
Abstract
An effective method for synthesising alkaloid-like compounds containing the 3-azabicyclo[3.3.1]non-3-ene core structure was successfully carried out in a stereoselective manner via the bridged-Ritter reactions. Important optically active 6-alkyl(aryl)amido-4-alkyl(aryl)-2,2,6-trimethyl-3-azabicyclo[3.3.1]non-3-enes (imino amides) and 4-alkyl(aryl)-2,2,6-trimethyl-3-azabicyclo[3.3.1]nona-3,6-dienes (imino alkenes) were obtained in one step from (−)-β-pinene and the respective nitriles in the presence of concentrated H2SO4. The relative compositions of these products were controlled by varying the reaction conditions. Kinetic studies were conducted to enable a mechanistic understanding of the reaction pathways.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
A typical Ritter reaction comprises the reaction of a carbenium ion (usually generated in situ) with a nitrile, and then quenching with water affords an amide product, as shown in Scheme 1. In its widest terms, however, the Ritter reaction is considerably more than this. It encompasses all reactions of a carbenium ion with a nitrile group, followed by subsequent event(s). When viewed in this light, it is seen to be one of the more versatile reactions in organic synthesis [1–9].

One fascinating variant is the bridged-Ritter reaction, examples of which are illustrated in Scheme 2. In such instances, the nitrile reagent essentially clips across a cyclic precursor in a transannular process to yield a cyclic imine (Schiff base) product [10, 11].

Our earlier work on bridged-Ritter reactions [11–17] clearly established that multi-cyclic imines, such as 2 and 4 chemical scaffolds, can be obtained in a single step from inexpensive starting materials (Scheme 2). For instance, some of these (like 4) are 3-azabicyclo[3.3.1]non-3-enes, nitrogen-containing molecules that are highly reminiscent of the natural alkaloid derivatives. For this reason, we refer to these compounds as alkaloid-like compounds. The uniqueness of these cyclic compounds lies in their structural similarity to that of the bioactive but rare Aristotelia alkaloids (five species are known so far) [18–20]. The 3-azabicyclo[3.3.1]nonane structure can be found in the core structure of known bioactive alkaloids such as (+)-aristolone, (+)-aristoteline, (−)-serratoline, and (+)-aristelone (Fig. 1).

Structures of bioactive Aristotelia alkaloids
Elegant examples of asymmetric bridged-Ritter reactions were demonstrated by Marschoff [21–23], where (+)-limonene underwent a bridged-Ritter reaction with CH3CN in the presence of HClO4 to produce an optically active (1R,5R,6R)-6-acetamido-2,2,4,6-tetramethy-3-azabicyclo[3.3.1]non-3-ene. The absolute stereochemistry of this compound was confirmed by X-ray analysis [24] with the reported specific optical rotation \([\alpha ]_{D}^{25}\) = −144.1° (c = 1.3, CHCl3) [25, 26]. Similar asymmetric bridged-Ritter processes also occurred using a variety of other terpenoid substrates [26].
As part of our alkaloid-like drug discovery program, we aim to develop feasible synthetic pathways that allow quick access to alkaloid-like chemical scaffolds that can be used to produce a library of alkaloid-like compounds for drug discovery.
In this article, we describe the detailed stereospecific synthesis of chiral 3-azabicyclo[3.3.1]non-3-enes via the bridged-Ritter reaction conditions, starting from (−)-β-pinene with acetonitrile, chloroacetonitrile, and benzonitrile. We further explore the selectivity outcome of the products under different reaction conditions and investigate the reaction mechanisms based on the study of time-dependent reactive intermediates and products, monitored by GC/MS analyses.
Results and discussion
Synthesis of chiral cyclic-imines from (−)-β-pinene and CH3CN
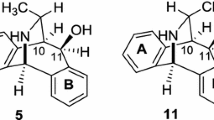
The use of (−)-β-pinene as the starting material was investigated in our bridged-Ritter reactions. When (−)-β-pinene was treated with acetonitrile, in the presence of concentrated H2SO4 [12], optically active imino amide (+)-5 and imino alkene (+)-6 were obtained in the ratio of 2.78:1 (Scheme 3). Compound (+)-5 was obtained in 64 % yield under concentrated H2SO4 conditions. This is an improvement of the isolated yield in comparison to the reaction of (−)-β-pinene with CH3CN, using HClO4 as the protic acid, where the yields were reported to be only between 12 and 36 % [27]. The assignment of configuration of (+)-5 was deducted on the grounds of chemical path stereospecificity [25] to be (1S,5S,6S) with the expected specific rotation of \([\alpha ]_{D}^{25}\) = +144.1°, based on the specific rotation of the corresponding opposite enantiomer [26]. The optical purity of (+)-5 was directly determined from its observed rotation (\([\alpha ]_{D}^{25}\) = +108°) to be 75 % e.e.

Imino alkene (+)-6 was also obtained under this study in which concentrated H2SO4 was used as an alternative to toxic mercury nitrate as previously reported by Delpech et al. [27, 28] for the synthesis of (±)-6 from (−)-α-pinene.
The 1H NMR spectrum reveals the alkene proton (H-7) at 5.35 ppm as a complex tdd. The C-6 methyl group appears at 1.80 ppm as a ddd. The 13C NMR spectrum showed two quaternary signals at 167.0 and 134.5, which are due to the imine carbon (C-4) and the alkene carbon (C-6), respectively. The optical rotation of compound (+)-6 was found to be \([\alpha ]_{D}^{25}\) = +9.1° (c = 0.95, CHCl3).
Polar solvents, such as acetic acid, were reported to increase the yields in Ritter reactions as they could increase the nucleophilicity of the nitrile and the stability of the carbenium ion [29, 30]. The polar reaction conditions reported by Bishop et al. [11] were adopted for the reaction of (−)-β-pinene and acetonitrile in two attempted methods (a and b). The conditions and outcome for each reaction are summarised in Scheme 4 and Table 1.

The results of these reaction conditions in method a and b revealed the formation of (+)-5 and (+)-6 in the ratios of 2.95:1 and 5:1. This clearly indicates that the use of acetic acid only provided a modest increase of (+)-5 over (+)-6 in method a. In the case of method b, although the formation of (+)-5 has somewhat improved over that of (+)-6. However, the downside was the reduction of the overall yields of the products. In attempts to optimise the formation of (+)-5, we also observed the formation of an unexpected isobornylacetamide 7 in both method a and b, in 10 and 4 % yields, respectively (Table 1).
The HRMS of 7 reveals the exact mass of 196.16593, which corresponds to C12H22NO ([M + H]+). NMR spectral data of 7 are in agreement to those previously reported in the literature [31]. The optical rotation of compound 7 was found to be \([\alpha ]_{D}^{25}\) = +20.3° (c = 0.47, CHCl3). Forster [32, 33] reported the specific optical rotation of (−)-exo-N-isobornylacetamide [(−)-7] to be \([\alpha ]_{D}^{25}\) = −19.5°. This stereochemical outcome was confirmed by the work by Zhou et al. [34]. On the basis of the optical rotation of compound 7, one would infer that this compound is the (+)-exo-N-isobornylacetamide. The X-ray analysis has revealed that the structure contained two molecules, and they are in the centrosymmetric space group C2/c. This indicates compound 7 is indeed a racemic mixture. The relative stereochemistry of 7 is shown in Fig. 2.

ORTEP diagram of the structure of compound 7, showing the relative stereochemistry of the compound
Under Ritter reaction conditions, Carman et al. [31] reported the formation of (±)-exo-N-isobornylacetamide 7 from the reaction of (+)-camphene with CH3CN in the presence of H2SO4/acetic acid. The racemic formation was further confirmed by Hanzawa et al. [35] on the reaction of chiral (−)-isobornyl acetate with benzonitrile to give the racemic (±)-exo-N-isobornylbenzamide. The racemisation was explained by the isobornyl carbocation intermediate that underwent the 6,2-hydride shift to form two equal carbocations.
In our case, the formation of compound 7 was unexpected, and its optical rotation contradicts the X-ray analysis. The mechanism in Scheme 5 has been proposed to explain the formation of (±)-7. The carbenium ion A underwent intermolecular cyclisation to give isobornyl carbocation B, which underwent the 6,2-hydride shift to form the minor carbocation intermediate C, in a lower proportion than that of B. Both intermediates underwent nucleophilic addition with CH3CN and then quenching with water afforded racemic mixture of 7.

The bridging Ritter reaction between (−)-β-pinene and chloroacetonitrile
When (−)-β-pinene was treated with chloroacetonitrile, under concentrated H2SO4 conditions, optically active imino amide (+)-8, imino alkene (+)-10, and the isobornylamide 11 were obtained (Scheme 6). The three compounds were isolated by silica column chromatography to give (+)-8, (+)-10, and 11 in the yields 31, 23, and 19 %, respectively.

The specific rotation of compound (+)-8 was found to be \([\alpha ]_{D}^{25}\) = +90.8° (c = 1.06, CHCl3). The 1H NMR spectrum revealed the presence of the two new CH2 groups; the group at C-4′ appears as two separate doublets at 4.18 and 4.03 ppm, while the chloroacetamide group (COCH 2Cl) as a broad singlet at 4.02 ppm. The carbons of these were also observed in the 13C spectrum at both 49.8 and 43.1 ppm, respectively. Other characteristic features of 8 were also observed, such as the three CH3 groups at 31.2, 26.5, and 25.9 ppm.
Structure of compound (+)-10 was fully characterised by spectral analysis. The 1H NMR spectrum showed the two terminal alkene protons (CH2-6′) to appear as two distinct singlets at 4.86 and 4.78 ppm. These two resonances were confirmed to correlate to the same C-6′ by HSQC. It is noteworthy that the H-5 appears at 3.36 ppm, further downfield than the corresponding proton (2.5 ppm) found in (+)-6. 13C NMR analysis confirms all the carbons required. The specific rotation of compound (+)-10 was found to be \([\alpha ]_{D}^{25}\) = +49.1° (c = 0.57, CHCl3).
It is suggested that (+)-10 was the rearranged product of 9 (Scheme 6). The presence of both compounds was initially observed in the 1H NMR spectrum of the crude mixture. The same sample was further monitored by obtaining its 1H NMR spectra over a period of time. The last 1H NMR spectrum was only shown to contain (+)-10. This finding suggests that compound 9 formed in the initial step of the bridged-Ritter reaction. It then underwent double-bond migration over time to form the less constrained (+)-10 [36].
The identity of isobornyl chloroacetamide 11 was confirmed by spectral analysis. The 1H NMR spectrum revealed the presence of the new CH2 group as a doublet at 4.04 ppm that was correlated to the 13C peak at 42.9 ppm by HSQC. It also shows H-2 at 3.89 ppm as a complex td. The two methyl groups at C-7′ appear as two singlets at 0.84 and 0.95 ppm, respectively. The third methyl at C-1′ was also observed at 0.84 ppm. The low specific optical rotation of 11 (\([\alpha ]_{D}^{25}\) = +1.4°) suggests that racemisation may also occur in the formation of compound 11, in a similar manner to that of compound 7 (Scheme 5).
In contrast to its counterpart compound 7, compound 11 was formed without the use of acetic acid. Despite this, the behaviour of the carbenium ion in chloroacetonitrile allowed for a much larger mole ratio of 11 to form in contrast to 7.
Bridged Ritter reaction of (−)-β-pinene and PhCN
The reaction of (−)-β-pinene with PhCN under our reaction conditions provided the imino amide (+)-12 and imino alkene (+)-13. The outcome of the reaction revealed that (+)-12 and (+)-13 were the major and minor products, in 42 and 24 % yields, respectively (Scheme 7).

The ratio of (+)-12 and (+)-13 was reversed by varying the reaction conditions. The cyclic imino alkene (+)-13 was obtained as the major product (74 %) over the cyclic imino amide (+)-12 (14 %) when the reaction mixture was left to stand at room temperature overnight without stirring, allowing the PhCN to diffuse slowly into the aqueous layer where the reaction took place. Beside (+)-12 and (+)-13, there was also a small trace of compound 14 with molecular mass of m/z = 257 as detected by GC/MS of the crude product. Due to its small quantity, it was not possible to isolate compound 14 for a full analysis, after a few attempts had been made.
Kinetic studies
Kinetic studies were undertaken to understand the reaction mechanisms for the formation of (+)-12, (+)-13 by investigating the rate at which intermediates and products formed over time. The reaction was monitored by direct removal of reaction aliquots at a time interval; these were then quenched with water and followed by GC–MS analysis. Figure 3 shows the composition of products (+)-12 and (+)-13, and the intermediate 14 (molecular weight of 257), in each of the GC/MS samples at specific times.

Reaction profile showing the composition of (+)-12, (+)-13, and 14 at specific times
At the 30-min mark, the studies show the amount of (+)-12 was 17 % and almost equal to that of 14 (16 %), while the concentration of (+)-13 was at the lowest (4 %). The composition of (+)-12 and (+)-13 increases almost in a linear fashion at a similar rate, while that of compound 14 proportionally decreased. The presence of intermediate alcohol 14 in the reaction mixture has provided a plausible reaction pathway, which suggests that the reaction proceeded through the intermediate E as shown in Scheme 7. Intermediate E came about via the nucleophilic addition of A to give the intermediate D. D further underwent intramolecular cyclisation to form the carbenium ion E from which the three compounds (+)-12, (+)-13, and 14 were derived. This was evidenced by (1) the composition of (+)-12, (+)-13, and 14 over the reaction time at 0 °C at the 30-min mark (4:1:4) and (2) the eventual increase in concentration of (+)-12 and (+)-13, corresponding to the decrease of 14. The formation of 14 dominated at low temperature. In contrast, at room temperature, the major products were eventually (+)-12 and (+)-13. The data suggest that the transformation of E into 14 becomes reversible at moderate temperature, and the rate-determining step (k 1) is the reaction of E with another mole of PhCN to form F and eventually (+)-12 (Scheme 8).

Conclusions
This study has led to a better understanding of the chemistry involved in the bridged-Ritter reaction and products that formed. The significance of the bridged-Ritter reaction conditions was determined. It was concluded that (1) a single enantiomer of either cyclic imine amide or imine alkene can be obtained in one step from the reaction of (−)-β-pinene and a nitrile and (2) the yields and the ratios of these products depend on the reaction conditions. It was observed that the stronger acidic conditions provide the highest yields. With more polar conditions, the yields were compromised while allowing for the formation of unexpected the isonorbornyl derivatives. The kinetic studies have allowed a proposed mechanism for the reaction to being uncovered.
Experimental
Reagents and analytical grade solvents were purchased from commercial sources. Progress of reactions was monitored by TLC analysis, performed on aluminium-backed Merck 60 GF254 silica gel or Merck 60 GF254 neutral alumina gel with UV detection at 254 nm and/or Dragendorff’s reagent. Compounds were purified by column chromatography using Merck flash either neutral alumina or silica gel (40–63 μm). Purity of compounds was determined by 1H NMR and GC/MS. 1H and 13C NMR spectra were recorded on an Agilent 500-MHz spectrometer (500 MHz 1H, 125 MHz 13C) in deuterated chloroform (CDCl3), unless otherwise specified. NMR assignments were based on COSY, HSQC, and DEPT experiments. 1H and 13C NMR assignments are based on the numbering system used on the systematic name. Gas chromatographer/mass spectrometer (GC/MS) analysis was carried out on an Agilent 6890 series gas chromatographer coupled to an Agilent 5973 network MS (EI) mass spectrometer. Separation was achieved on a Zebron ZB-5mS (5 % polysilphenylene, 95 % polydimethylsiloxane) capillary column (30 m × 0.25 mm × 0.25 μm). Helium was used as the carrier gas at a flow rate of 1.2 cm3 min−1. The injection was done by three front inlet washes, three samples pumps, and three post washes. The injection port was in split mode, with a split flow rate of 1.7 cm3 min−1. The mass spectrometer was operated from 50 to 290 amu, in positive mode, and a solvent delay of 2 min was applied. The following temperature program was used: 50 °C maintained for 2 min, then ramped at 10 °C min−1 to 290 °C and held for 4 min. High-resolution mass spectra were obtained on an Agilent 6510 Accurate Mass Q-TOF Mass Spectrometer, equipped with an ESI source.
Bridged Ritter reaction of (−)-β-pinene with CH3CN
Sulfuric acid (18 M, 4.2 cm3, 78.7 mmol) was stirred at 0 °C in a flask fitted with a reflux condenser and a drying tube. To the reaction flask was added 21 cm3 acetonitrile (0.4 mol). A solution of 1.0 g (−)-β-pinene (1.15 cm3, 7.34 mmol) in 5 cm3 benzene was added drop-wise, via the condenser, into the reaction mixture. A further 0.5 cm3 of benzene was used to wash all traces of (−)-β-pinene into the reaction flask. The mixture was stirred at 0 °C for 30 min and then at room temperature for 24 h. Water (30 cm3) was added into the reaction mixture and stirred for 30 min. The resulting mixture was washed with light petroleum ether, and then the aqueous layer was basified with 2 M solution of sodium hydroxide to pH > 10. The aqueous layer was extracted with chloroform (40 cm3 × 3). The combined extracts were washed with 50 cm3 water, dried over anhydrous Na2CO3, and filtered. The solvent was removed under reduced pressure to yield green viscous oil as the crude product. The crude product was purified by silica column chromatography using gradient elution from hexane to ethyl acetate/hexane (1:1) to provide (+)-5, (+)-6 in 64 and 23 % yields, respectively.
N-((1S,5S,6S)-2,2,4,6-Tetramethyl-3-azabicyclo[3.3.1]non-3-en-6-yl)acetamide (5, C14H24N2O)
A green wax (1.106 g, 4.67 mmol, 64 % yield); R f = 0.73 (neutral alumina, 10 % ethyl acetate/hexane); \([\alpha ]_{D}^{25}\) = +108.0° (c = 1, CH2Cl2); IR (film): \(\bar{\nu }\) = 3,295, 3,073, 2,969, 2,935, 1,644, 1,544, 1,460, 1,372, 1,289, 1,210, 1,168, 1,110, 1,037, 894, 752, 665 cm−1; 1H NMR: δ = 5.32 (br s, 1H, NH), 3.17 (br d, J = 2.0 Hz, 1H, H-5), 2.09 (s, 3H, CH3-4′), 1.99 (s, 3H, COCH 3 ), 1.81 (ddd, J = 2.0, 2.0, 11.0 Hz, 1H, Ha-9), 1.75 (dd, J = 2.0, 2.0 Hz, 1H, H-1), 1.71 (ddd, J = 2.5, 14.0 Hz, 1H, Ha-8), 1.57 (ddd, J = 4.0, 6.0, 10 Hz, 1H, Hb-9), 1.54 (dd, J = 4.5, 8.5 Hz, 1H, Ha-7), 1.47 (s, 3H, CH3-6′), 1.44 (dd, J = 4.5, 7.5 Hz, 1H, Hb-7), 1.40 (ddd, J = 4.5, 7.5, 10.0 Hz, 1H, Hb-8), 1.28 (s, 3H, CH3-2′), 1.15 (s, 3H, CH3-2′) ppm; 13C NMR: δ = 169.8 (C=O), 166.7 (C-4), 60.4 (C-6), 57.0 (C-2), 55.7 (CH-1), 40.3 (CH-5), 33.2 (CH2-7), 31.7 (CH3-4′), 29.8 (CH3-6′), 24.6 (COCH3), 24.6 (CH2-9), 24.1 (CH2-8), 21.1 (CH3-2′), 14.2 (CH3-2′) ppm; GC–MS (EI): R t = 17.18 min, m/z (%) = 236 (60, M+), 221 (25), 177 (70), 136 (68); HR-ESI–MS: [M + H]+ found 237.19780, C14H25N2O requires 237.19669.
(1S,5S)-2,2,4,6-Tetramethyl-3-azabicyclo[3.3.1]nona-3,6-diene (6, C12H19N)
A yellow oil (0.311 g, 1.76 mmol, 23 % yield); R f = 0.61 (silica, 50 % ethyl acetate/hexane, trace amount of Et3N); \([\alpha ]_{D}^{25}\) = +9.07° (c = 0.95, CHCl3); IR (film): \(\bar{\nu }\) = 2,965, 2,926, 2,870, 1,665, 1,454, 1,359, 1,330, 1,296, 1,253, 1,054, 1,035, 1,018, 884, 821, 799 cm−1; 1H NMR: δ = 5.35 (tdd, J = 1.5, 2.5, 3.0 Hz, 1H, H-7), 2.50 (br s, 1H, H-5), 2.22 (ddd, J = 1.5, 2.5, 3.0 Hz, 1H, Ha-8), 2.18 (ddd, J = 2.5, 5.0, 7.5 Hz, 1H, Hb-8), 2.04 (s, 3H, CH3-4′), 1.86-1.85 (m, 2H, Ha-9, H-1), 1.80 (ddd, 1.5, 2.5, 5.0 Hz, 3H, CH3-6′), 1.56 (ddd, J = 2.5, 5.0, 11.5 Hz, 1H, Hb-9), 1.20 (s, 3H, CH3-2′), 1.15 (s, 3H, CH3-2′) ppm; 13C NMR: δ = 167.0 (C-4), 134.5 (C-6), 122.2 (CH-7), 58.5 (C-2), 40.4 (CH-5), 33.4 (CH-1), 31.8 (CH3-4′), 28.9 (CH2-8), 28.2 (CH3-6), 28.0 (CH3-2′), 25.5 (CH2-9), 23.9 (CH3-2′) ppm; GC–MS (EI): R t = 10.95 min, m/z (%) = 177 (15, M+), 136 (35); HR-ESI–MS: [M + H]+ found 178.15790, C12H20N requires 178.15903.
Bridged-Ritter reaction of (−)-β-pinene with CH3CN in acetic acid
Method a: Sulfuric acid (18 M, 4.5 cm3) was diluted with 0.5 cm3 water and stirred in a flask fitted with a reflux condenser and a drying tube at room temperature. To the reaction flask, 21 cm3 acetonitrile (0.40 mol) was added. A solution of 1.0 g (−)-β-pinene (1.15 cm3, 7.34 mmol) in 5 cm3 glacial acetic acid was added drop-wise, via the condenser, into the reaction mixture. A further 0.5 cm3 of acetic acid was used to wash all traces of (−)-β-pinene into the reaction flask. The reaction was then heated to ~50 °C for 24 h. The reaction mixture was worked up by the addition of 20 cm3 water and then basified with 4 M NaOH until pH was >10. The reaction mixture was worked up to give the crude product (1.857 g) and purified as previously described to give 1.121 g 5 (65 %), 0.295 g 6 (22 %), and 0.146 g 7 (10 %). Compound 7 was further purified by recrystallization from hexane to give pale yellow crystals.
Method b: (−)-β-pinene (1.46 g, 10.7 mmol) was added to a stirred mixture of 1.5 cm3 98 % sulfuric acid, 10 cm3 acetonitrile, and 15 cm3 glacial acetic acid at 0 °C. The reaction was stirred for 30 min at 0 °C and then overnight at room temperature. The reaction was then worked up to give the crude product (2.174 g), purified as previously described to give 1.292 g 5 (51 %), 0.22 g 6 (11 %), and 0.086 g 7 (4 %).
N-((1S,2S,4S)-1,7,7-Trimethylbicyclo[2.2.1]heptan-2-yl)acetamide (7, C12H21NO)
A pale yellow solid (58.2 mg, 0.298 mmol, 4 %); m.p.: 125 °C; R f = 0.68 (silica, 50 % ethyl acetate/hexane); \([\alpha ]_{D}^{25}\) = +20.3° (c = 0.47, CHCl3); IR (film): \(\bar{\nu }\) = 3,314, 2,955, 2,875, 1,651, 1,547, 1,461, 1,372, 1,330, 1,291 cm−1; 1H NMR: δ = 5.35 (br s, 1H, NH), 3.90 (ddd, J = 4.5, 4.5, 9.0 Hz, 1H, H-2), 1.96 (s, 3H, NCOCH 3 ), 1.86 (dd, J = 9.0, 13.0 Hz, 1H, H-3), 1.73 (dd, J = 4.0, 8.0 Hz, 1H, H-4), 1.70 (dddd, J = 4.0, 8.0, 12.0, 13.5 Hz, 1H, H-5), 1.56 (ddd, J = 4.5, 12.0, 13.0 Hz, 1H, H-3), 1.53 (ddd, J = 4.0, 8.5, 8.5 Hz, 1H, H-6), 1.23 (ddd, J = 4.0, 9.0, 13.5 Hz, H-6), 1.15 (ddd, J = 4.0, 9.5, 13.5 Hz, H-5), 0.90 (s, 3H, CH3-1′), 0.83 (s, 3H, CH3-7′), 0.82 (s, 3H, CH3-7′) ppm; 13C NMR: δ = 169.2 (C=O), 56.8 (CH-2), 48.4 (C-7), 47.10 (C-1), 44.9 (CH-4), 39.2 (CH2-3), 36.0 (CH2-6), 27.0 (CH2-5), 23.6 (COCH3), 20.3 (CH3-1′), 20.3 (CH3-7′), 11.7 (CH3-7′) ppm; GC–MS (EI): R t = 14.19 min, m/z (%) = 195 (30, M+), 180 (10), 136 (40); HR-ESI–MS: [M + H]+ found 196.16593, C12H22NO requires 196.16567.
Bridging Ritter reaction between (−)-β-pinene and chloroacetonitrile
Sulfuric acid (18 M, 4.2 cm3, 78.7 mmol) was stirred in a flask fitted with a reflux condenser and a drying tube at 0 °C. To the reaction flask, 25 cm3 chloroacetonitrile (0.40 mol) was added. A solution of 1.0 g (−)-β-pinene (1.15 cm3, 7.34 mmol) in 5 cm3 benzene was added drop-wise into the reaction mixture, via the condenser. A further 0.50 cm3 of benzene was used to wash all traces of (−)-β-pinene into the reaction flask. After 30 min the reaction was allowed to reach room temperature and left to run for 2.5 h. The reaction was then quenched by the addition of 30 cm3 water. The mixture was then basified with 2 M NaOH (until pH > 10). The aqueous layer was washed with petroleum spirits (2 × 50 cm3) to remove any remaining pinene before extraction with CHCl3 (2 × 15 cm3) and drying with Na2SO4.
Within the petroleum spirits fraction, crystals that formed were filtered to give pure (+)-8 (0.704 g). Petroleum spirits was removed from the filtrate to give a crude mixture (0.390 g) that was analysed by GC/MS to be mainly (+)-10 and 11 with small quantities of (+)-8 remaining. The chloroform extraction was also analysed by GC/MS after the solvent had been removed via reduced pressure to show it contained mainly 8, with small quantities of (+)-10 and 11. The two crudes were therefore combined and purified via separation on an alumina column using a mobile phase of 15 % EtOAc and 85 % petroleum spirits. Elution from the column was monitored by TLC, using the same mobile phase, developed in I 2(g) atmosphere. This resulted in isolation of a further 8 (0.306 g), as well as 0.511 g 10 and 0.468 g 11.
2-Chloro-N-[(1S,5S,6S)-4-(chloromethyl)-2,2,6-trimethyl-3-azabicyclo[3.3.1]non-3-en-6-yl]acetamide (8, C14H22Cl2N2O)
A pale orange solid (1.011 g, 3.32 mmol, 31 %); m.p.: 152 °C; R f = 0.18 (1.5:8.5 EtOAc/petroleum spirits); \([\alpha ]_{D}^{25}\) = +90.8° (c = 1.06, CHCl3); IR (neat): \(\bar{\nu }\) = 3,280, 3,098, 2,974, 2,953, 2,898, 2,872, 1,683, 1,653, 1,573, 1,459, 1,432, 1,343, 1,262, 1,107, 972, 890, 806, 725, 706, 692, 660 cm−1; 1H NMR: δ = 6.55 (s, 1H, NH), 4.18 (d, J = 11.0 Hz, H1, H-4a’) 4.03 (d, J = 11.0 Hz, H1, H-4b’), 4.02 (br s, 2H, COCH 2Cl), 3.41 (br s, 1H, H-5), 1.87 (br m, 1H, H-9a), 1.84-1.81 (m, 1H, H-8a), 1.76-1.70 (m, 3H, H-8b, H-9b, H-1), 1.60 (tt, J = 4.0, 14.0 Hz, H, H-7a), 1.46 (s, 3H, CH3-6′), 1.35 (td, J = 5.0, 14.0 Hz, H-7b), 1.32 (s, 3H, CH3-2′), 1.19 (s, 3H, CH3-2′) ppm; 13C NMR: δ = 165.0 (C=O), 164.4 (C = N, C-4), 59.2 (C-6), 56.1 (C, C-2), 49.8 (CH2, C-4′), 43.1 (CH2, COCH2Cl), 36.9 (CH, C-5), 34.0 (CH, C-1), 32.3 (CH2, C-7), 31.2 (CH3, C-2′), 26.5 (CH3, C-6′), 25.9 (CH3, C-2′), 24.6 (CH2, C-9), 24.4 (CH2, C-8) ppm; GC–MS (EI): R t = 20.43 min, m/z (%) = 304, 289 (11), 269 (100), 211 (13), 176 (23); HR-ESI–MS: [M + H]+ found 305.11109, C14H23Cl2N2O requires 305.11092.
(1S,5S)-4-(Chloromethyl)-2,2-dimethyl-6-methylene-3-azabicyclo[3.3.1]non-3-ene (10, C12H18ClN)
A dark brown oily substance (0.511 g, 2.42 mmol, 23 %); R f = 0.75 (1.5:8.5 EtOAc/petroleum spirits); \([\alpha ]_{D}^{25}\) = +49.1° (c = 0.57, CHCl3); IR (neat): \(\bar{\nu }\) = 2,970, 2,935, 2,872, 2,209, 1,562, 1,460, 1,382, 1,262, 1,141, 909, 810 cm−1; 1H NMR: δ = 4.86 (s, 1H, H-6a′), 4.78 (s, 1H, H-6b′), 4.13 (d, J = 12.0 Hz, 1H, H-4a′), 4.04 (d, J = 12.0 Hz, 1H, H-4b′), 3.36 (br s, 1H, H-5), 2.27–2.25 (m, 2H, CH2-8), 2.01–1.88 (m, 1H, H-7a), 1.82 (s, 3H, CH3-2′), 1.77 (1H, H1), 1.73–1.63 (1H, H-7b), 1.58–1.53 (2H, CH2-9), 1.25 (s, 3H, CH3-2′) ppm; 13C NMR: δ = 164.5 (C-4), 122.3 (C-6), 110.5 (CH2, C-6′), 60.2 (C-2), 45.0 (CH2, C-4′), 41.5 (CH, C-5), 35.3 (CH, C-1), 30.9 (CH3, C-2′), 29.7 (CH2, C-9), 28.7 (CH2, C-8), 25.0 (CH2, C-7), 23.5 (CH3, C-2′) ppm; GC–MS (EI): R t = 13.02 min, m/z (%) = 211, 196 (6), 176 (100), 93 (44); HR-ESI–MS: [M + H]+ found 212.11262, C12H19ClN requires 212.11278.
2-Chloro-N-((1R,4S)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl)acetamide (11, C12H20ClNO)
A white solid with red flecks (0.468 g, 2.04 mmol, 19 %); m.p.: 70 °C; R f = 0.51 (1.5:8.5 EtOAc/petroleum spirits); \([\alpha ]_{D}^{25}\) = + 1.4° (c = 1.78, CHCl3); IR (neat): \(\bar{\nu }\) = 3,324, 2,952, 2,877, 1,654, 1,542, 1,458, 1,393, 1,371, 1,242, 1,032, 931, 793, 680 cm−1; 1H NMR: δ = 6.63 (1H, NH), 4.04 (d, J = 4.0 Hz, 2H, COCH 2Cl), 3.89 (td, J = 5.5, 9.0 Hz, 1H, H-2), 1.88 (dd, J = 9.0, 13.0 Hz, 1H, H-3a), 1.78 (t, J = 5.0 Hz, 1H, H-4), 1.73 (dddd, J = 5.0, 4.0 Hz, 1H, H-5a), 1.64–1.62 (m, 1H, H-3b), 1.59 (dd, J = 5.0, 12.0 Hz, 1H, H-6a), 1.30 (ddd, J = 4.0, 4.5, 12.0 Hz, 1H, H-6b), 1.18 (ddd, J = 2.5, 4.0, 12.0 Hz, 1H, H-5b), 0.95 (s, 3H, CH3-7′), 0.84 (s, 6H, CH3-7′, CH3-1′) ppm; 13C NMR: δ = 164.9 (C=O), 57.0 (CH, C-2), 54.3 (C-7), 51.8 (C-1), 44.9 (CH, C-4), 42.9 (CH2Cl, C-4′), 38.9 (CH2, C-3), 35.8 (CH2, C-6), 27.0 (CH2, C-5), 20.2 (CH3, C-7′), 20.1 (CH3, C-7′), 11.7 (CH3, C-1′) ppm; GC–MS (EI): R t = 15.65 min, m/z (%) = 229 (2, M+), 194 (4), 136 (20), 121 (49), 95 (100); HR-ESI–MS: [M + H]+ found 230.12343, C12H21ClNO requires 230.12334.
Bridged Ritter reaction of (−)-β-pinene with PhCN
A mixture of 10 cm3 PhCN (97 mmol) and 2 cm3 18 M H2SO4 (37 mmol) in round-bottom flask fitted with a condenser and a drying tube was stirred at 0 °C. To the reaction mixture was added drop-wise a solution of 0.94 g (−)-β-pinene (1.1 cm3, 6.9 mmol) in 8 cm3 benzene via the condenser. The reaction mixture was stirred at 0 °C for 30 min and then left to stand, without stirring, at room temperature for 24 h. Water (2 cm3) was added to the reaction flask, followed by 100 cm3 diethyl ether. The mixture was then set to stir for 10 min. The ether layer was removed and the aqueous layer basified with 4 M NaOH until pH 12. The aqueous layer was then extracted with chloroform (30 cm3 × 3). The combined extracts were washed with 30 cm3 saturated NaCl solution, dried over anhydrous Na2CO3, and filtered. The solvent was removed under reduced pressure, and the crude product was purified by a neutral aluminium oxide column chromatography (EtOAc/light petroleum, 1:9) to give 0.337 g (+)-12 (14 %) and 1.193 g (+)-13 (73 % yield).
The reaction was repeated, and after at 0 °C for 30 min, the reaction was further stirred at RT for 24 h. After the usual workup and purification, the reaction gave 1.03 g (+)-12 (42 %) and 0.39 g (+)-13 (24 %).
N-((1S,5S,6S)-2,2,6-Trimethyl-4-phenyl-3-azabicyclo[3.3.1]non-3-en-6-yl)benzamide (12, C24H28N2O)
A yellow solid (1.03 g, 42 %); m.p.: 59 °C; \([\alpha ]_{D}^{25}\) = +147.6° (c = 1, CH2Cl2); R f = 0.54 (neutral alumina, 10 % EtOAc/light petroleum); IR (film): \(\bar{\nu }\) = 3,300, 2,969, 2,933, 1,641,1,570, 1,525 cm−1; 1H NMR: δ = 7.86 (dd, J = 1.5, 7.5 Hz, 2H, 2HAr), 7.77 (dd, J = 1.5, 7.5 Hz, 2H, 2HAr), 7.48 (td, J = 1.5, 7.5, 7.5 Hz, 1H, HAr), 7.45 (td, J = 1.5, 7.5, 7.5 Hz, 1H, 2HAr), 7.34–7.40 (m, 4H, 4HAr), 6.13 (s, 1H, HN), 4.40 (s, 1H, H-5), 2.03 (ddd, J = 2.5, 2.5, 13.0 Hz, 1H, H-9), 1.95–1.88 (m, 3H, H-9, 2H-8), 1.76–1.72 (m, 1H, H-1), 1.55–1.52 (m, 2H, 2H-7), 1.41 (s, 3H, CH3-4′), 1.27 (s, 3H, CH3-2′), 1.10 (s, 3H, CH3-2′) ppm; 13C NMR: δ = 167.2 (CO), 165.7 (C-4), 142.2 (C), 135.7 (C), 131.4 (2CH), 129.1 (2CH), 128.7 (2CH), 128.4 (2CH), 127.1 (CH), 126.7 (CH), 58.8 (C-2), 56.6 (C-8), 34.9 (CH-5), 34.3 (CH-1), 33.8 (CH2-7), 31.6 (COCH3), 27.4 (CH3-2′), 26.6 (CH3-2′), 24.9 (CH2-9), 24.3 (CH2) ppm; GC–MS (EI): R t = 27.20 min, m/z (%) = 360 (60, M+), 345 (25), 239 (70), 196 (85); HR-ESI–MS: [M + H]+ found 361.22760, C24H29N2O requires 361.22744.
(4S,8S)-4,4,8-Trimethyl-2-phenyl-3-azabicyclo[3.3.1]nona-2,7-diene (13, C17H21N)
A waxy solid (0.39 g, 24 %); \([\alpha ]_{D}^{25}\) = +33.4° (c = 1, CH2Cl2); R f = 0.83 (neutral alumina, 10 % EtOAc/light petroleum); IR (film): \(\bar{\nu }\) = 2,965, 2,937, 2,888, 2,834, 1,631, 1,550 cm−1; 1H NMR: δ = 7.76 (dd, J = 1.5, 7.5 Hz, 2H, 2HAr), 7.61 (td, J = 1.5, 7.5, 7.5 Hz, 1H, HAr), 7.48 (td, J = 1.5, 7.5, 7.5 Hz, 2H, 2HAr), 5.32 (dd, 1H, J = 1.5, 1.5 Hz, H-7), 3.35 (br s, 1H, H-5), 2.27–2.23 (m, 2H, 2H-8), 2.40–1.96 (m, 2H, Ha-9, H-1), 1.78 (ddd, J = 2.0, 2.0, 12.1 Hz, 1H, H-9), 1.42 (qd, J = 1.5, 3.5 Hz, 3H, CH3-6′), 1.32 (s, 3H, CH3-2), 1.29 (s, 3H, CH3-2) ppm; 13C NMR: δ = 166.6 (C-4), 140.9 (C-6), 135.1 (C), 132.1 (CH), 129.1 (2CH), 128.2 (2CH), 121.3 (CH-7), 59.5 (C-2), 36.5 (CH-5), 33.8 (CH-1), 31.9 (CH3-6′), 29.0 (CH2), 27.7 (CH3-2′), 25.8 (CH2), 23.5 (CH3-2′) ppm; GC–MS (EI): R t = 17.65 min, m/z (%) = 239 (35), 198 (20); HR-ESI–MS: [M + H]+ found 240.17740, C17H22N requires 240.17522.
Kinetic study
A mixture of 10 cm3 PhCN (97 mmol) and 2 cm3 18 M H2SO4 (37 mmol) was stirred in a round-bottom flask fitted with a condenser and a drying tube at 0 °C. To the reaction mixture was added drop-wise a solution of 0.94 g (−)-β-pinene (1.1 cm3, 6.9 mmol) in 8 cm3 benzene via the condenser. The reaction mixture was stirred at 0 °C for 30 min; an aliquot (0.5 cm3) of the reaction mixture was taken and transferred into a sample vial that contained 1 cm3 water. While stirring, the mixture in the sample vial was basified with 4 M NaOH until pH > 10. The aqueous mixture was extracted with CHCl3 (by adding 1–2 cm3 of CHCl3 while stirring). After the two layers were separated, a small volume of the CHCl3 was removed and set aside and kept at 0 °C for the GC/MS analysis. The process was repeated at the 30-min mark at 0 °C. After the reaction had been allowed to stir at room temperature, the process was repeated at an hour interval for another five aliquots (see the time intervals below).
After the last aliquot (at 5 h and 30 min mark), the reaction mixture was left to stir at room temperature for another 18 h and 30 min. Upon completion, the reaction was worked up following the general procedure. A sample of the crude product was prepared as part of the GC/MS analysis for the kinetic study.
X-ray crystallographic data
Suitable single crystals of compound 7 were selected under the polarizing microscope (Leica M165Z) and were picked up on a MicroMount (MiTeGen, USA) consisting of a thin polymer tip with a wicking aperture. The X-ray diffraction measurements were carried out on a Bruker kappa-II CCD diffractometer at 150 K by using graphite-monochromated Mo-Kα radiation (λ = 0.710723 Å). The single crystals, mounted on the goniometer using cryo loops for intensity measurements, were coated with paraffin oil and then quickly transferred to the cold stream using an Oxford Cryo stream attachment. Symmetry-related absorption corrections using the program SADABS [37] were applied, and the data were corrected for Lorentz and polarisation effects using Bruker APEX2 software [38]. All structures were solved by direct methods, and the full-matrix least-square refinements were carried out using SHELXL [39]. The non-hydrogen atoms were refined anisotropically. The molecular graphic was generated using Mercury [40]. Key crystallographic data and refinement details are presented in Tables 1 and 2. Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 960059. The data can be obtained free of charge via http://www.ccdc.cam.ac.uk, by e-mailing data_request@ccdc.cam.ac.uk or by contacting The Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223/336-033, Tel.: (+44) 1223/336-408.
References
Krimen LI, Cota DJ (1969) Org React 17:213
Bishop R (1991) Ritter-type reactions. In: Trost BM, Fleming I (eds), Comprehensive organic synthesis: selectivity, strategy and efficiency in modern organic chemistry, vol 6. In: Winterfeld E (ed), Heteroatom manipulation. Chapter 6.1.9. Comprehensive organic synthesis. Pergamon, Oxford, p 261. 59299_v06_01_09a.pdf and 59299_v06_01_09b.pdf. Available via http://www.knovel.com
Guérinot A, Reymond SJ, Cossy J (2012) Eur J Org Chem 77:19
Ganesh V, Sureshkumar D, Chandrasekaran S (2011) Angew Chem Int Ed 50:5878
Justribo V, Pellegrinet SC, Colombo MI (2007) J Org Chem 72:3702
Martınez AG, Vilar ET, Moreno-Jiminez F, Garcıa AMA (2006) Tetrahedron Asymmetry 17:2970
Reddy UC, Raju BR, Kumar EKP, Saikia AK (2008) J Org Chem 73:1628
Yadav JS, Reddy BVS, Aravind S (2008) Tetrahedron 64:3025
Reddy BVS, Ramesh K, Ganesh AV (2011) Tetrahedron Lett 52:495
Lamenec TR, Bender DR, DeMarco AM, Karady S, Reamer RA, Weinstock LM (1988) J Org Chem 53:1768
Bishop R, Hawkins SC, Ibaba IC (1988) J Org Chem 53:427
Bong ICC, Ung AT, Craig DC, Scudder ML, Bishop R (1989) Aust J Chem 42:1929
Pich KC, Bishop R, Graig DC, Scudder ML (1994) Aust J Chem 47:837
Lin Q, Ball GE, Bishop R (1997) Tetrahedron 53:10899
Lin Q, Djaidi D, Bishop R, Craig DC, Scudder ML (1998) Aust J Chem 51:799
Djaidi D, Bishop R, Craig DC, Scudder ML (1996) J Chem Soc Perkin Trans 1:1859
Hemtasin C, Ung AT, Kanokmedhakul S, Kanokmedhakul K, Bishop R, Satraruji T, Bishop D (2012) Monatsh Chem 143:955
Borschberg HJ (1992) In: Atta-ur-Rahman (ed) Aristotelia alkaloids: synthetic and biomimetic studies, vol 11. Elsevier, Amsterdam, p 277
Borschberg HJ (1996) In: Cordell GA (ed) The alkaloids: chemistry and pharmacology. Chapt 3. Academic Press, Inc, San Diego
Brick IR, Hai MA, Preston NW (1985) Tetrahedron 41:3127
Caram JA, Martins ME, Marschoff CM, Cafferta LFR, Gross EG (1984) Z Naturforsch B 39:972
Samaniego WN, Baldessari A, Ponce MA, Rodriguez JB, Gros EG, Caram JA, Marschoff CM (1994) Tetrahedron Lett 35:6967
Rodriguez JB, Gros EG, Caram JA, Marschoff CM (1995) Tetrahedron Lett 36:7825
Caram JA, Rivero BE, Piro OE, Gros EG, Marschoff CM (1990) Can J Chem 68:334
Viturro HR, Rivero BE, Piro OE, Poro E, Caram JA, Martins ME, Marschoff CM (1987) Can J Chem 65:2000
Yarovaya OI, Korchagina DV, Rybalova TV (2004) Russ J Org Chem Engl Transl 40:1593
Delpech B, Khuong-Huu Q (1978) J Org Chem 43:4898
Pancrazi A, Kabore I, Delpech B, Khuong-Huu Q (1998) Tetrahedron Lett 39:3729
Christol HS, Solladié G (1966) Bull Soc Chim Fr 1299–1307
Parris CL, Christenson RM (1960) J Org Chem 25:331
Carman RM, Greenfield KL (1984) Aust J Chem 37:1785
Forster MO (1898) J Chem Soc 73:386
Forster MO, Hart-Smith J (1900) J Chem Soc 77:1152
Zhou Y, Dong J, Zhang F, Gong Y (2011) J Org Chem 76:588
Hanzawa Y, Kasashima Y, Tomono K, Mino T, Sakamoto M, Fujita T (2012) J Oleo Sci 61:393
Roth HD, Weng H, Zhoua D, Lakkarajub PS (1997) Acta Chem Scand 997:626
Bruker (2001) SADABS. Bruker AXS Inc, Madison
Bruker (2007) APEX2 and SAINT. Bruker AXS Inc, Madison
Sheldrick GM (2008) Acta Cryst A 64:112
Macrae CF, Bruno IJ, Chisholm JA, Edgington PR, McCabe P, Pidcock E, Rodriguez-Monge L, Taylor R, van de Streek J, Wood PA (2008) J Appl Cryst 41:466
Acknowledgments
We are grateful to the Faculty of Science, UTS, for providing financial support for this project. We thank Dr. Ronald Shimmon and Dr. Linda Xiao for providing NMR and GC/MS trainings to Steven Williams and Alexander Angeloski.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ung, A.T., Williams, S.G., Angeloski, A. et al. Formation of 3-azabicyclo[3.3.1]non-3-enes: imino amides vs. imino alkenes. Monatsh Chem 145, 983–992 (2014). https://doi.org/10.1007/s00706-014-1185-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-014-1185-x