
Abstract
Homopolymer of 3-(cyanomethyl)-1-vinylimidazolium hexafluorophosphate was synthesized and used as a support for Pd nanoparticles. The Pd@poly-CN-PF6 catalyst (2 mol % Pd) selectively catalyzes the reductive homocoupling reaction of aryl iodides and bromides at 100 °C in water, and the catalyst can be recycled and reused several times with slight loss of activity.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
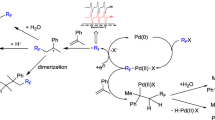
The reductive dimerization of aryl halides is one of the most important C–C bond-forming methods in organic synthesis because biaryls are useful building blocks for many biologically significant pharmaceuticals and agrochemicals. In general, the biaryls are synthesized by copper-mediated Ullmann homocoupling of aryl halides [1]. However, the need for a stoichiometric amount of copper catalysts and high temperature limited its wide application. In recent years, researchers have made improvements to this methodology by utilizing other transition metals, including palladium, zinc [2], indium [3], and nickel [4] to promote the Ullmann homocoupling reactions under milder conditions. For the palladium-catalyzed reductive homocoupling of aryl halides, various reducing agents, including hydroquinone [5, 6], alcohols [7–10], benzoin [11], poly(ethylene glycol) [12], glucose [13], ascorbic acid [14], hydrogen gas [15], amines [16], formate salt [17, 18], zinc [19–22], and indium [23] have been utilized as hydrogen donor and/or electron source to regenerate the reductive Pd0 active species from the oxidative Pd2+ species to complete the catalytic redox cycle. But most studies focused on homogeneous catalysts, and the recycling and reuse of the palladium catalysts were rarely discussed [12, 13, 18, 24, 25]. Thus, the development of recyclable heterogeneous palladium catalysts is highly desirable.
Polymerized ionic liquids (PILs) [26, 27], which combine the unique properties of ionic liquids and macromolecular architectures, have recently attracted more and more attention. The application of PILs as supports in noble-metal-catalyzed reactions is an important aspect of this chemistry [28–32]. Exploiting the fact that the nitrile group is a good stabilizer of Pd nanoparticles [33, 34], we report herein the reductive homocoupling of aryl halides catalyzed by Pd nanoparticles supported and stabilized by a nitrile functionalized ionic liquid homopolymer. The nitrile functionalized ionic liquid moieties in the polymer act not only as stabilizer to Pd nanoparticles, but also make the catalyst more compatible with the organic substrates. The immobilized Pd nanoparticles can efficiently catalyze the homocoupling reactions of aryl halides in water at 100 °C with good yields, and the catalyst can be recycled and reused several times.
Results and discussion
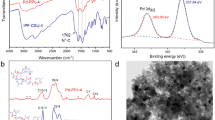
As shown in Scheme 1, the functionalized ionic liquid monomer 3 was synthesized by the reaction of 1-vinylimidazole (1) and 2-chloroacetonitrile (2), and then treated with potassium hexafluorophosphate to obtain the monomer. The azodiisobutyronitrile (AIBN)-catalyzed free radical homo-polymerization of the monomer afforded poly-CN-PF6. Finally, the polymer was added to the aqueous H2PdCl4 solution with vigorous stirring, followed by reduction of Pd2+ to Pd0 with NaBH4 and drying under vacuum to afford the catalyst Pd@poly-CN-PF6. A high loading amount of Pd on the catalyst (10 wt %) was achieved as determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES). The as-synthesized catalyst was found to be insoluble in water and hardly soluble in commonly used organic solvents, such as methanol, ethanol, acetonitrile, acetone, dichloromethane, toluene, and ethyl acetate. Therefore, the catalyst can be easily recycled by simple filtration after the reaction. The catalyst was also characterized by transmission electron microscopy (TEM). The Pd nanoparticles were well dispersed in the polymer with an average diameter of 3 nm as shown in Fig. 1a.


TEM images of freshly prepared Pd@poly-CN-PF6 (a) and reused four times (b)
The synthesized catalyst was then applied in the reductive homocoupling of aryl halides. The catalytic activities of Pd@poly-CN-PF6 under different conditions were evaluated in the model reductive homocoupling reaction of 1-iodo-4-methoxybenzene (4c, Scheme 2). The results are listed in Table 1. A low yield of 10 % was obtained with 1 mol % Pd as catalyst and without any additive (Table 1, entry 1). When one equivalent of glucose, ascorbic acid, or hydroquinone was used as an additive, the yields of product were dramatically improved to 51, 58, and 45 %, respectively (Table 1, entries 2–4). Other additives, such as ethylene glycol, glycerol, and β-CD, gave no product and the starting material remained unchanged (Table 1, entries 5–7). We therefore chose ascorbic acid as the most effective additive for further investigation. The yield of reaction product went up smoothly as the reaction temperature increased and a yield of 85 % after reaction for 9 h was achieved at 100 °C (Table 1, entries 8–10). Further investigation demonstrated that 2 mol % Pd based upon the amount of 4c could afford the homocoupling product 5c in a high yield of 97 % (Table 1, entry 11). It was also noted that little reductive deiodination product was observed according to GC analysis, implying good selectivity of the catalyst for reductive homocoupling over reductive deiodination. Finally, we chose the following optimized reaction conditions: 2 mol % Pd in Pd@poly-CN-PF6 as catalyst, water as solvent, NaOH as base, ascorbic acid as additive, and reaction at 100 °C.

Under the optimized conditions, the palladium-catalyzed reductive homocoupling reaction was extended to various aryl halides, and the results are summarized in Table 2 (Scheme 2). The reactions proceeded smoothly with both electron-deficient and electron-rich aryl iodides in good yields (Table 2, entries 1–4). For the less active aryl bromides, longer reaction times were required to get good yields (Table 2, entries 6–10). The highly sterically demanding 1-iodo-2-(trifluoromethyl)benzene gave the product in a yield of 10 % after 9 h. The homocoupling of 1-iodo-4-chlorobenzene and 1-bromo-4-chlorobenzene delivered the dichloro products with good yields, which exhibited good selectivity of iodo and bromo groups over the chloro group (Table 2, entries 4 and 9). Poor catalytic activity for chlorobenzene was observed as only a yield of 8 % was obtained even with 6 mol % Pd and reacting for 24 h (Table 2, entry 11). On the whole, the Pd@poly-CN-PF6 catalyst system showed better catalytic activity than the reported PdCl2/EDTA/ascorbic acid catalyst system with respect to higher yields and lower amount of catalyst [14].
The homocoupling reaction of 1-iodo-4-methoxybenzene (4c) was used to test the reusability of the Pd@poly-CN-PF6 catalyst. After each cycle, the catalyst was recovered by filtration, washed with ethyl acetate and water, and dried ready for the next run. As shown in Table 3, the catalyst activity decreased slightly as the cycle times increased. To identify the reason for this catalytic activity decline, palladium leaching was determined by ICP-AES as 20 ppm in 3 cm3 water, which was consistent with the loading amount of palladium of the first recycled catalyst (9.7 wt %, ICP-AES). The TEM image of the catalyst after being reused four times (Fig. 1b) showed that bigger Pd nanoparticles than the original ones were formed with an average diameter of about 5 nm. In a comparative experiment, the reaction was carried out at room temperature, but no product was detected and the palladium leaching in the reaction was also negligible (below detection limit of ICP-AES). Thus, owing to the combined action of palladium leaching at high temperature and aggregation of palladium nanoparticles [35, 36], a moderate yield of 74 % was obtained in the fourth cycle even after prolonging the reaction time to 11 h.
In conclusion, a nitrile-functionalized ionic liquid polymer was synthesized and applied to the immobilization of Pd nanoparticles. The Pd@poly-CN-PF6 catalyst (2 mol % Pd) selectively catalyzes the reductive homocoupling of aryl iodides and bromides at 100 °C in water. The catalyst can be easily recycled and its activity decreases slightly as the recycling times increased owing to the combined action of palladium leaching at high temperature and aggregation of palladium nanoparticles. Further application of the catalyst to other palladium-catalyzed reactions at lower temperature is ongoing in our lab.
Experimental
All chemicals and reagents were purchased from standard commercial suppliers. All the homocoupling products are known compounds; their physical and spectroscopic data were compared with those reported in the literature [9, 25, 37–39] and found to be identical. Melting points were recorded on an EZ-melt MPA120 (Stanford Research Systems, Inc., USA). 1H NMR and 13C NMR spectra were recorded on a Bruker AM-400 (400 MHz) spectrometer with CDCl3 or DMSO-d 6 as the solvent and TMS as the internal standard. Chemical shifts are reported in δ (parts per million) values. Coupling constants (J) are reported in Hz. Electrospray ionization (ESI) analyses were recorded by a Voyager RP MALDI-TOF spectrometer (Micromass LCT™, Mass Spectrometry Instruments Ltd.). Elemental analyses were conducted on an Elementar Vario EL III (Elementar Analysensysteme GmbH, Germany). ICP-AES was measured by a Varian 710-ES. Gel permeation chromatography was conducted on a Waters Breeze HPLC system. Analytical thin-layer chromatography (TLC) was carried out on precoated plates (silica gel 60 F254), and spots were visualized under ultraviolet light.
Gas chromatography was performed on an Agilent 7890A equipped with an HP-5 capillary column (30.0 m × 0.32 mm × 0.25 μm). The injector and flame ionization detector (FID) temperatures were set to 280 and 300 °C, respectively. Gas flow through the column was set at 8 cm3/min. The oven initial temperature was held at 70 °C for 2 min, increased to 300 °C at a rate of 20 °C/min, then cooled to 60 °C and held at that temperature until the next injection. The response peak area ratios of product 5c and internal standard dodecane (A 5c /A d = x) were obtained from Agilent 7890A. The GC yields of 5c (y) were calculated by the formula of curve fitting (y = 0.59223x − 0.00307).
3-(Cyanomethyl)-1-vinylimidazolium hexafluorophosphate (3, C7H8F6N3P)
A mixture of 4.7 g 1-vinylimidazole (50 mmol) and 3.75 g 2-chloroacetonitrile (50 mmol) was heated with stirring at 75 °C for 3 h. Then, the product was washed with diethyl ether (50 cm3 × 2) and EtOAc (50 cm3 × 2). After filtration and drying, 3-(cyanomethyl)-1-vinylimidazolium chloride was obtained as a light yellow solid powder (8.1 g, 96 %). 1H NMR (400 MHz, D2O): δ = 9.32 (s, 1H), 7.91 (s, 1H), 7.79 (s, 1 H), 7.29 (dd, J 1 = 7.2 Hz, J 2 = 4.4 Hz, 1H), 5.88 (dd, J 1 = 7.6 Hz, J 2 = 4.0 Hz, 1H), 5.55 (s, 2H), 5.51 (dd, J 1 = 4.8 Hz, J 2 = 4.0 Hz, 1H) ppm; 13C NMR (100 MHz, D2O): δ = 135.81, 128.08, 123.22, 120.46, 113.63, 110.98, 37.45 ppm; HRMS (ESI): calcd for C7H8N3 134.0718 [M − Cl]+, found 134.0711, calcd. for C14H16ClN6 303.1125 [2M − Cl]+, found 303.1121.
Then, 5.07 g 3-(cyanomethyl)-1-vinylimidazolium chloride (30 mmol) was dissolved in 50 cm3 water, and 5.53 g potassium hexafluorophosphate (30 mmol) was added with vigorous stirring. After reaction for 48 h at room temperature, the reaction mixture was filtered. Then, the filter cake was washed with water (50 cm3 × 2) and EtOAc (50 cm3 × 2), and dried under vacuum to obtain 3-(cyanomethyl)-1-vinylimidazolium hexafluorophosphate as a light yellow powder (8.0 g, 95 %). 1H NMR (400 MHz, DMSO-d 6): δ = 9.57 (s, 1H), 8.26 (t, J = 1.6 Hz, 1H), 8.02 (s, 1H), 7.35 (dd, J 1 = 8.0 Hz, J 2 = 4.4 Hz, 1H), 6.00 (dd, J 1 = 9.6 Hz, J 2 = 1.2 Hz, 1H), 5.59 (s, 2H), 5.49 (dd, J 1 = 4.4 Hz, J 2 = 1.2 Hz, 1H) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 137.21, 129.18, 123.92, 120.02, 114.85, 110.14, 37.64 ppm; HRMS (ESI): calcd for C7H8N3 134.0718 [M − PF6]+, found 134.0715, calcd for C14H16F6N6P 413.1078 [2M − PF6]+, found 413.1075.
Synthesis of poly-CN-PF 6
In a 100-cm3 round-bottom flask, 5.58 g 3-(cyanomethyl)-1-vinylimidazolium hexafluorophosphate (20 mmol) was dissolved in 50 cm3 acetonitrile and the reaction was heated to 80 °C. Then, 32.8 mg AIBN (1 mol %) was added and the reaction was continued for 72 h under nitrogen. On completion, the polymer was collected by filtration, washed with acetonitrile (50 cm3 × 2), and dried under vacuum to afford the product as a light yellow solid powder (4.9 g, 80 %). The M n and M w of the polymer were 23,766 and 39,848, respectively, as determined by gel permeation chromatography (GPC).
Preparation of Pd@poly-CN-PF 6
A mixture of 1 g poly-CN-PF6 and 100 cm3 H2PdCl4 (0.01 M) was stirred at room temperature for 72 h, then filtered and washed with water (20 cm3 × 3). After drying, the obtained gray solid was re-dispersed in water (50 cm3) and reduced by a freshly prepared solution of 185 mg (5 mmol) NaBH4 in 10 cm3 water. Then the reaction mixture was filtered, washed with water (20 cm3 × 3) and ethanol (20 cm3 × 3), and dried under vacuum to afford the desired Pd@poly-CN-PF6 as a black powder. The content of Pd in the catalyst was 10 wt % as determined by ICP-AES.
General procedure for reductive homocoupling of aryl halides
Pd@poly-CN-PF6 catalyst (21.2 mg, 2 mmol % Pd based on aryl halide), aryl halide (1 mmol), 80 mg NaOH (2 mmol), 176 mg ascorbic acid (1 mmol), and 3 cm3 water were added into a 25-cm3 round-bottom flask, which was then heated to 100 °C for several hours. The reaction was monitored by TLC. After the reaction was finished, the catalyst was filtered and washed with ethyl acetate (4 cm3 × 3) and water (4 cm3 × 3), which was then dried ready for the next use. The filtrate was extracted with EtOAc (4 cm3 × 3). To obtain the GC yield of 5c for optimization of reaction conditions, the combined organic phase was analyzed by GC on an Agilent 7890A using 100 mg dodecane as an internal standard. To obtain the isolated yield, the combined EtOAc extracts were evaporated to leave the crude products which were purified by column chromatography over silica gel to afford the pure product. It was noted that in some cases, such as 1-iodo-4-chlorobenzene and 1-bromo-4-chlorobenzene, the products may sublimate to the bottom of the condenser tube wall; this sublimation could be avoided by adding three drops of toluene (about 0.1–0.15 cm3) into the reaction mixture.
References
Hassan J, Sévugnon M, Gozzi C, Schulz E, Lemaire M (2002) Chem Rev 102:1359
Abiraj K, Srinivasa GR, Gowda DC (2004) Tetrahedron Lett 45:2081
Ranu BC, Dutta P, Sarkar A (1998) Tetrahedron Lett 39:9557
Peng J, Liu X, Kishi Y (2011) Tetrahedron Lett 52:2172
Hennings DD, Iwama T, Rawal VH (1999) Org Lett 1:1205
He HS, Zhang C, Ng CKW, Toy PH (2005) Tetrahedron 61:12053
Hassan J, Penalva V, Lavenot L, Gozzi C, Lemaire M (1998) Tetrahedron 54:13793
Penalva V, Hassan J, Lavenot L, Gozzi C, Lemaire M (1998) Tetrahedron Lett 39:2559
Zeng M, Du Y, Shao L, Qi C, Zhang XM (2010) J Org Chem 75:2556
Shao L, Du Y, Zeng M, Li X, Shen W, Zuo S, Lu Y, Zhang XM, Qi C (2010) Appl Organomet Chem 24:421
Park BR, Kim KH, Kim TH, Kim JN (2011) Tetrahedron Lett 52:4405
Wang L, Zhang Y, Liu L, Wang Y (2006) J Org Chem 71:1284
Monopoli A, Calò V, Ciminale F, Cotugno P, Angelici C, Cioffi N, Nacci A (2010) J Org Chem 75:3908
Ram RN, Singh V (2006) Tetrahedron Lett 47:7625
Mukhopadhyay S, Rothenberg G, Wiener H, Sasson Y (1999) Tetrahedron 55:14763
Kuroboshi M, Waki Y, Tanaka H (2003) J Org Chem 68:3938
Mukhopadhyay S, Rothenberg G, Qafisheh N, Sasson Y (2001) Tetrahedron Lett 42:6117
Wan Y, Chen J, Zhang D, Li H (2006) J Mol Catal A Chem 258:89
Venkatraman S, Li CJ (1999) Org Lett 1:1133
Li CJ, Venkatraman S (2000) Tetrahedron Lett 41:4831
Venkatraman S, Huang T, Li CJ (2002) Adv Synth Catal 344:399
Li JH, Xie YX, Yin DL (2003) J Org Chem 68:9867
Lee PH, Senmoon D, Lee K (2005) Org Lett 7:343
Iranpoor N, Firouzabadi H, Azadi R (2008) J Organomet Chem 693:2469
Iranpoor N, Firouzabadi H, Ahmadi Y (2012) Eur J Org Chem 2012:305
Yuan J, Antonietti M (2011) Polymer 52:1469
Lu J, Yan F, Texter J (2009) Prog Polym Sci 34:431
Mu XD, Meng JQ, Li ZC, Kou Y (2005) J Am Chem Soc 127:9694
Qiao K, Sugimura R, Bao Q, Tomida D, Yokoyama C (2008) Catal Commun 9:2470
Liu G, Hou M, Song J, Jiang T, Fan H, Zhang Z, Han B (2010) Green Chem 12:65
Li S, Wang J, Kou Y, Zhang S (2010) Chem Eur J 16:1812
Wang J, Song G, Peng Y (2011) Tetrahedron Lett 52:1477
Zhao D, Fei Z, Geldbach TJ, Scopelliti R, Dyson PJ (2004) J Am Chem Soc 126:15876
Fei Z, Zhao D, Pieraccini D, Ang WH, Geldbach TJ, Scopelliti R, Chiappe C, Dyson PJ (2007) Organometallics 26:1588
Wang Z, Shen B, Zou A, He N (2005) Chem Eng J 113:27
Wang Z, Xiao P, Shen B, He N (2006) Colloids Surf A 276:116
Chen X, Wang L (2009) Chin J Chem 27:2037
Ma N, Zhu Z, Wu Y (2007) Tetrahedron 63:4625
Xu X, Cheng D, Pei W (2006) J Org Chem 71:6637
Acknowledgments
Financial support for this work from the National Natural Science Foundation of China (NSFC, Grant 20676033), National Basic Research Program of China (973 Program) (Grant 2010CB126101), National Key Technology R and D Program (Grant No. 2011BAE06B05-4), and DC Foundation +is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, J., Li, Y., Li, P. et al. Polymerized functional ionic liquid supported Pd nanoparticle catalyst for reductive homocoupling of aryl halides. Monatsh Chem 144, 1159–1163 (2013). https://doi.org/10.1007/s00706-013-0925-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-013-0925-7