
Abstract
A simple route for the synthesis of new polyfluorinated 4-thiazolidinones and 1,4-bis(4-thiazolidinone)phenylenes was achieved from one-pot three-component reaction of fluorinated aniline, mono- and dialdehyde and thioglycolic acid followed by fluorination with trifluoroacetamide and 4-fluorobenzaldehyde. Also, some new fluorinated α-aminophosphonates, 1,4-bis(α-aminophosphonate)phenylenes and their corresponding acids were synthesized via one-pot three-component reaction of fluorinated aniline, mono- and dialdehyde and diethyl phosphite followed by fluorination with trifluoroacetic anhydride. All the synthesized compounds were screened for their antifungal activity against Alternaria alternate and Fusarium oxysporium. Also, their effects on the enzymatic effect of cellobiase produced from Alternaria alternate and Fusarium oxysporium were estimated. Compounds that have trifluoromethyl groups recorded good inhibition against the fungal strains and also enhanced the enzymatic effect of cellobiase.
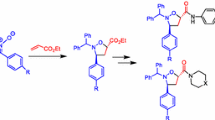
Graphical abstract

.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Fluorine occupies a unique place among all elements of the periodic classification because of its high electronegativity and its specific properties. Thus, organofluorine chemistry is of great importance because of this singular nature of the fluorine atom combined with the unique physical and chemical properties that fluorine imparts to compounds that contain it. The strong electronic contribution and negligible steric demands of fluorine present interesting and unusual properties. Indeed, the specific physicochemical properties of fluorinated organic compounds are of huge interest in a wide range of applications [1–5]. Consequently, organofluorine chemistry has been steadily growing and today possesses a distinctive role in highly diverse technological developments (fluoropolymers, pharmaceutical and agrochemical products, materials science, etc.) [6, 7]. Therefore, the development of new methods to introduce fluorine into molecules is of great interest to organic and medicinal chemists.
4-Thiazolidinones are key structures in numerous compounds of therapeutic importance as antimicrobial, plant protection, anticancer and anti-AIDS activities [8–15]. On the other hand, α-aminophosphonate derivatives are structural mimics of α-amino acids, and some of these compounds exhibit very high potency in inhibiting the enzymes that are involved in the metabolism of the corresponding amino acids. These compounds have already been found to act as antibacterial agents, neuroactive compounds, anticancer drugs and pesticides, with some of them already being commercialized [16–22].
Keeping in view the diverse biological activities associated with organofluorine compounds, 4-thiazolidinones and α-aminophosphonates, it was intended to construct novel fluorinated 4-thiazolidinones and α-aminophosphonates hoping to achieve additive effects toward their biological activities. As an extension of our research work on the synthesis of bioactive heterocycles [23–27], we wish to report herein the synthesis of some new fluorinated 4-thiazolidinines and α-aminophosphonates and evaluation of their fungicidal properties and enzymatic effect on cellobiase.
Results and discussion
Chemistry
The most convenient method for the synthesis of 2,3-disubstituted-4-thiazolidinones is the one-pot three-component reaction of a primary amine, carbonyl compound and thioglycolic acid. The different reaction conditions, such as long-term heating with a dehydrant, using an acylation agent or microwave-assistant organic synthesis, were described [28, 29]. Based on the above-mentioned approach, we have synthesized 2,3-bis(fluorinated substituted phenyl)thiazolidin-4-one (1a,b) from the reaction of fluorinated substituted aniline, fluorinated substituted benzaldehyde and thioglycolic acid in dry dioxane in the presence of anhydrous zinc chloride (Scheme 1).

Compounds 1a,b contain an active methylene group in position C-5 of the core ring, which opens wide opportunities for their modification, taking into consideration the critical influence of the presence and the nature of the C-5 position moieties on biological activity. The polyfluorinated 4-thiazolidione derivatives 2a,b and 3a,b have been achieved via fluorination of compounds 1a,b with fluorine reagents such as trifluoroacetamide and 4-fluorobenzaldehyde. Thus, fusion of compounds 1 a,b with trifluoroacetamide in the presence of glacial acetic acid afforded 2,3-bis(fluorinated substituted phenyl)-5-(trifluoroacetyl)thiazolidin-4-ones (2a,b), while their condensation with 4-fluorobenzaldehyde in ethanolic sodium ethoxide under Knoevenagel reaction condition gave 5-(4-fluorobenzylidene)-2,3-bis(fluorinated substituted phenyl)thiazolidin-4-ones (3a,b) (Scheme 1).
The main aim of this work is the combination of many fluorinated rings together in a single molecular structure hoping to achieve additive biocidal effects toward their antifungal activity, where the fluorine atoms leads to a strong polarization and changes various physical and biological properties of the molecules. Thus, one-pot three-component reaction of 4-fluoroaniline, 1,4-terephthalaldehyde and thioglycolic acid in dry dioxane in the presence of anhydrous zinc chloride as a catalyst under reflux afforded 2,2′-(1,4-phenylene)bis[3-(4-fluorophenyl)thiazolidin-4-one] (4) (Scheme 2). Furthermore, the latter bis-thiazolidinone 4 reacted with trifluoroacetamide and 4-fluorobenzaldehyde under the same previously described conditions to yield the interesting 2,2′-(1,4-phenylene)bis[3-(4-fluorophenyl)-5-(2,2,2-trifluoroacetyl)thiazolidin-4-one] (5) and 2,2′-(1,4-phenylene)bis[5-(4-fluorobezylidene)-3-(4-fluorophenyl)thiazolidin-4-one] (6), respectively (Scheme 2). The structures of the new thiazolidinone derivatives 1–6 were assigned on the basis of their analytical and spectral data. The characteristic C=O bands of the thiazolidinone rings appeared in the region 1,656–1,696 cm−1 in the IR spectra of the thiazolidinones 1–6, while C=O of the trifluoroacetyl moiety in 2a,b and 5 appeared at 1,729, 1,730 and 1,731 cm−1, respectively. Furthermore, the compounds 1a,b and 4 display doublets at 3.85–3.95 ppm because of the HA and HB systems in their 1H NMR spectra. In the compounds 2a,b, 3a,b, 5 and 6. These HA and HB systems were absent, confirming that trifluoroacetylation and condensation reactions had taken place on the 5-CH2 groups. Regarding compounds 1–6, their 1H-NMR spectra showed singlets at 5.92–6.03 ppm because of a proton of 2-CH. Also, the 13C-NMR spectra recorded the characteristic C=O bands of the thiazolidinone rings at 170.6–171.4 ppm, while C=O of the trifluoroacetyl moiety in 2a,b and 5 appeared at 195.0, 193.5 and 195.1 ppm, respectively.

Introduction of the fluorine-containing group in a biomolecule may change the ‘normal’ route of its interaction with a biological target or the direction of binding. Therefore, such an approach is very popular for the construction of new biologically active molecules. However, in contrast to the rather well-developed α-aminophosphonic acid area, only limited representatives of fluorine containing α-aminophosphonates are currently known [30–38]. Thus, the synthesis of new fluorinated α-amino-phosphonates as potential drug candidates is of current interest. One-pot three-component reaction of fluorinated substituted anilines, fluorinated substituted benzaldehydes and diethyl phosphite in the presence of BF3.Et2O as a catalyst at 70 °C under Kabachink-Fields reaction condition afforded the corresponding diethyl[(fluorinated substituted phenylamino) (fluorinated substituted phenyl)methyl]phosphonates (7a,b) in high yield [37, 38] (Scheme 3). The fluorination of the α-aminophosphonates 7a,b with trifluoroacetic anhydride in the presence of anhydrous sodium acetate gave [2,2,2-trifluoro-N-(fluorinated substituted phenyl)acetamido] [fluorinated substituted phenyl]methyl phosphonic acids (8a,b) and not the diesters 9a,b (Scheme 3). This reaction underwent trifluoroacetylation of the NH group of compounds 7a,b followed by hydrolysis of the diester because of the presence of the formed trifluoroacetic acid. Similarly, applying the Kabachnik-Fields reaction to 4-fluoroaniline, 1,4-terephthalaldehyde and diethyl phosphite in the presence of BF3.Et2O as a catalyst at 70 °C afforded diethyl 1,4-phenylene-bis[N-(4-fluorophenyl)aminomethylphosphonate] (10) (Scheme 4). The latter bis-aminophosphonate also underwent trifluoroacetylation followed by acid hydrolysis to give the novel interesting 1,4-phenylene-bis[N-(2,2,2-trifluoroacetyl)-N-(4-fluorophenyl)aminomethylphosphonic acid] (11) (Scheme 4). The chemical structures of α-aminophosphonic acid derivatives 7a,b, 8a,b, 10 and 11 were confirmed by elemental analysis, IR, 1H-, 13C- and 31P-NMR spectra. Compounds 7a,b and 10 exhibited characteristic IR stretching frequencies in the region 3,339–3,100, 1,281–1,238 and 1,013–1,092 cm−1 for NH, P=O and P–O–C functions, respectively. The P–CH protons of 7a,b and 10 resonated as doublets at δ 4.69, 4.12 and 5.06 ppm (J PCH = 23.9, 15.6 and 17.2 Hz), respectively, because of their coupling with phosphorus. The ethyl groups of the diethyl phosphonate moieties resonated as two distinct triplets and quartets at δ 1.19–1.28 and 3.69–4.20 ppm [38]. Also, the P–CH carbon atom shift signals for 7b and 10 appeared as doublets at δ 44.0 and 43.3 ppm (J PCH = 143.8 and 150 Hz), respectively, in their 13C-NMR spectra, while the phosphorus atoms were resonated in the 31P-NMR spectrum as singlets at δ 22.3 and 20.5 ppm, respectively. Similarly, the spectroscopic data of 8a,b and 11 were in complete agreement with the proposed structures. The appearance of OH, C=O and P=O was at 3,200–3,365, 1,709–1,731 and 1,220–1,257 cm−1, respectively, in their IR spectra. Also, in their 1H-NMR spectra, the P–CH protons appeared as doublets at δ 3.65, 4.00 and 4.61 (J PCH = 16.8, 19.2, and 19.2 Hz) for 8a,b and 11, respectively. Also, the 13C-NMR spectra were supported for the proposed structures, which exhibited the resonances of P–CH and C=O at the region δ 51.5, 50.6, 48.5 and 193, 195, 190.3 ppm, respectively. The 31P-NMR spectra of 8b and 11 recorded singlets for them at δ 14.4 and 15.3 ppm, respectively. Although the compounds 10 and 11 have two chiral centers, they exist in only one diastereomeric form because of the presence of one peak for each carbon and phosphorus of the P–CH moiety in their NMR spectra [39].


Biological evaluation
In vitro antifungal activity
The newly synthesized compounds were evaluated for their in vitro antifungal activity against two fungal strains, Alternaria alternate and Fusarium oxysporium, by the poisoned food method [40, 41]. Fluconazole was used as the reference drug, and the results were recorded as the percentage of growth inhibition (Table 1). The results revealed that, in general, all the tested compounds possessed good antifungal activity. On the basis of percentage of growth inhibition against the tested fungal strains, it seems that replacement of the active methylene group in compounds 1a,b and 4 with trifluoroacetyl and 4-fluorobenzylidene exhibited a noticeable increase in the antifungal activity. Also, trifluoroacetylation of the NH groups of compounds 7a,b and 10 and transformation of ethoxy groups into hydroxyl caused improvement the antifungal activity.
The effect of the synthesized compounds on the enzymatic effect on cellobiase
The effect of synthesized compounds on cellobiase activity produced by Alternaria alternate and Fusarium oxysporium was estimated [42, 43]. The obtained results were recorded in Table 2. Generally, at lower concentration, the enzymatic effect of the tested compounds increased while the higher concentration of the synthesized compounds inhibited that enzymatic effect. Only compounds 2a,b, 5, 8b and 11 exhibited highly enzymatic effects at low concentration because of the hydrophobic character of CF3 groups that essentially improves the pharmacokinetic profiles of potential drugs.
Conclusions
In this article, some new fluorinated 4-thiazolidinone and α-aminophosphonic acid derivatives were synthesized. All the synthesized compounds were screened for their antifungal activity and the enzymatic effect on cellobiase. Of the tested compounds, 2a,b, 5 and 11 exhibited good antifungal activity against the tested fungi and also enhanced the enzymatic effect of cellobiase. However, none of the newly synthesized compounds was found to be superior compared to the used references.
Experimental
Melting points of the products were determined on Stuart SMP3. A Perkins-Elmer model RXI-FT-IR system 55529 was used for recording the IR spectra of the prepared compounds using CDCl3 and D2O solvents. 1H-, 13C- and 31P-NMR spectra were recorded on a Bruker 600 MHz spectrometer operating at 600, 150 and 242 MHz, respectively. The chemical shifts are reported in ppm with respect to the references and were stated relative to tetramethylsilane (TMS) for 1H- and 13C-NMR and to 85 % phosphoric acid for 31P-NMR. Mass spectra were recorded on a gas chromatographic DI analysis Shimadzu instrument Q-2010 Plus at 70 eV. The purity of the synthesized compounds was checked by thin layer chromatography (TLC) and elemental analysis. The biological activities were measured at the Department of Biochemistry, Faculty of Science, King Abdulaziz University.
2,3-Bis(fluorinated substituted phenyl)thiazolidin-4-one (1a,b)
A mixture of fluorinated substituted aniline (2.5 mmol) and fluorinated substituted benzaldehyde (2.5 mmol) was heated in dry dioxane (15 cm3) under reflux for 10 min, followed by addition of a solution of thioglycolic acid (3 mmol) in dry dioxane (5 cm3) and 1.5 g freshly fused zinc chloride. The reaction mixture was heated under reflux for an additional 6 h. The mixture was cooled then neutralized with aqueous NaHCO3 (10 %, 30 cm3). The formed solid was filtered off, washed with water three times and crystallized from ethanol to give the pale yellow crystals.
2,3-Bis(4-fluorophenyl)thiazolidin-4-one (1a, C15H11F2NOS)
Yield 0.45 g (62 %). M.p.: 98–100 °C; IR (CDCl3): \( \bar{v} \) = 1,656 (C=O), 1,239 (C–F) cm−1; 1H NMR (600 MHz, CDCl3): δ = 3.88, 3.91 (dd, 2H, 5-CH2, AB system), 6.02 (s, 1H, 2-CH), 6.96–6.99 (m, 4H, Ar–H), 7.02–7.09 (m, 2H, Ar–H), 7.27–7.29 (m, 2H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 33.4 (5-CH2), 65.1 (2-CH), 116.0, 116.3, 127.8, 127.9, 133.0, 134.7, 160.4, 162.0, 171.0 (C=O) ppm; anal. calcd. for C15H11F2NOS (291.32): C, 61.84; H, 3.81; N, 4.81; S, 11.01. Found: C, 61.66; H, 3.59; N, 4.71; S, 10.70.
2-(2-Chloro-6-fluorophenyl)-3-(3,4-difluorophenyl)thiazolidin-4-one (1b, C15H9ClF3NOS)
Yield 0.51 g (60 %). M.p.: 70–72 °C; IR (CDCl3): \( \bar{v} \) = 1,660 (C=O), 1,203 (C–F) cm−1; 1H NMR (600 MHz, CDCl3): δ = 3.85, 3.88 (dd, 2H, 5-CH2, AB system), 6.00 (s, 1H, 2-CH), 6.99–7.10 (m, 6H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 33.5 (5-CH2), 58.5 (2-CH), 114.8, 115.6, 117.5, 121.1, 125.6, 127.6, 130.7, 130.9, 133.4, 160.6, 161.0, 162.3, 170.6 (C=O) ppm; MS (70 eV): m/z = 344 (M+); anal. calcd. for C15H9ClF3NOS (343.75): C, 52.41; H, 2.64; N, 4.07; S, 9.33. Found: C, 52.19; H, 2.53; N, 3.82; S, 9.11.
5-(2,2,2-Trifluoroacetyl)-2,3-bis(fluorinated substituted phenyl)thiazolidin-4-one (2a,b)
A mixture of 2,3-bis(fluorinated substituted phenyl)thiazolidin-4-one (1a,b) (2.5 mmol) and 0.28 g trifluoroacetamide (2.5 mmol) in the presence of few drops of glacial acetic acid was fused at 120 °C for 1 h. The cooled reaction mixture was poured onto 100 g of crushed ice. The formed solid was filtered off, washed with water three times then crystallized from aqueous ethanol to give the yellowish crystals.
5-(2,2,2-Trifluoroacetyl)-2,3-bis(4-fluorophenyl)thiazolidin-4-one (2a, C17H10F5NO2S)
Yield 0.78 g (81 %). M.p.: 122–124 °C; IR (CDCl3): \( \bar{v} \) = 1,729 (C=O), 1,669 (C=O), 1,222 (C–F) cm−1; 1H NMR (600 MHz, CDCl3): δ = 4.20 (s, 1H, 5-CH), 6.03 (s, 1H, 2-CH), 6.95–6.99 (m, 4H, Ar–H), 7.00–7.10 (m, 2H, Ar–H), 7.27–7.30 (m, 2H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 45.3 (5-CH), 65.1 (2-CH), 113.9 (CF3), 116.0, 116.3, 127.8, 127.9, 133.0, 134.7, 160.4, 162.0, 171.0 (C=O), 195.0 (C=O) ppm. MS (70 eV): m/z = 387 (M+); anal. calcd. for C17H10F5NO2S (387.33): C, 52.72; H, 2.60; N, 3.62; S, 8.28. Found: C, 52.47; H, 2.39; N, 3.36; S, 8.02.
2-(2-Chloro-6-fluorophenyl)-3-(3,4-difluorophenyl)-5-(2,2,2-trifluoro acetyl)thiazolidin-4-one (2b, C17H8ClF6NO2S)
Yield 0.6 g (55 %). M.p.: 101–103 °C; IR (CDCl3): \( \bar{v} \) = 1,730 (C=O), 1,696 (C=O), 1,220 (C–F) cm−1; 1H NMR (600 MHz, CDCl3): δ = 4.10 (s, 1H, 5-CH), 6.02 (s, 1H, 2-CH), 6.98–7.10 (m, 6H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 44.5 (5-CH), 64.7 (2-CH), 110.0 (CF3), 114.9, 115.7, 117.5, 121.1, 125.6, 127.6, 130.8, 130.9, 133.4, 160.6, 161.0, 162.3, 170.6 (C=O), 193.5 (C=O) ppm; anal. calcd. for C17H8ClF6NO2S (439.75): C, 46.43; H, 1.83; N, 3.19; S, 7.29. Found: C, 46.26; H, 1.54; N, 2.87; S, 7.08.
5-(4-Fluorobenzylidene)-2,3-bis(fluorinated substituted phenyl)thiazolidin-4-one (3a,b)
A mixture of 2,3-bis(fluorinated substituted phenyl)thiazolidin-4-one (1a,b) (2.5 mmol) and 0.31 g 4-fluorobenzaldehyde (2.5 mmol) in ethanolic sodium ethoxide (0.2 g Na in 15 cm3 of ethanol) was heated under reflux for 10 h. The cooled reaction mixture was poured onto ice cold water, then acidified with diluted acetic acid. The formed solid was filtered off and crystallized from ethanol to give the yellowish products.
5-(4-Fluorobenzylidene)-2,3-bis(4-fluorophenyl)thiazolidin-4-one (3a, C22H14F3NOS)
Yield 0.73 g (74 %). M.p.: 242–244 °C; IR (CDCl3): \( \bar{v} \) = 1,682 (C=O), 1,221 (C–F) cm−1; 1H NMR (600 MHz, CDCl3): δ = 5.95 (s, 1H, 2-CH), 6.90–7.30 (m, 13H, Ar–H and =CH) ppm; 13C NMR (150 MHz, CDCl3): δ = 65.1 (2-CH), 116.0, 116.1, 116.3, 127.8, 127.9, 129.1, 129.2, 133.0, 133.1, 134.7, 162.0, 162.1, 163.7, 171.0 (C=O) ppm; anal. calcd. for C22H14F3NOS (397.41): C, 66.49; H, 3.55; N, 3.52; S, 8.07. Found: C, 66.21; H, 3.38; N, 3.34; S, 7.94.
2-(2-Chloro-6-fluorophenyl)-3-(3,4-difluorophenyl)-5-(4-fluorobenzylidene) thiazolidin-4-one (3b, C22H12ClF4NOS)
Yield 0.84 g (75 %). M.p.: 264–265 °C; IR (CDCl3): \( \bar{v} \) = 1,667 (C=O), 1,600 (C=C), 1,231 (C–F) cm−1; 1H NMR (600 MHz, CDCl3): δ = 5.92 (s, 1H, 2-CH), 6.78–7.23 (m, 11H, Ar–H and =CH) ppm; 13C NMR (150 MHz, CDCl3): δ = 58.6 (2-CH), 115.4, 115.5, 115.6, 117.5, 121.2, 121.3, 124.3, 124.4, 125.5, 125.6, 127.6, 130.7, 130.8, 130.9, 133.3, 133.4, 160.5, 161.0, 162.0, 162.2, 171.40 (C=O) ppm; MS (70 eV): m/z = 450 (M+); anal. calcd. for C22H12ClF4NOS (449.84): C, 58.74; H, 2.69; N, 3.11; S, 7.13. Found: C, 58.42; H, 2.42; N, 3.01; S, 6.92.
2,2′-(1,4-Phenylene)bis[3-(4-fluorophenyl)thiazolidin-4-one] (4, C24H18F2N2O2S2)
A mixture of 0.22 g 4-fluoroaniline (2 mmol) and 0.134 g 1,4-terephthalaldehyde (1 mmol) was heated in dry dioxane (15 cm3) under reflux for 15 min, followed by addition of a solution of thioglycolic acid (3 mmol) in dry dioxane (5 cm3) and 1.5 g freshly fused zinc chloride. The reaction mixture was heated under reflux for an additional 10 h. The mixture was cooled then neutralized with aqueous NaHCO3 (10 %, 30 cm3). The formed solid was filtered off, washed with water three times and crystallized from tetrahydrofuran to give pale yellow crystals. Yield 0.41 g (88 %). M.p.: 247–249 °C. IR (CDCl3): \( \bar{v} \) = 1,665 (C=O), 1,227 (C–F); 1H NMR (600 MHz, CDCl3): δ = 3.89, 3.95 (dd, 4H, 5-CH2, AA′BB′ system), 5.95 (s, 2H, 2-CH), 6.90–6.93 (m, 4H, Ar–H), 6.94–6.98 (m, 4H, Ar–H), 7.21–7.22 (m, 4H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 33.3 (5-CH2), 65.2 (2-CH), 116.1, 127.8, 127.9, 132.9, 140.0, 162.0, 170.9 (2 C=O) ppm; MS (70 eV): m/z = 468 (M+); anal. calcd. for C24H18F2N2O2S2 (468.55): C, 61.52; H, 3.87; N, 5.98; S, 13.69. Found: C, 61.32; H, 3.64; N, 5.77; S, 13.48.
2,2′-(1,4-Phenylene)bis[3-(4-fluorophenyl)-5-(2,2,2-trifluoroacetyl) thiazoldin-4-one] (5, C28H16F8N2O4S2)
A mixture of 0.468 g 2,2′-(1,4-phenylene)bis[3-(4-fluorophenyl) thiazolidin-4-one) (4) (1 mmol) and 0.226 g trifluoroacetamide (2 mmol) in the presence of a few drops of glacial acetic acid was fused at 250 °C for 2 h. The cooled reaction mixture was poured onto 100 g of crushed ice. The formed solid was filtered off, washed with water three times then crystallized from methanol to give white crystals. Yield 0.48 g (74 %). M.p.: 202–204 °C. IR (CDCl3): \( \bar{v} \) = 1,731 (C=O), 1,667 (C=O), 1,222 (C–F) cm−1; 1H NMR (600 MHz, CDCl3): δ = 4.23 (s, 2H, 5-CH), 5.96 (s, 2H, 2-CH), 6.90–6.99 (m, 8H, Ar–H), 7.21–7.23 (m, 4H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 48.1 (5-CH), 65.2 (2-CH), 114.4 (CF3), 116.2, 127.7, 127.9, 133.0, 140.0, 162.0, 171.0 (C=O), 195.1 (C=O) ppm; anal. calcd. for C28H16F8N2O4S2 (660.57): C, 50.91; H, 2.44; N, 4.24; S, 9.71. Found: C, 50.66; H, 2.24; N, 3.97; S, 9.53.
2,2′-(1,4-Phenylene)bis[5-(4-fluorobenzylidene)-3-(4-fluorophenyl) thiazolidin-4-one] (6, C38H24F4N2O2S2)
A mixture of 0.468 g 2,2′-(1,4-phenylene)bis[3-(4-fluorophenyl) thiazolidin-4-one) (4) (1 mmol) and 0.248 g 4-fluorobenzaldehyde (2 mmol) in ethanolic sodium ethoxide (0.2 g Na in 30 cm3 of ethanol) was heated under reflux for 10 h. The cooled reaction mixture was poured onto ice cold water then acidified with diluted acetic acid. The formed solid was filtered off and crystallized from ethanol to give yellow crystals. Yield 0.53 g (79 %). M.p.: 263–265 °C; IR (CDCl3): \( \bar{v} \) = 1,670 (C=O), 1,597 (C=C), 1,227 (C–F) cm−1; 1H NMR (600 MHz, CDCl3): δ = 5.95 (s, 2H, 2-CH), 6.92–6.97 (m, 8H, Ar–H), 7.10–7.30 (m, 14H, Ar–H and =CH) ppm; 13C NMR (150 MHz, CDCl3): δ = 65.1 (2-CH), 116.1, 116.3, 127.83, 127.9, 128.1, 129.2, 133.0, 133.1, 134.7, 162.0, 163.7, 171.0 (2 C=O) ppm; anal. calcd. for C38H24F4N2O2S2 (680.75): C, 67.05; H, 3.55; N, 4.12; S, 9.42. Found: C, 66.83; H, 3.28; N, 3.94; S, 9.20.
Diethyl [(fluorinated substituted phenylamino)(fluorinated substituted phenyl)methyl]phosphonate (7a,b)
A mixture of fluorinated substituted anilines (1 mmol), fluorinated substituted benzaldehyde (1 mmol) and diethyl phosphite (1.5 mmol) in the presence of BF3.Et2O (0.1 cm3) was heated under reflux at 70 °C for 2 h. The mixture was concentrated to dryness under reduced pressure, and the residue was taken up with ethyl acetate. The organic layer was washed with anhydrous sodium sulfate, and the solvent was removed under vacuum to give the crude product, which was recrystallized from ethanol to give white crystals.
Diethyl [(4-fluorophenylamino)(4-fluorophenyl)methyl]phosphonate (7a, C17H20F2NO3P)
Yield 0.34 g (96 %). M.p.: 80–82 °C (Lit [37]. 80–81 °C); IR (CDCl3): \( \bar{v} \) = 3,331 (NH), 1,273 (P=O), 1,231 (C–F), 1,014 (P–O–C) cm−1; 1H NMR (600 MHz, CDCl3): 1.19 (t, 3H, J = 7.14 Hz, CH3), 1.28 (t, 3H, J = 7.14 Hz, CH3), 3.69–4.20 (m, 4H, CH2), 4.69 (d, 1H, J = 23.9 Hz, CH–P), 6.49–6.60 (m, 2H, Ar–H), 6.78–6.86 (m, 2H, Ar–H), 6.99–7.49 (m, 5H, Ar–H and NH) [38]; anal. calcd. for C17H20F2NO3P (355.32): C, 57.47; H, 5.67; N, 3.94. Found: C, 57.12; H, 5.55; N, 3.71.
Diethyl [(2-chloro-6-fluorophenyl)(3,4-difluorophenylamino)methyl] phosphonate (7b, C17H18ClF3NO3P)
Yield 0.33 g (83 %). M.p.: 90–92 °C; IR (CDCl3): \( \bar{v} \) = 3,339 (NH), 1,281 (P=O), 1,225 (C–F), 1,013 (P–O–C) cm−1; 1H NMR (600 MHz, CDCl3): δ = 1.25 (t, 6H, J = 6 Hz, CH3), 3.90 (q, 4H, J = 6 Hz, CH2), 4.12 (d, 1H, J = 15.6 Hz, CH–P), 6.78–7.20 (m, 6H, Ar–H), 8.00 (br, 1H, NH) ppm; 13C NMR (150 MHz, CDCl3): δ = 13.8 (CH3), 44.0 (d, J = 143.8 Hz, CH–P), 62.4 (CH2), 114.9, 115.6, 117.5, 121.1, 125.5, 127.6, 130.7, 130.8, 133.4, 160.6, 161.0, 162.3 ppm; 31P NMR (242 MHz, CDCl3): δ = 22.3 ppm. MS (70 eV): m/z = 407 (M+); anal. calcd. for C17H18ClF3NO3P (407.75): C, 50.08; H, 4.45; N, 3.44. Found: C, 49.82; H, 4.18; N, 3.13.
[2,2,2-Trifluoro-N-(aryl)acetamido][aryl]methylphosphonic acid (8a,b)
A mixture of diethyl α-aminophosphonates 7a,b (1 mmol), 1 g freshly fused sodium acetate and 5 cm3 of trifluoroacetic anhydride was stirred and heated under reflux for 4 h. The cooled reaction mixture was poured onto 100 g of crushed ice. The formed solid was filtered off, washed with water three times then crystallized from ethyl acetate to give white crystals.
[2,2,2-Trifluoro-N-(4-fluorophenyl)acetamido][4-fluorophenyl]methyl phosphonic acid (8a, C15H11F5NO4P)
Yield 0.27 g (69 %). M.p.: 206–208 °C; IR (KBr): \( \bar{v} \) = 3,365 (br, OH), 1,729 (C=O), 1,248 (C–F) 1,220 (P=O) cm−1; 1H NMR (600 MHz, D2O): δ = 3.56 (d, 1H, J = 16.8 Hz, CH–P), 4.75 (br, 2H, OH), 6.96–6.99 (m, 4H, Ar–H), 7.00–7.08 (m, 2H, Ar–H), 7.26–7.29 (m, 2H, Ar–H) ppm; 13C NMR (150 MHz, D2O): δ = 51.5 (d, J = 149 Hz, CH–P), 113.9 (CF3), 116.0, 116.3, 127.8, 127.9, 133.1, 134.7, 160.4, 162.0, 193.0 (C=O) ppm; MS (70 eV): m/z = 395 (M+); anal. calcd. For C15H11F5NO4P (395.23): C, 45.59; H, 2.81; N, 3.54. Found: C, 45.21; H, 2.63; N, 3.37.
{(2-Chloro-6-fluorophenyl)[(3,4-difluorophenyl)(2,2,2-trifluoroacetyl)amino]methyl}phosphonic acid (8b, C15H9ClF6NO4P)
Yield 0.32 g (72 %). M.p.: 232–234 °C; IR (KBr): \( \bar{v} \) = 3,200 (br, OH), 1,709 (C=O), 1,265 (C–F) 1,257 (P=O) cm−1; 1H NMR (600 MHz, D2O): δ = 4.00 (d, 1H, J = 19.2 Hz, CH–P), 6.78–7.19 (m, 6H, Ar–H) ppm; 13C NMR (150 MHz, D2O): δ = 50.6 (d, J = 158.7 Hz, CH–P), 109.1 (CF3), 114.8, 115.6, 117.5, 121.1, 125.6, 127.6, 130.8, 130.9, 133.2, 160.7, 161.0, 162.5, 195.0 (C=O) ppm; 31P NMR (242 MHz, D2O): δ = 14.4 ppm; anal. calcd. for C15H9ClF6NO4P (447.65): C, 40.25; H, 2.03; N, 3.13. Found: C, 40.03; H, 1.86; N, 2.97.
Diethyl 1,4-phenylene bis[N-(4-fluorophenyl)aminomethylphosphonate] (10, C28H36F2N2O6P2)
A mixture of 0.22 g 4-fluoroaniline (2 mmol), 0.134 g 1,4-terephthalaldehyde (1 mmol) and diethyl phosphite (4 mmol,) in the presence of BF3.Et2O (0.15 cm3) was heated under reflux at 70 °C for 2 h. The mixture was concentrated to dryness under reduced pressure, and the residue was taken up with ethyl acetate. The organic layer was washed with anhydrous sodium sulfate, and the solvent was removed under vacuum to give the crude product, which was recrystallized from ethanol to give white crystals. Yield 0.47 g (80 %). M.p.: 158–159 °C; IR (CDCl3): \( \bar{v} \) = 3,100 (NH), 1,238 (C–F), 1,230 (P=O), 1,092 (P–O–C) cm−1; 1H NMR (600 MHz, CDCl3): δ = 1.25 (t, 12H, J = 7.2 Hz, CH3), 3.91–3.96 (m, 8H, CH2), 5.06 (d, 2H, J = 17.2 Hz, CH–P), 6.00 (br, 2H, NH), 6.96–6.99 (m, 8H, Ar–H), 7.00–7.10 (m, 4H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 13.1 (CH3), 43.3 (d, J = 150 Hz, CH–P), 62.3 (CH2), 116.2, 127.7, 127.9, 133.0, 140.0, 162.0 ppm; 31P NMR (242 MHz, CDCl3): δ = 20.5 ppm; anal. calcd. for C28H36F2N2O6P2 (596.55): C, 56.38; H, 6.08; N, 4.70. Found: C, 56.13; H, 5.83; N, 4.39.
1,4-Phenylene bis[N-(2,2,2-trifluoroacetyl)-N-(4-fluorophenyl)amino methylphosphonic acid] (11, C24H18F8N2O8P2)
A mixture of 0.596 g diethyl 1,4-phenylenebis[N-(4-fluorophenyl)aminomethylphosphonate] (9) (1 mmol), 2 g freshly fused sodium acetate and 5 cm3 of trifluoroacetic anhydride was stirred and heated under reflux for 5 h. The cooled reaction mixture was poured onto 100 g of crushed ice. The formed solid was filtered off, washed with water three times then crystallized from ethyl acetate to give white crystals. Yield 0.52 g (77 %). M.p.: 243–245 °C; IR (KBr): \( \bar{v} \) = 3,357 (br, OH), 1,731 (C=O), 1,244 (C–F), 1,230 (P=O) cm−1; 1H NMR (600 MHz, D2O): δ = 4.61 (d, 2H, J = 19.2 Hz, CH–P), 6.96–6.99 (m, 8H, Ar–H), 7.00–7.10 (m, 4H, Ar–H) ppm; 13C NMR (150 MHz, D2O): δ = 48.5 (d, J = 163.5 Hz, CH–P), 110.2 (2 CF3), 116.2, 127.8, 127.9, 133.0, 140.0, 160.3, 190.3 (2 C=O) ppm; 31P NMR (242 MHz, D2O): δ = 15.3 ppm; anal. calcd. for C24H18F8N2O8P2 (676.35): C, 42.62; H, 2.68; N, 4.14. Found: C, 42.38; H, 2.46; N, 3.89.
Biological assay
In vitro antifungal assay
The antifungal activity of newly synthesized compounds was evaluated against the fungal strains Alternaria alternate and Fusarium oxysporium in three replications by the food poison technique [40, 41]. The molds were grown on to potato-dextrose Agar (PDA) medium at 25 °C for 7 days and used as inocula. Then 100 μM3 volume of each compound, having three concentrations of 250, 500 and 1,000 ppm, was reconstituted in dry acetone and poured into a sterile petri plate. The poisoned agar plates were incubated at the center with fungal plugs obtained from the actively growing colony and incubated at 25 °C for 7 days. Dry acetone was used as solvent, and fluconazole was used as the reference drug. The experiments were performed in triplicates. The diameter of the fungal colonies was measured and expressed as percent inhibition determined by applying the following formula: percent of growth inhibition % = (dc − dt)/dc × 100 where dc = average diameter of fungal colony in control plates; dt = average diameter of fungal colony in experimental plates.
The effect of the synthesized compounds on the enzymatic effect on cellobiase
The effect of synthesized compounds on cellobiase activity produced by Alternaria alternate and Fusarium oxysporium was estimated. The procedure for determining cellobiase activity was according to Tolan and Foody [42, 43]. Each compound was dissolved in an appropriate amount of DMF at concentrations of 10, 50 and 100 μg/cm3, then added separately to the assay mixture consisting of 1 cm3 of 15 mM cellobiose solution (prepared in 0.05 M citrate buffer pH 4.8) and 1 cm3 of the diluted enzyme solution at 50 °C for 1 h. The released reducing sugar (glucose) was estimated colorimetrically at 540 nm as an indication for the enzyme activity. The obtained results are recorded in Table 2.
References
Smart BE (2001) J Fluorine Chem 109:3
Dunitz JD (2004) ChemBioChem 5:614
Biffinger JC, Kim HW, Di Magno SG (2004) ChemBioChem 5:622
Filler R, Kobayashi Y, Yagulpolskii LM (1993) Organofluorine Compounds in Medicinal Chemistry and Biomedical Applications. Elsevier, Amsterdam
Hiyama T (2000) Organofluorine compounds: chemistry and properties, Chapter 5. Springer, Berlin, pp 137–182
Dolbier WR Jr (2005) J Fluorine Chem 126:157
Schofield HJ (1999) J Fluorine Chem 100:7
Gürsoy A, İyİköşker T, Terzİoğlu N, OTük G (2005) Turkish J Pharm Sci 2:1
Patel NB, Patel SD (2009) Der Pharma Chem 1:199
Srivastava T, Haq W, Katti SB (2008) Tetrahedron 58:7619
Karali N, Gürsoy A, Kandemirli F, Shvets N, Kaynak FB, Özbey S, Kovalishyn V, Dimoglo A (2007) Bioorg Med Chem 15:5888
Rawal RK, Prabhakar YS, Katti SB, De Clercq E (2005) Bioorg Med Chem 13:6771
Rawal RK, Tripathi R, Katti SB, Pannecouque C, De Clercq E (2007) Bioorg Med Chem 15:1725
Gududuru V, Hurh E, Dalton JT, Miller DD (2005) J Med Chem 48:2584
Gududuru V, Hurh E, Dalton JT, Miller DD (2004) Bioorg Med Chem Lett 14:5289
Kuhkar VP, Hudson HR (eds) (2000) Aminophosphonic and Aminophosphinic Acids. Wiley, Chichester
Kafarski P, Lejczak B (2001) Curr Med Chem 1:301
Berlicki L, Kafarski P (2005) Curr Org Chem 9:1829
Strater N, Lipscomb WN (1995) Biochemistry 34:9200
Umezawa H (1980) Cancer Res 75:115
Taylor A, Daims MA, Lee J, Surgenor T (1982) Curr Eye Res 2:47
Pulido-Cejudo G, Conway B, Proulx P, Brown R, Izaguirre CA (1997) Anti Viral Res 36:167
Makki MST, Abdel-Rahman RM, El-Shahawi MS (2011) Eur J Chem 8:887
Ali TE, Abdel-Rahman RM, Hanafy FI, El-Edfawy SM (2008) Phosphorus, Sulfur Silicon Relat Elem 183:2565
Bakhotmah DA, Abdel-Rahman RM, Makki MST (2011) Int J Chem Res 2:65
Abdel-Rahman RM, Makki MS, Ali TE, Ibrahim MA (2010) Eur J Chem 1:236
Ali TE (2009) Eur J Med Chem 44:4539
Srivastava T, Haq W, Katti SB (2002) Tetrahedron 58:7619
Bolognese A, Correale G, Manfra M, Lavecchia A, Novellino E, Barone V (2004) Org Biomol Chem 2:2809
Haas AM, Haegele G (1996) J Fluorine Chem 78:75
De Medina P, Ingrassia LS, Mulliez ME (2003) J Org Chem 68:8424
Kudzin Z, Majchrzak MW (1989) J Organomet Chem 376:245
Onys’ko P, Sinitsa AD (1990) Zhurn Obshch Khim 60:966
Huang W, Zhang Y, Yuan C (1995) Phosphorus, Sulfur Silicon Relat Elem 107:21
Osipov SN, Artyushin OI, Kolomiets AF, Bruneau C, Picquet M, Dixneuf PH (2001) Eur J Org Chem 20:3891
Odinets IL, Artyushin OI, Lyssenko KA, Shevchenko NE, Petrovskii PV, Nenaidenko VG, Roschenthaler GV (2009) J Fluorine Chem 130:662
Gruss U, Haegele G (1994) Phosphorus, Sulfur Silicon Relat Elem 97:209
Lv XY, Zhang JM, Xing CH, Du WQ, Zhu SZ (2007) Synth Commun 37:743
Failla S, Finocchiaro P, Hagele G, Kalchenko VI (1997) Phosphorus, Sulfur Silicon Relat Elem 128:63
Othana R, Maccari R, Barreca ML, Bruno G, Rotondo A, Rossi A, Chiricosta G, Paola RD, Sautebin L, Cuzzocrea S, Vigorita MG (2005) Bioorg Med Chem 13:4243
Al-Burtmani SKS, Fatope MO, Marwah RG, Onifade AK, Al-Saidi SH (2005) J Ethnopharmaco 96:107
Tolan JS, Foody B (1999) Adv Biochem Eng Biotech 65:41
Chen M, Xia L, Xue P (2007) Int Biodeterior Biodegradation 59:85
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Abdel-Rahman, R.M., Ali, T.E. Synthesis and biological evaluation of some new polyfluorinated 4-thiazolidinone and α-aminophosphonic acid derivatives. Monatsh Chem 144, 1243–1252 (2013). https://doi.org/10.1007/s00706-013-0934-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-013-0934-6