
Abstract
A sensitive and selective procedure for the determination of silver(I) at p-isopropylcalix[6]arene modified carbon paste electrode has been developed. Silver(I) was accumulated in open-circuit design on the electrode surface via complex formation with the modifier at pH 9.00. After electrochemical reduction of silver(I), the anodic stripping wave of silver(0) appeared at +0.04 V versus reference electrode on scanning the potential in the positive direction in 0.1 M H2SO4 + 2.0 × 10−2 M KBr. The effects of various parameters such as preconcentration time, stripping medium, reduction potential, etc. were investigated. In the optimum condition, a linear calibration curve was obtained in the range of 5.0 × 10−8 M to 2 × 10−6 M with detection limit (3δ) of 4.8 × 10−8 M. Many coexisting metal ions had little effect on the determination of silver(I). Also, the proposed method was applied to the determination of silver(I) in X-ray photographic film samples. The results were in good agreement with those obtained by atomic absorption spectroscopy (AAS).
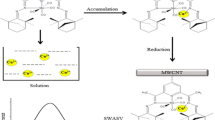
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The development and application of chemically modified carbon paste electrodes (CMCPEs) have received considerable attention in recent years. CMCPEs are characterized by purposefully altering their surface characteristics (states) to display new qualities that can be exploited for analytical purposes. These kinds of electrodes have many advantages such as easy manufacture, low price, no toxicity, low background current, wider operational potential window, rapid renewal surface, and stability in various solvents. One of the main reasons for modification is to improve the selectivity and sensitivity of electrochemical measurements by preconcentrating the analytical target from a dilute solution on the electrode surface. The accumulation step preceding the measurement can be performed with or without the use of an applied potential (closed- or open-circuit condition) depending on the nature of the preconcentration. However, the latter approach makes it possible to separate the species under consideration and eliminate interference from other components by proceeding to a medium exchange after the preconcentration step [1, 2].
During this century, a large increase of silver quantities has been observed in environmental samples due to the expansion of industrial and medicinal activities [3]. Silver is most toxic to microorganisms or larval forms of aquatic animals [4–6]. Thus, preconcentration and sensitive determination of the silver ion is of increasing interest.
A number of studies on the use of CMCPEs for determination of silver(I) by stripping voltammetric measurements have been reported. There are CPEs modified with alizarin violet [7], ethylenediamine tetraacetic acid (EDTA) [8], S2O2 donor dopant [9], Dowex 50 W-12 [10], thiacrown compounds [11], N,N′-diphenyloxamide [12], vermiculite clay mineral [13], zeolites [14], keratin [15], imine-thioether polymer [16], 2,2′-dithiopyridine [17], and 2,3-dicyano-1,4-naphthoquinone (CYNQ) [18].
Calix[n]arenes are macrocyclic compounds formed from phenol–formaldehyde condensations, in which n refers to the number of phenolic units. Values of n = 4–14 are known, but the most common are n = 4, 6, and 8. These compounds have received much attention in the last two decades because of their potential as enzyme mimics and their molecular and ion binding capability [19]. Therefore, they have been used in various fields of analytical chemistry such as optical sensors, chiral recognition devices, mobile phase modifiers in chromatography, and electrochemical sensors [20]. Electrochemical sensors are based on potentiometric or voltammetric methods. Ion-selective electrodes for many metal ions such as potassium, cesium, lead(II), and copper(II) have been prepared with calixarene ionophores [21–23]. However, a few papers have been published about the use of calixarene in a voltammetric sensor. These voltammetric sensors have been used for determination of lead(II), copper(II), and mercury(II) [24, 25]. To the best of our knowledge, there are no reports about the use of calix[6]arene for determination of silver(I). Due to the importance of silver, we investigated the ability of a p-isopropylcalix[6]arene (Scheme 1) modified carbon paste electrode (CAMCPE) for its determination.

Results and discussion
Electrochemical behavior of Ag(I) at CAMCPE
The ability of CAMCPE for accumulation of Ag(I) was studied using cyclic voltammetry experiments. The cyclic voltammogram of bare CPE in blank solution (0.1 M H2SO4 + 2 × 10−2 M KBr) does not show any anodic or cathodic peaks (Fig. 1a). Figure 1b, c shows cyclic voltammograms of unmodified CPE and CAMCPE in blank solution after accumulation in solution containing 1.0 × 10−7 M Ag(I) at pH 9.00, respectively. As can be seen, when the CAMCPE was dipped into the accumulation medium containing 1 × 10−7 M Ag(I) for 3 min followed by cycling in the stripping medium, a well-defined stripping anodic peak with peak potential of +0.076 V versus Ag|AgCl|KClsat appeared (Fig. 1c). This peak was due to the reoxidation of Ag(0) to Ag(I) produced by reduction of accumulated Ag(I) at −0.25 V versus Ag|AgCl|KClsat on the reverse potential scan. A small cathodic peak was obtained at −0.08 V versus Ag|AgCl|KClsat due to reduction of Ag(I) to Ag(0). The results demonstrated the preconcentration of Ag(I) at the surface of CAMCPE.

Cyclic voltammograms of (a) bare CPE in blank solution (0.1 M H2SO4 + 2.0 × 10−2 M KBr), (b) bare CPE, and (c) CAMCPE after 3 min of open-circuit accumulation in solution (pH 9.00) containing 1.0 × 10−7 M Ag(I) in blank solution at scan rate of 150 mV s−1
Effect of p-isopropylcalix[6]arene loading
The accumulation of Ag(I) at the CAMCPE was based on the complex formation reaction between Ag(I) and p-isopropylcalix[6]arene as a modifier. Therefore, the effect of p-isopropylcalix[6]arene concentration in the carbon paste on the voltammetric response of the modified electrode was investigated. The obtained results showed that increasing the concentration of this modifier in the CPE matrix led to an increase of the peak current to a maximum magnitude at 1% (w/w), followed by a decline. The current decrease at higher p-isopropylcalix[6]arene loading could presumably be ascribed to decrease of the conductive area at the electrode surface.
pH of accumulation medium
We examined some solutions with various buffered and nonbuffered pH values as accumulation media. Na2HPO4/NaH2PO4 and NH +4 /NH3 buffers were used as acidic and basic buffers, respectively. The solutions with nonbuffered pH values in the range of 2.00–11.00 were prepared with HNO3 and tetramethylammonium hydroxide. The results showed that the peak currents of cyclic voltammograms obtained in buffers were smaller than those obtained in solutions with nonbuffered pH values. Also, the greatest peak current was obtained at pH 9.00 (Fig. 2).

Effect of pH on anodic stripping peak current. Accumulation medium contains 1.0 × 10−5 M Ag(I). Open-circuit condition, accumulation time 3 min, stripping medium 0.1 M H2SO4 + 2 × 10−2 M KBr, reduction potential −0.25 V versus Ag|AgCl|KClsat, reduction time 25 s, scan rate 20 mV s−1
Thus, pH 9.00 was used as optimum pH in all measurements. The lack of peak currents in the cyclic voltammograms obtained in buffered solution can be explained as follows: sodium ions in Na2HPO4/NaH2PO4 buffer medium can interfere with Ag(I); and in NH +4 /NH3 buffer, NH3 molecules can compete with p-isopropylcalix[6]arene molecules in the coordination of Ag(I) ions.
Accumulation time
The dependence of the anodic peak current on the preconcentration time for 1.0 × 10−5 M Ag(I) was also investigated (Fig. 3). As can be seen, the peak current increased with accumulation time from 0 to 3 min and then remained nearly constant. Thus, accumulation time of 3 min was chosen for further experiments.

Effect of preconcentration time on anodic stripping peak current of Ag(0). Other conditions are as in Fig. 2
Choice of stripping medium
We examined 0.1 M HNO3, 0.1 M H3PO4, and 0.1 M H2SO4 as stripping medium, with 0.1 M H2SO4 giving the best cyclic voltammetric response. Because sensitivity and peak shape depend on the type of supporting electrolyte, we investigated the influence of different salts such as KNO3, KBr, and KCl at various concentrations ranging from 2.0 × 10−4 M to 2.0 × 10−2 M (Fig. 4).

Effect of different salts: (a) 2.0 × 10−2 M KBr, (b) 2.0 × 10−2 M KCl, and (c) 2.0 × 10−2 M KNO3 on cyclic voltammograms of accumulated CAMCPE in 0.1 M H2SO4. Accumulation concentration of Ag(I) is 1.0 × 10−5 M. Accumulation time is 3 min and scan rate is 50 mV s−1
As shown in Fig. 4, the anodic peak current is the highest in the presence of KBr. Also, the best result was obtained at KBr concentration of 2.0 × 10−2 M. Therefore 0.1 M H2SO4 + 2.0 × 10−2 M KBr was chosen as the stripping medium.
A possible mechanism for the greater sensitivity of anodic stripping peak current is that Ag(I) produced during the anodic potential scan reacts with halide ion (X−), forming first a related silver halide precipitate [AgX(s)] and then a [AgX −2 ] complex. Because the formation constant of AgBr −2 is larger that that of AgCl −2 [26], the anodic reaction is more accelerated in the presence of Br− ion, and consequently the increase in the stripping peak current and shift in the peak potential to more negative potential are more pronounced.
Effect of reduction potential and reduction time
According to Fig. 5, the anodic stripping peak current of silver increased as the reduction potential became more negative, reaching the highest point at −0.25 V versus Ag|AgCl|KClsat. At more negative potentials the peak current decreased due to hydrogen evolution. A reduction potential of −0.25 V versus Ag|AgCl|KClsat was used for all subsequent measurements.

Effect of reduction potential on anodic stripping peak current of Ag(0). Other conditions are as in Fig. 2
Dependence of peak current on the reduction time was also investigated (Fig. 6). As can be seen, the peak current increased for reduction time from 0 to 25 s and then remained constant. Consequently, a reduction time of 25 s was selected as optimum.

Effect of reduction time on anodic stripping peak current. Other conditions are as in Fig. 2
Analytical aspects
Standard solutions containing different concentrations of Ag(I) were prepared in buffered solution at pH 9.00. Figure 7 shows the obtained differential pulse voltammograms of these solutions at CAMCPE. A linear calibration plot (y = 0.0687x + 5.4989 with R 2 = 0.9969) was obtained over the range of 5.0 × 10−8 M to 2.0 × 10−6 M. The detection limit (3δ) was estimated to be 4.8 × 10−8 M for 3 min of accumulation time. The relative standard deviation for four successive determinations of 1.0 × 10−7 M and 5.0 × 10−7 M Ag(I) was 4.8% and 6.5%, respectively.

Differential pulse anodic stripping voltammograms of Ag(0) accumulated onto CAMCPE after accumulation in various Ag(I) concentrations: a 5.0 × 10−8 M, b 1.0 × 10−7 M, c 2.0 × 10−7 M, d 5.0 × 10−7 M, e 1.0 × 10−6 M, and f 2.0 × 10−6 M. Inset shows the related calibration plot. Conditions are as in Fig. 2
Interference studies
Some metal ions were examined as interfering ions in the determination of 1 × 10−7 M Ag(I). The results showed that a 10-fold excess of Pb(II), Cd(II), Zn(II), Mg(II), or NH +4 , 5-fold excess of Cu(II), and 50-fold excess of Na+ did not interfere in the determination of Ag(I) with CAMCPE. Figure 8 shows the results obtained for some of these studied ions.

Differential pulse anodic stripping voltammograms of Ag(0) accumulated onto CAMCPE after accumulation in 1.0 × 10−7 M Ag(I) in the absence (a) and presence (b) of various interfering ions: (a) 1.0 × 10−6 M Cd(II), (b) 1.0 × 10−6 M Zn(II), and (c) 5.0 × 10−7 M Cu(II). Conditions are as in Fig. 2
Analytical application
The proposed method was applied to the determination of silver in an X-ray photography film. The film was washed with distilled water, dried for 20 min in an oven at 40 °C, and then cut into small pieces, 2 g of which were dissolved in 25 cm3 nitric acid (5.0 M), and then the mixture was filtered. The solution was diluted to 250 cm3 with distilled water. Then 200 mm3 of the latter solution was taken, pH was adjusted to 9.00 by addition of tetramethylammonium hydroxide, and it was diluted to 50 cm3. The content of silver was then determined by the standard addition method according to the procedure described above. The measured value (50.74 μg) was in agreement with that obtained by atomic absorption spectrometry (50 μg). Accuracy was examined by comparison of data obtained from this method with the AAS method for determination of Ag(I). The results from the statistical calculation indicate good agreement between the mean values (t-test) and precision (F-test) for two methods (Table 1).
Experimental
Apparatus and reagents
The electrochemical experiments were performed using a BHP potentiostat–galvanostat (BHP 2601-C Electrochemical Analysis system, Behpajooh, Iran) coupled with a Pentium III personal computer with a standard three-electrode configuration. CAMCPE served as working electrode; a platinum wire electrode provided the counterelectrode, with an Ag|AgCl|KClsat (Metrohm) reference electrode completing the cell assembly. A pH-meter (Ion Analyzer 250, Corning) was used to read pH values of solutions.
A 1,000 ppm silver(I) stock solution (Fluka) was used. The experimental solutions were prepared by diluting the stock solution with distilled water. Tetramethylammonium hydroxide was used for adjusting pH. p-Isopropylcalix[6]arene, graphite powder (particle diameter 0.1 mm), and Nujol (as pasting oil) from Merck were used to prepare the modified electrode. All other chemicals were of analytical-reagent grade.
Working electrode preparation
Unmodified carbon paste was prepared by thoroughly mixing 1.0 g graphite powder with 0.6 cm3 pasting oil with a mortar and pestle. Modified carbon paste was prepared similarly, except that p-isopropylcalix[6]arene (1% w/w) was dissolved in diethyl ether and hand-mixed with 99 times its weight of graphite powder with a mortar and pestle. The solvent was evaporated by stirring and then 0.6 cm3 Nujol was added and blended by hand-mixing. A portion of the resulting paste was packed into a glass tube (internal radius 3.4 mm), in which a copper wire was inserted to establish electrical contact. The electrode surface was polished on a piece of tracing paper until it had a shiny appearance.
Procedure
The modified electrode was immersed in a magnetically stirred solution containing Ag(I) at pH 9.00 for 3 min. Then, the electrode was removed and rinsed with water. The electrode was transferred into a solution containing 0.1 M H2SO4 + 2 × 10−2 M KBr. A potential of −0.25 V was applied for 35 s to reduce Ag(I) to Ag(0). Immediately after the reduction time expired, the potential was scanned in the positive direction by differential pulse stripping voltammetry (pulse amplitude 50 mV, pulse interval 1 s, scan rate 20 mV s−1). All experiments were carried out at ambient temperature and it was not necessary to remove oxygen from the solution.
References
Wang J (1994) Analytical electrochemistry. VCH, New York
Kalcher K, Kauffmann J-M, Wang J, Švancara I, Vytřas K, Neuhold C, Yang Z (1995) Electroanalysis 7:5
Kolthoff IM, Elving PJ (1966) Treatise on analytical chemistry. Interscience, New York, p 4
Bury NR, Grosell M, Grover AK, Wood CM (1999) J Toxic Appl Pharm 159:1
Environment Protection Agency, Ambient Water Quality Criteria for Silver (1980) EPA 4405-80-071, Office of water regulations, Washington DC
Meian E (1991) Metals and their compounds in environment. VCH, New York
Li YH, Xie HQ, Zhou FQ (2005) Talanta 67:28
Zhang SB, Zhang XJ, Lin XQ (2002) Chin J Anal Chem 30:745
Ha KS, Kim JH, Ha YS, Lee SS, Seo ML (2001) Anal Lett 34:675
Gellon H, Gonzales PS, Fontal CA (2003) Anal Lett 36:2749
Tanaka S, Yoshida H (1989) Talanta 36:1044
Won MS, Yeom JS, Yoon JH, Jeong ED, Shim YB (2003) Bull Korean Chem Soc 24:948
Svegl IG, Kolar M, Ogorevc B, Pihlar B (1998) Fresenius J Anal Chem 361:358
Wang J, Martinez T (1988) Anal Chim Acta 207:95
Sugawara K, Matsui H, Hoshi S, Akatsuka K (1998) Analyst 123:2013
Li P, Gao Z, Xu Y, Wang G, Zhao Z (1990) Anal Chim Acta 229:213
Sugawara K, Tanaka S, Taga M (1991) J Electroanal Chem 304:249
Khodari M, About-Krish MM, Fandy R (1994) Talanta 41:2179
O’Conner KM, Arrigan DWM, Svehla G (1995) Electroanalysis 7:205
McMahon G, Nolan SK, Diamond D (2003) Arkivoc vii:23
King AM, Moore CP, Sandanayake KS, Sutherland IO (1992) J Chem Soc Chem Commun 582
Fanni S, Arnaud-Neu F, McKervey MA, Schwing-Weill MJ, Ziat K (1996) Tetrahedron Lett 37:7975
Arnaud-Neu F, Barrett G, Corry D, Cermin S, Ferguson G, Gallagher JF, Harris SJ, McKervey MA, Schwing-Weill MJ (1997) J Chem Soc Perkin Trans 575
Arrigan DWM, Svehla G, Harris SJ, McKervey MA (1994) Electroanalysis 6:97
Arrigan DWM, Svehla G, Harris SJ, McKervey MA (1992) Analyt Proc 29:27
Dean JA (1973) Lange’s handbook of chemistry, 11th edn. McGraw-Hill, New York, p 5:46
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Raoof, J.B., Ojani, R., Alinezhad, A. et al. Differential pulse anodic stripping voltammetry of silver(I) using p-isopropylcalix[6]arene modified carbon paste electrode. Monatsh Chem 141, 279–284 (2010). https://doi.org/10.1007/s00706-010-0258-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-010-0258-8