
Abstract
Synthesis of novel benzo[h]-1,6-naphthyridines was successfully achieved by cyclocondensation of ethyl 4-aminoquinoline-3-carboxylates with malononitrile. The pyrazolo[4,3-c]quinolines were synthesized by nucleophilic substitution and subsequent addition reaction of ethyl 4-chloroquinoline-3-carboxylates with different hydrazines. All new compounds were characterized by spectral and analytical methods.
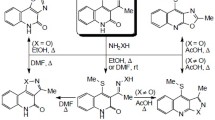
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Functionalized quinolines and their benzo/hetero-fused analogs are an important class of organic molecules that have attracted much attention from synthetic and medicinal chemists because of their presence in numerous natural products and their wide range of physiological activity [1]. The quinolines, particularly those substituted at position 4, have marked antimalarial, antibacterial, and anti-inflammatory activity [2–4]. It is also apparent from the literature that benzo[h]-1,6-naphthyridine derivatives functionalized at position 4 have been used as candidates for antimalarials [5]. Recently, Hinschberger et al. [6] reported the synthesis of new benzo[h]naphthyridines as selective antagonists of 5-HT4 receptors. The remarkable applications of these compounds has not only prompted many chemists to synthesize this type of compound, they have also become an active research area of continuing interest [7].
Pyrazoles and their derivatives are also important constituents of biologically active synthetic compounds [8–12], because these systems have been associated with useful biological activity for example antiviral [13], antimalarial [14, 15], antibacterial [16, 17], anticancer [18], and antimicrobial [19] activity. Recently, pyrazolo[4,3-c]quinolines were found to be highly fluorescent materials in the blue region of the spectrum [20] and promising materials for electroluminescence applications [21, 22]. These literature reports prompted us to develop a new synthetic route to novel quinoline fused heterocycles. As a part of our ongoing interest in this area [23–27], we have reported the synthesis of 4-aryl/4-aminopyrimidines, fused pyrimidines, benzo[h]quinolines, chromenes, quinolines, and pyrazolo[3,4-b]pyridines. Recently, we have reported the synthesis of pyrazolo[3,4-b]pyrrolo[2,3-d]pyridines from 5-aminopyrazoles and cyclic β-ketoesters, and pyrazolo[3,4-b]pyridines present in biological molecules [28, 29]. In this communication we report a simple and efficient route for the synthesis of novel benzo[h]-1,6-naphthyridines and pyrazolo[4,3-c]quinolines.
Results and discussion
The intermediate ethyl 4-chloroquinoline-3-carboxylates 1a–1c required for the synthesis of quinoline fused heterocycles were synthesized by a Gould–Jacobs reaction between primary aromatic amines and diethyl ethoxymethylenemalonate via a chlorination reaction using phosphorus oxychloride [30, 31]. The bifunctional compounds 1a–1c were then used as precursors for syntheses of benzo[h]-1,6-naphthyridines and pyrazolo[4,3-c]quinolines. Compounds 1a–1c on reaction with sodium azide in DMF at 20–25 °C furnished a mixture of azidoquinoline derivatives 2a–2c and triazinoquinolines 3a–3c in 70 and 10% yields, respectively (Scheme 1) [32], which were separated by column chromatography. The structures of 2 and 3 were established on the basis of spectral data and formulae confirmed by elemental analysis. For instance, the IR spectrum of 2a showed absorption bands at 1,708 and 2,100 cm−1 for carbonyl and azido groups, respectively. However, in the IR spectrum of 3a a broad absorption band was observed at 3,425 cm−1 for the OH group. The 1H NMR spectrum of 2a in CDCl3 showed a triplet and a quartet at 1.38 and 4.39 ppm, respectively, for ethoxy protons. In the 1H NMR spectrum of 3a these signals disappeared and the broad singlet for a phenolic OH proton was observed at 10.52 ppm. The 13C NMR spectrum of 2a exhibited peaks at 14.1 and 60.2 ppm corresponding to OCH2CH3 carbons, which are absent in 3a. Furthermore the molecular formulae of 2a and 3a were confirmed by elemental analyses, which are in agreement with the proposed structures.

Reduction of the N3 group was achieved successfully by heating compounds 2a–2c in methanol at reflux temperature with a stoichiometric amount of sodium dithionite to give the expected 4-aminoquinoline-3-carboxylates 4a–4c in 60–65% yield (Scheme 1). The structures of 4 were confirmed by spectroscopic and analytical data, for example the IR spectrum of 4a showed bands of the elongation vibrations of the C=O group at 1,695 cm−1 and two bands for the NH2 group at 3,375–3,420 cm−1.
The unexpected decrease of the ester C=O frequency by 50–60 cm−1 might be because of intramolecular hydrogen bonding between a hydrogen atom of the NH2 group and the oxygen of C=O [33]. In addition to ethoxy and aromatic signals, the 1H NMR of compound 4a in DMSO-d6 contained a broad singlet at 8.39 ppm for NH2 protons, which underwent a facile hydrogen deuterium exchange upon addition of deuterium oxide. The mass spectrum of 4a revealed a molecular ion peak at m/z = 250. Furthermore this structure was supported by the 13C NMR spectrum, which was in agreement with the proposed structure.
The compounds 4 containing amino and ester functionality ortho to each other were utilized for the synthesis of novel quinoline fused heterocycles. However, the reactions of 4 with active methylene compounds proved to be difficult, which might be because of the strong intramolecular hydrogen bonding between the ester carbonyl and the 4-amino group, as reported earlier by Bare et al. [33] for pyrazolopyridines. Reaction of 4a and 4b only with malononitrile in ethanol containing a catalytic amount of KOH at reflux temperature for about 15 h was successfully carried out to yield benzo[h]-1,6-naphthyridines 5a and 5b in 56–60% yield (Scheme 1). The structures of compounds 5 were confirmed on the basis of spectral data and elemental analysis. For example, the IR spectrum of 5a contained strong absorption bands for the CN and OH groups at 2,215 and 3,496 cm−1 and two bands for the NH2 group at 3,335–3,413 cm−1. The 1H NMR spectrum of compound 5a contained two broad singlets at 4.74 and 5.46 ppm corresponding to the protons of the NH2 and OH groups. Aromatic protons appeared in the range 7.18–8.63 ppm. The mass spectrum of 5a contained characteristics peaks for M+ at m/z = 270 and 272, because of the presence of chlorine. This structure was also confirmed by the 13C NMR spectrum and elemental analysis in agreement with the proposed structure.
In continuation of our research on the well known intermediate compound 1 we carried out the reaction with a primary aromatic amine and different hydrazines. In our study we performed the SNAr reaction on 1 using the primary aromatic amine as nucleophile. Thus, reaction of 1a and 1b with aniline at 130–140 °C furnished an open chain derivative 6a and 6b in quantitative yield, and reaction of 1a and 1b with substituted hydrazines in ethanol containing a catalytic amount of triethylamine or piperidine at reflux for 2–3 h yielded the targeted pyrazolo[4,3-c]quinolines 7a–7f in 70–80% yield (Scheme 2). IR, 1H NMR, 13C NMR, MS, and elemental analysis were used to deduce the structures of 6 and 7. For example, the IR of compound 7a contained NH stretching bands at 3,310 and 3,400 cm−1. The 1H NMR spectrum of 7a in DMSO-d6 contained broad singlets at 11.43 and 12.49 ppm corresponding to two NH protons. The mass spectrum of this compound contained M+ peaks at m/z = 219 and 221, because of the presence of chlorine. Furthermore the 13C NMR and elemental analysis data of this compound were in agreement with the proposed structure.

Conclusion
We have used a simple and convenient method for synthesis of novel quinoline fused heterocycles such as benzo[h]-1,6-naphthyridines and pyrazolo[4,3-c]quinolines starting from ethyl 4-chloroquinoline-3-carboxylates with simple work up and clean products.
Experimental
Melting points were determined on a Barnstead Electro Thermal melting point apparatus, Mod. No. IA-9200 in open capillary tubes. The 1H NMR (300 MHz) and 13C NMR (75 MHz) spectra were recorded on a Varian XL-300 spectrometer. Chemical shifts were reported in δ units in ppm from the internal standard tetramethylsilane. The solvent for NMR spectra was deuterochloroform unless otherwise stated. Infrared spectra were taken on a Shimadzu IR-408 instrument in potassium bromide pellets unless otherwise stated. Elemental analyses were performed on a Hosli CH-Analyzer and results were within ±0.3% of the calculated values. High-resolution mass spectra were obtained with a Mat 112 Varian Mat Bremen (70 eV) mass spectrometer. Column chromatography was carried out on silica gel (SD Fine Chemicals, 60–80 mesh). Solutions were concentrated in a rotary evaporator under reduced pressure. All reactions were monitored by thin layer chromatography (TLC), carried out on 0.2 mm silica gel 60 F254 (Merck) plates using UV light (254 and 366 nm) for detection. Common reagent-grade chemicals are either commercially available and were used without further purification or were prepared by standard literature procedures.
General procedure for synthesis of ethyl 4-azidoquinoline-3-carboxylates 2a–2c and triazino[5,4-c]quinolines 3a–3c
A mixture of ethyl quinoline-3-carboxylate 1 (0.01 mol) and 0.650 g sodium azide (0.01 mol) in 15 cm3 DMF was stirred at 20–25 °C until the starting material had disappeared (2 h, checked by TLC monitoring). The solution was then poured into 50 cm3 cold water and stirred for 30 min. The solid obtained was isolated by filtration, washed with 100 cm3 cold water, and dried. It afforded a mixture of compounds 2 and 3 which was separated by column chromatography using 8:2 toluene–acetone as eluent to afford compounds 2 in 70% yield and 3 in 10% yield.
Ethyl 4-azido-6-chloroquinoline-3-carboxylate (2a, C12H9ClN4O2)
Yield 1.93 g (70%); m.p.: 291 °C (ethanol); R f = 0.64 (toluene–acetone 8:2); IR (KBr): \( \bar{v} = 3{,}087, \) 2,918, 2,100, 1,708, 1,581, 1,484, 1,367, 1,307, 1,204, 1,020, 980, 835, 720, 625 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.38 (t, J = 7 Hz, 3H, CH3), 4.39 (q, J = 7 Hz, 2H, OCH2), 7.69 (dd, J = 8.7, 2.4 Hz, 1H, C7–H), 7.72 (d, J = 2.4 Hz, 1H, C5-H), 8.24 (d, J = 8.7 Hz, 1H, C8–H), 9.10 (s, 1H, C2–H) ppm; 13C NMR (75 MHz, CDCl3): δ = 14.1 (CH3), 60.2 (OCH2), 124.1, 127.4, 129.4, 130.4, 133.2, 133.9, 138.0, 146.9, 147.7, 166.3 ppm.
Ethyl 4-azido-6-bromoquinoline-3-carboxylate (2b, C12H9BrN4O2)
Yield 2.18 g (68%); m.p.: 277 °C (ethanol); R f = 0.66 (toluene–acetone 8:2); IR (KBr): \( \bar{v} = 3{,}065, \) 2,910, 2,110, 1,711, 1,583, 1,486, 1,367, 1,304, 1,202, 1,025, 983, 836, 725, 620 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.35 (t, J = 6.9 Hz, 3H, CH3), 4.33 (q, J = 6.9 Hz, 2H, OCH2), 7.80 (dd, J = 8.7, 2.4 Hz, 1H, C7-H), 7.91 (d, J = 2.4 Hz, 1H, C5–H), 8.29 (d, J = 8.7 Hz, 1H, C8–H), 9.22 (s, 1H, C2–H) ppm; 13C NMR (75 MHz, CDCl3): δ = 15.3 (CH3), 62.9 (OCH2), 122.0, 123.2, 126.7, 130.4, 130.7, 135.9, 138.0, 147.7, 149.5, 168.2 ppm.
Ethyl 4-azido-7-chloroquinoline-3-carboxylate (2c, C12H9ClN4O2)
Yield 1.76 g (64%); m.p.: 281 °C (ethanol); R f = 0.62 (toluene–acetone 8:2); IR (KBr): \( \bar{v} = 3{,}065, \) 2,910, 2,115, 1,703, 1,588, 1,490, 1,368, 1,309, 1,201, 1,029, 988, 838, 720, 626 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.30 (t, J = 7.1 Hz, 3H, CH3), 4.29 (q, J = 7.1 Hz, 2H, OCH2), 7.60 (dd, J = 8.7, 2.4 Hz, 1H, C6–H), 7.82 (d, J = 8.7 Hz, 1H, C5–H), 8.10 (d, J = 2.4 Hz, 1H, C8–H), 9.50 (s, 1H, C2–H) ppm; 13C NMR (75 MHz, CDCl3): δ = 14.9 (CH3), 60.9 (OCH2), 123.0,126.8, 129.0, 129.5, 131.2, 137.6, 139.0, 148.6, 149.5, 170.3 ppm.
9-Chloro[1,2,3]triazino[5,4-c]quinolin-4-ol (3a, C10H5ClN4O)
Yield 0.232 g (10%); m.p.: 198 °C (ethanol); R f = 0.28 (toluene–acetone 8:2); IR (KBr): \( \bar{v} = 3{,}425, \) 2,920, 2,850, 1,624, 1,535, 1,444, 1,356, 1,273, 1,020, 833, 808, 653 cm−1; 1H NMR (300 MHz, CDCl3): δ = 7.60 (dd, J = 8.5, 2.4 Hz, 1H, C8–H), 7.83 (d, J = 2.4 Hz, 1H, C10-H), 7.94 (d, J = 8.5 Hz, 1H, C7-H), 8.70 (s, 1H, C5–H), 10.52 (bs, 1H, OH) ppm; 13C NMR (75 MHz, CDCl3): δ = 121.9, 126.0, 127.0, 130.5, 131.5, 132.3, 134.6, 145.0, 148.2, 153.3 ppm.
9-Bromo[1,2,3]triazino[5,4-c]quinolin-4-ol (3b, C10H5BrN4O)
Yield 0.332 g (12%); m.p.: 179 °C (ethanol); R f = 0.30 (toluene–acetone 8:2); IR (KBr): \( \bar{v} = 3{,}410, \) 2,900, 2,820, 1,614, 1,532, 1,440, 1,352, 1,271, 1,021, 830, 801, 652 cm−1; 1H NMR (300 MHz, CDCl3): δ = 7.75 (dd, J = 8.2, 2.2 Hz, 1H, C8–H), 7.91 (d, J = 2.2 Hz, 1H, C10–H), 7.96 (d, J = 8.2 Hz, 1H, C7–H), 8.86 (s, 1H, C5–H), 10.15 (bs, 1H, OH) ppm; 13C NMR (75 MHz, CDCl3): δ = 121.1, 121.6, 124.3, 129.3, 130.5, 133.5, 134.6, 147.6, 148.6, 151.5 ppm.
8-Chloro[1,2,3]triazino[5,4-c]quinolin-4-ol (3c, C10H5ClN4O)
Yield 0.301 g (13%); m.p.: 165 °C (ethanol); R f = 0.26 (toluene–acetone 8:2); IR (KBr): \( \bar{v} = 3{,}400, \) 2,933, 2,800, 1,624, 1,502, 1,445, 1,352, 1,278, 1,023, 830, 803, 650 cm−1; 1H NMR (300 MHz, CDCl3): δ = 7.44 (dd, J = 8.6, 2.3 Hz, 1H, C9–H), 7.62 (d, J = 8.6 Hz, 1H, C10-H), 8.06 (d, J = 2.3 Hz, 1H, C7–H), 8.81 (s, 1H, C5–H) 10.24 (bs, 1H, OH) ppm; 13C NMR (75 MHz, CDCl3): δ = 122.0, 124.4, 128.6, 129.1, 129.8, 135.2, 135.5, 147.6, 149.5, 155.5 ppm.
General procedure for synthesis of ethyl 4-aminoquinoline-3-carboxylates 4a–4c
A solution of 2 (0.01 mol) and sodium dithionite (0.01 mol) in 15 cm3 methanol was heated under reflux for 4 h. The solution was then allowed to cool and poured into 30 cm3 ice–cold water, and the mixture was stirred for 30 min. The precipitated solid was isolated by filtration, dried, and recrystallized from an appropriate solvent to afford 4 in good yield.
Ethyl 4-amino-6-chloroquinoline-3-carboxylate (4a, C12H11ClN2O2)
Yield 1.62 g (65%); m.p.: 281 °C (DMF–ethanol); R f = 0.35 (toluene–ethyl acetate 8:2); IR (KBr): \( \bar{v} = 3{,}420, \) 3,375, 3,095, 2,985, 2,904, 1,695, 1,624, 1,554, 1,523, 1,465, 1,384, 1,292, 1,195, 1,028, 8,68, 827, 650, 603 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 1.33 (t, J = 6.9 Hz, 3H, CH3), 4.30 (q, J = 6.9 Hz, 2H, OCH2), 7.73 (dd, J = 8.1, 2.3 Hz, 1H, C7–H), 7.80 (d, J = 2.3 Hz, 1H, C5–H), 8.39 (bs, 2H, NH2), 8.55 (d, J = 8.1 Hz, 1H, C8–H), 8.89 (s, 1H, C2–H) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 15.2 (CH3), 59.2 (OCH2), 114.3, 120.1, 121.3, 130.7, 131.9, 133.6, 147.5, 148.0, 155.6, 165.3 ppm; MS (70 eV): m/z = 252 ([M+2]+), 250 (M+), 236, 222, 204, 190, 177, 161, 150, 138, 114, 102, 87, 75, 63, 43.
Ethyl 4-amino-6-bromoquinoline-3-carboxylate (4b, C12H11BrN2O2)
Yield 1.82 g (62%); m.p.: 262 °C (DMF–ethanol); R f = 0.39 (toluene–ethyl acetate 8:2); IR (KBr): \( \bar{v} = 3{,}425, \) 3,380, 3,098, 2,975, 2,904, 1,695, 1,629, 1,559, 1,529, 1,469, 1,389, 1,290, 1,191, 1,026, 866, 828, 655, 609 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 1.40 (t, J = 6.9 Hz, 3H, CH3), 4.36 (q, J = 6.9 Hz, 2H, OCH2), 7.60 (dd, J = 8.1, 2.3 Hz, 1H, C7–H), 7.86 (d, J = 2.3 Hz, 1H, C5–H), 7.95 (d, J = 8.1 Hz, 1H, C8–H), 8.50 (bs, 2H, NH2), 9.03 (s, 1H, C2–H) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 14.1 (CH3), 60.2 (OCH2), 114.0, 118.6, 120.7, 123.4, 130.7, 135.6, 148.0, 150.1, 155.6, 169.1 ppm.
Ethyl 4-amino-7-chloroquinoline-3-carboxylate (4c, C12H11ClN2O2)
Yield 1.50 g (60%); m.p.: 269 °C (DMF–ethanol); R f = 0.34 (toluene–ethyl acetate 8:2); IR (KBr): \( \bar{v} = 3{,}422, \) 3,350, 3,094, 2,977, 2,910, 1,690, 1,620, 1,555, 1,529, 1,469, 1,380, 1,290, 1,191, 1,026, 866, 828, 624, 604 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 1.30 (t, J = 7 Hz, 3H, CH3), 4.29 (q, J = 7 Hz, 2H, OCH2), 7.54 (dd, J = 8.1, 2.3 Hz, 1H, C6-H), 7.78 (d, J = 8.1 Hz, 1H, C5–H), 8.09 (d, J = 2.3 Hz, 1H, C8–H), 8.55 (bs, 2H, NH2), 9.30 (s,1H, C2–H) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 14.6 (CH3), 60.8 (OCH2), 113.4, 118.7, 123.9, 128.2, 129.3, 137.3, 148.9, 150.1, 156.5, 168.1 ppm.
General procedure for synthesis of 2-amino-4-hydroxybenzo[h]-1,6-naphthyridine-3-carbonitriles 5a and 5b
A solution of 4 (0.01 mol) and 0.65 g malononitrile (0.01 mol) in 15 cm3 ethanol containing a catalytic amount of potassium hydroxide was heated under reflux for 15 h. The solution was then cooled to room temperature and the precipitated solid was isolated by filtration, dried, and recrystallized from an appropriate solvent to afford 5 in good yield.
2-Amino-9-chloro-4-hydroxybenzo[h]-1,6-naphthyridine-3-carbonitrile (5a,C13H7ClN4O)
Yield 1.62 g (60%); m.p.: 246 °C (DMF); R f = 0.32 (toluene–ethyl acetate 6:4); IR (KBr): \( \bar{v} = 3{,}496, \) 3,413, 3,335, 3,091, 2,215, 1,617, 1,553, 1,524, 1,470, 1,358, 1,295, 1,188, 1,032, 866, 825, 800, 604 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 4.74 (bs, 2H, NH2), 5.46 (bs, 1H, OH), 7.18–8.03 (m, 3H, Ar–H), 8.63 (s, 1H, C5–H) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 82.6, 109.1, 117.7, 126.0, 127.0, 130.5, 131.5, 132.3, 136.5, 145.0, 148.6, 162.8, 171.2 ppm; MS (70 eV): m/z = 272 ([M + 2]+), 270 (M+), 252, 250, 236, 222, 206, 193, 178, 163, 154, 140, 116, 100, 89, 76, 66, 46.
2-Amino-9-bromo-4-hydroxybenzo[h]-1,6-naphthyridine-3-carbonitrile (5b, C13H7BrN4O)
Yield 1.76 g (56%); m.p.: 257 °C (DMF); R f = 0.30 (toluene–ethyl acetate 6:4); IR (KBr): \( \bar{v} = 3{,}480, \) 3,435, 3,380, 3,096, 2,220, 1,617, 1,556, 1,522, 1,470, 1,358, 1,295, 1,190, 1,036, 865, 822, 802, 602 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 4.86 (bs, 2H, NH2), 6.13 (bs, 1H, OH), 7.75–8.13 (m, 3H, Ar–H), 9.05 (s, 1H, C5–H) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 82.4, 108.8, 116.8, 121.1, 124.3, 129.3, 130.5, 133.5, 136.3, 147.6, 148.6, 162.8, 171.8 ppm.
General procedure for synthesis of ethyl 4-(phenylamino)quinoline-3-carboxylates 6a and 6b
A mixture of 1 (0.01 mol) and aniline (0.04 mol) was heated at 130–140 °C for about 1 h, until TLC showed the starting material had disappeared. The mixture was then cooled to 20 °C, 20 cm3 methanol was added, and the resulting solid was isolated by filtration under vacuum, washed with methanol, dried, and recrystallized from an appropriate solvent to afford 6 in good yield.
Ethyl 6-chloro-4-(phenylamino)quinoline-3-carboxylate (6a, C18H15ClN2O2)
Yield 2.15 g (66%); m.p.: 178 °C (DMF–ethanol); R f = 0.47 (toluene–acetone 8:2); IR (KBr): \( \bar{v} = 3{,}325, \) 3,095, 2,985, 2,904, 1,697, 1,624, 1,554, 1,523, 1,465, 1,384, 1,292, 1,195, 1,028, 868, 827, 650, 603 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 1.14 (t, J = 7 Hz, 3H, CH3), 4.05 (q, J = 7 Hz, 2H, OCH2), 7.32–8.24 (m, 8H, Ar–H), 8.30 (bs, 1H, NH), 9.06 (s, 1H, C2–H) ppm.
Ethyl 6-bromo-4-(phenylamino)quinoline-3-carboxylate (6b, C18H15BrN2O2)
Yield 2.52 g (68%); m.p.: 146 °C (DMF–ethanol); R f = 0.49 (toluene–acetone 8:2); IR (KBr): \( \bar{v} = 3{,}335, \) 3,090, 2,989, 2,908, 1,697, 1,620, 1,552, 1,520, 1,465, 1,384, 1,298, 1,195, 1,020, 866, 827, 650, 603 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 1.16 (t, J = 7 Hz, 3H, CH3), 4.10 (q, J = 7 Hz, 2H, OCH2), 7.45–8.15 (m, 8H, Ar–H), 8.36 (bs, 1H, NH), 9.08 (s, 1H, C2–H) ppm.
General procedure for synthesis of 3H-pyrazolo[4,3-c]quinolin-3-ones7a–7f
A solution of compound 1 (0.01 mol) and different hydrazines (0.01 mol) in 15 cm3 ethanol containing 0.5 cm3 triethylamine was heated under reflux for 2–3 h. The excess solvent was removed under reduced pressure. The solid obtained was filtered, washed with ethanol, dried, and recrystallized from an appropriate solvent to afford 7 in 70–80% yield.
8-Chloro-1,2-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (7a, C10H6ClN3O)
Yield 1.66 g (76%); m.p.: 356 °C (DMF); R f = 0.70 (toluene–acetone 8:2); IR (KBr): \( \bar{v} = 3{,}400, \) 3,310, 3,091, 2,925, 2,853, 1,692, 1,612, 1,527, 1,459, 1,336, 1,295, 1,095, 813, 720, 621, 551 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 7.60–7.96 (m, 3H, Ar–H), 8.50 (s,1H, C4–H), 11.43 (bs, 1H, NH), 12.49 (bs, 1H, NH) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 121.9, 126.0, 127.0, 130.5, 131.5, 132.3, 138.1, 145.0, 148.6, 155.4 ppm; MS (70 eV): m/z = 221 ([M + 2]+), 219 (M+), 206, 190, 176, 162, 152, 137, 126, 99, 98, 74, 63, 50, 38.
8-Chloro-1,2-dihydro-2-phenyl-3H-pyrazolo[4,3-c]quinolin-3-one (7b, C16H10ClN3O)
Yield 2.30 g (78%); m.p.: 368 °C (DMF); R f = 0.61 (toluene–acetone 8:2); IR (KBr): \( \bar{v} = 3{,}422, \) 3,091, 2,852, 2,800, 1,694, 1,619, 1,527, 1,450, 1,360, 1,104, 784, 581 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 7.14–7.71 (m, 5H, Ar–H), 8.15–8.20 (m, 3H, Ar–H), 8.75 (s, 1H, C4-H), 12.20 (bs, 1H, NH) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 120.2, 121.6, 126.0, 126.3, 127.3, 129.4, 130.5, 131.5, 132.3, 134.6, 139.7, 141.0, 145.0, 148.6 ppm; MS (70 eV): m/z = 297 ([M + 2]+), 295 (M+), 294, 268, 266, 238, 215, 204, 189, 176, 162, 147, 126, 98, 77, 63, 51, 39.
8-Chloro-1,2-dihydro-2-(2-hydroxyethyl)-3H-pyrazolo[4,3-c]quinolin-3-one (7c, C12H10ClN3O2)
Yield 1.89 g (72%); m.p.: 323 °C (DMF); R f = 0.72 (toluene–acetone 8:2); IR (KBr): \( \bar{v} = 3{,}395, \) 3,153, 3,091, 2,903, 1,695, 1,618, 1,554, 1,524, 1,469, 1,379, 1,359, 1,295, 1,188, 1,151, 1,105, 1,031, 957, 866, 825, 800, 645, 605 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 2.40 (bs, 1H, OH), 3.92 (t, J = 7 Hz, 2H, CH2), 4.01 (t, J = 7 Hz, 2H, OCH2), 7.62-7.98 (m, 3H, Ar–H), 8.81 (s, 1H, C4–H), 11.90 (bs, 1H, NH) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 57.4, 61.5, 121.9, 126.0, 127.0, 130.5, 131.5, 132.3, 134.6, 138.0, 145.0, 148.6 ppm.
8-Bromo-1,2-dihydro-3H-pyrazolo[4,3-c]quinolin-3-one (7d, C10H6BrN3O)
Yield 1.95 g (74%); m.p.: 338 °C (DMF); R f = 0.68 (toluene–acetone 8:2); IR (KBr): \( \bar{v} = 3{,}420, \) 3,330, 3,096, 2,925, 2,850, 1,690, 1,612, 1,527, 1,459, 1,366, 1,294, 1,095, 813, 710, 631, 541 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 7.65–7.94 (m, 3H, Ar–H), 8.45 (s, 1H, C4-H), 11.48 (bs, 1H, NH), 12.52 (bs, 1H, NH) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 121.2, 126.5, 127.8, 131.1, 131.9, 133.1, 139.1, 146.2, 149.2, 156.2 ppm.
8-Bromo-1,2-dihydro-2-phenyl-3H-pyrazolo[4,3-c]quinolin-3-one (7e, C16H10BrN3O)
Yield 2.61 g (77%); m.p.: 320 °C (DMF); R f = 0.63 (toluene–acetone 8:2); IR (KBr): \( \bar{v} = 3{,}433, \) 3,096, 2,848, 2,800, 1,691, 1,615, 1,529, 1,455, 1,369, 1,104, 781, 585 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 7.19–7.78 (m, 5H, Ar–H), 8.20–8.35 (m, 3H, Ar–H), 8.78 (s, 1H, C4–H), 12.08 (bs, 1H, NH) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 120.8, 121.2, 126.5, 126.9, 127.8, 129.5, 130.9, 131.9, 132.1, 134.7, 139.1, 141.5, 145.0, 148.6 ppm.
8-Bromo-1,2-dihydro-2-(2-hydroxyethyl)-3H-pyrazolo[4,3-c]quinolin-3-one (7f, C12H10BrN3O2)
Yield 2.24 g (73%); m.p.: 304 °C (DMF); R f = 0.74 (toluene–acetone 9:1); IR (KBr): \( \bar{v} = 3{,}400, \) 3,175, 3,095, 2,901, 1,690, 1,615, 1,554, 1,514, 1,460, 1,377, 1,355, 1,298, 1,185, 1,151, 1,108, 1,021, 959, 869, 825, 810, 645, 605 cm−1; 1H NMR (300 MHz, DMSO-d6): δ = 2.45 (bs, 1H, OH), 3.96 (t, J = 7 Hz, 2H, CH2), 4.09 (t, J = 7 Hz, 2H, OCH2), 7.65–8.10 (m, 3H, Ar–H), 8.93 (s, 1H, C4–H), 11.55 (bs, 1H, NH) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 58.2, 62.1, 122.1, 126.6, 128.1, 130.8, 131.9, 132.8, 135.2, 138.2, 145.0, 149.2 ppm.
References
Michael J (1997) Nat Prod Rep 14:605
Gebbhardt L, Bachtold J (1955) Proc Soc Exptl Biol Med 88:103
Balasubramanian M, Keay J (1996) In: Katritzky A, Rees C, Scriven E (eds) Comprehensive heterocyclic chemistry II, vol 5. Pergamon Press, Oxford, p 245
Bezuglyi P, Ukrainets I, Skaif N, Gorokhova O, Sidorenko L (2003) Farmakon 3:23
Roseman K, Gould M, Linfield M, Edwards B (1970) J Med Chem 13:230
Hinschberger A, Butt S, Lelong V, Boulouard M, Dumuis A, Dauphin F, Bureau R, Pfeiffer B, Renard P, Rault S (2003) J Med Chem 46:138
Gorlitzer K, Bode M, Jones P, Jomaa H, Wiesner J (2007) Pharmazie 62:15
Penning T, Talley J, Bertenshaw S, Carter J, Colins P, Graneton J, Lee L, Malecha J (1997) J Med Chem 40:1347
Menozzi G, Mosti L, Fossa P, Maltioli F, Ghia M (1997) J Heterocycl Chem 34:963
Naik R, Desai K (1998) Orient J Chem 14:161
Kidwai M, Negi N (1997) Monatsh Chem 128:85
El-Sayed O, El-Bieh F, El-Aqeel, Al-Bassam B, Hussein M (2002) Bull Chim Farm 141:461
Simrnoff P, Crenshaw R (1977) Antimicrob Agent Chemother 11:571, Chem Abstr (1977) 85:153844d
Stein R, Beil J, Singh T (1970) J Med Chem 13:153
Joshi A, Narkhede S, Viswanathan C (2005) Bioorg Med Chem Lett 15:73
Suresh T, Nandha Kumar R, Magesh S, Mohan P (2003) Ind J Chem 42B:2133
Suresh T, Nandha Kumar R, Magesh S, Mohan P (2003) Ind J Chem 42B:688
Dlugosz A, Dus D (1996) Farmaco 51:367
Selvi S, Nadaraj V, Mohan S, Sasi R, Hema M (2006) 14:3896
He Z, Milburn G, Baldwin K, Smith D, Danel A, Tomasik P (2000) J Lumin 86:1
He Z, Milburn G, Danel A, Puchala A, Tomasik P, Rasala D (1997) J Mater Chem 7:2323
Danel A, He Z, Milburn G, Tomasik P (1999) J Mater Chem 9:339
Jachak M, Tantak C, Toche R, Badgujar N (2004) Monatsh Chem 135:1529
Toche R, Ghotekar B, Kazi M, Kendre D, Jachak M (2007) Tetrahedron 63:8157
Jachak M, Kendre D, Avhale A, Toche R, Sabnis R (2007) J Heterocycl Chem 44:1525
Kendre D, Toche R, Jachak M (2008) J Heterocycl Chem 45:667
Jachak M, Avhale A, Ghotekar B, Kendre D, Toche R (2008) J Heterocycl Chem 45:1221
Toche R, Ghotekar B, Kendre D, Kazi M, Jachak M (2008) J Heterocycl Chem 45:1711
Kendre D, Toche R, Jachak M (2008) J Heterocycl Chem 45:1281
Shah K, Coats E (1977) J Med Chem 20:1001
Fryer R, Zhang P, Rios R, Gu Z, Basile A, Skolnick P (1993) J Med Chem 36:1669
Kendre D, Toche R, Jachak M (2007) Tetrahedron 63:11000
Bare T, McLaren C, Campbell J, Firor J, Resch J, Walters C, Salama A, Meiners B, Patel J (1989) J Med Chem 32:2561
Acknowledgments
The authors thank the University Grants Commission (UGC), New Delhi, India, for financial support and Professor D.D. Dhavale, Department of Chemistry, University of Pune, India for his encouragement and useful discussion.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ghotekar, B.K., Ghagare, M.G., Toche, R.B. et al. Synthesis of new quinoline fused heterocycles such as benzo[h]-1,6-naphthyridines and pyrazolo[4,3-c]quinolines. Monatsh Chem 141, 169–175 (2010). https://doi.org/10.1007/s00706-009-0236-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-009-0236-1