063
Balintova J, Plucnara M, Vidlakova P, Pohl R, Havran L, Fojta M, Hocek M (2013) Chem Eur J 19:12720
Simonova A, Balintova J, Pohl R, Havran L, Fojta M, Hocek M (2014) ChemPlusChem 79:1703
Hocek M (2014) J Org Chem 79:9914
Fojta M, Kostecka P, Trefulka MR, Havran L, Palecek E (2007) Anal Chem 79:1022
Ajloo D, Yoonesi B, Soleymanpour A (2010) Int J Electrochem Sci 5:459
Batchelor-McAuley C, Li Q, Dapin SM, Compton RG (2010) J Phys Chem B 114:4094
Quan M, Sanchez D, Wasylkiw MF, Smith DK (2007) J Am Chem Soc 129:12847
Mahajan S, Richardson J, Ben Gaied N, Zhao Z, Brown T, Bartlett PN (2009) Electroanalysis 21:2190
Wettig SD, Bare GA, Skinner RJS, Lee JS (2003) Nano Lett 3:617
Zhang Y-J, He X-P, Hu M, Li Z, Shi X-X, Chen G-R (2011) Dyes Pigm 88:391
Abou-Elkhair RAI, Dixon DW, Netzel TL (2009) J Org Chem 74:4712
Gorodetsky AA, Barton JK (2007) J Am Chem Soc 129:6074
Jacobsen MF, Ferapontova EE, Gothelf KV (2009) Org Biomol Chem 7:905
Hocek M, Fojta M (2008) Org Biomol Chem 6:2233
Peckova K, Barek J, Navratil T, Yosypchuk B, Zima J (2009) Anal Lett 42:2339
Zuman P (1993) Collect Czech Chem Commun 58:41
Beckett EL, Lawrence NS, Davis J, Compton RG (2002) Anal Lett 35:339
Boateng A, Brajter-Toth A (2012) Analyst 137:4531
Cordero-Rando MD, Barea-Zamora M, Barbera-Salvador JM, Naranjo-Rodriguez I, Munoz-Leyva JA, de Cisneros J (1999) Mikrochim Acta 132:7
De Souza D, Mascaro LH, Fatibello-Filho O (2011) Int J Anal Chem 2011:726462
Gupta S, Agarwal H, Gupta M, Verma PS (2010) J Indian Chem Soc 87:481
Gupta S, Gupta M, Verma PS (2009) Asian J Chem 21:7316
Chu L, Han L, Zhang X (2011) J Appl Electrochem 41:687
Kawde A-N, Aziz MA (2014) Electroanalysis 26:2484
Liu Z, Zhang H, Ma H, Hou S (2011) Electroanalysis 23:2851
Danhel A, Peckova K, Cizek K, Barek J, Zima J, Yosypchuk B, Navratil T (2007) Chem List 101:144
Dejmkova H, Stoica A-I, Barek J, Zima J (2011) Talanta 85:2594
Deylova D, Yosypchuk B, Vyskocil V, Barek J (2011) Electroanalysis 23:1548
Fischer J, Vanourkova L, Danhel A, Vyskocil V, Cizek K, Barek J, Peckova K, Yosypchuk B, Navratil T (2007) Int J Electrochem Sci 2:226
Niaz A, Fischer J, Barek J, Yosypchuk B, Sirajuddin, Bhanger MI (2009) Electroanalysis 21:1786
Acknowledgments
This work was supported by the Czech Science Foundation (grant P206/12/G151 to M.F. and 206/12/2378 to L.H.) and by the ASCR (RVO 68081707). The authors thank Jana Balintová, Hana Macíčková-Cahová, and Michal Hocek (Institute of Organic Chemistry and Biochemistry, ASCR, Prague, Czech Republic) for providing the modified nucleoside triphosphates used in this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Vidláková, P., Pivoňková, H., Fojta, M. et al. Electrochemical behavior of anthraquinone- and nitrophenyl-labeled deoxynucleoside triphosphates: a contribution to development of multipotential redox labeling of DNA. Monatsh Chem 146, 839–847 (2015). https://doi.org/10.1007/s00706-015-1435-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-015-1435-6