
Abstract
Since 1948, pale yellow wheat spike have been reported in southern Brazil. This symptom was associated with tenuiviruses due to the observation of cytoplasmic inclusions constituted by a mass of filamentous particles (7–10 nm in diameter) with indeterminate length, identical to those found in “leaf dip” preparations. Such symptoms are still seen in wheat crops; however, there is a lack of information regarding this pathosystem. Decades after the first report, the first sequences of wheat white spike virus were characterized. Wheat plants with symptoms such as pale yellowing, chlorotic streaks, and leaf mosaic were collected in Paraná State, Southern Brazil. High-throughput sequencing was used to determine the nearly complete nucleotide sequence of the viral genome. The genome is composed of five RNAs with a total size of 18,129 nucleotides, with eight open reading frames (ORFs). The virus identified in this study can be included in a new species in the family Phenuiviridae, genus Tenuivirus, and we have tentatively named this virus “wheat white spike virus”.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
In 1948, the first symptoms of white spike virus in wheat plants (Triticum aestivum L.) were reported in the Pelotas region of Rio Grande do Sul state, Brazil [1]. Since the 1970s, the viral etiology of this disease was determined, and its occurrence in other Brazilian states was demonstrated [2,3,4] through the observation of symptomatic leaf tissues by conventional and electron microscopy. In infected tissues, intracellular inclusions with a fibrous and twisted appearance were observed in the cytoplasm. These inclusions were formed by a mass of filamentous particles of 7–10 nm in diameter with indeterminate length, which is identical to those found in “leaf dip” preparations and similar to those in plants infected by viruses of the genus Tenuivirus [2, 5].
Symptoms observed in infected plants range from pale yellowing to whitening in bands on the leaves, chlorotic streaks, and striated mosaic, which can cause plant death before tillering. The spike of affected plants are partially and/or entirely pale yellow in color, showing malformation on the edges, no grains, and sometimes an abnormal downward twist. Due to the observed symptoms as well as the viral particle morphology, the causal agent was named “wheat white spike virus” by the researchers [2]. Although it was later referred to as “Brazilian wheat spike virus” (BWSpV) [6], it was more recently named “wheat white spike virus” [7]. Viruses of the genus Tenuivirus are transmitted by planthoppers in a persistent propagative manner. In a study by Costa in 1973 [8], planthoppers of the species Sogatella kolophon K. demonstrated the ability to transmit the virus associated with white spike symptoms in wheat. This virus has occurred with variable frequency in Brazilian wheat regions and is generally limited to crop edges and is mainly seen in experimental plots. However, larger crop areas have been affected recently in Paraná State, Brazil. Despite this, there is little information regarding this pathosystem.
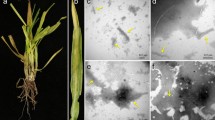
In this study we sequenced the genome of the virus causing wheat white spike disease using high-throughput sequencing (HTS). In 2018, a sample comprising leaves and stalks of symptomatic wheat plants of the cultivars TBIO Sinuelo, TBIO Itaipu, and BRS Reponte was collected in the municipality of Ponta Grossa, Paraná State, Brazil (25°00'50''S; 50°09'07''W; 886 m altitude) (Fig. 1A). The sample was stored in an ultrafreezer (−80°C) for subsequent extraction of double-stranded RNA (dsRNA), as described by Valverde et al. [9], with some modifications [10].

(A) Typical virus symptoms in different wheat cultivars (Triticum aestivum) (1, 2, and 3, wheat cultivars TBIO Sinuelo, TBIO Itaipu, and BRS Reponte, respectively) collected in Ponta Grossa, Paraná State, Brazil (25°00'50''S; 50°09'07''W; 886 m altitude) and symptom of white spike (4). (B) Schematic representation of the positions of open reading frames (ORFs) on the viral and viral-complementary sequences of RNA1, RNA2, RNA3, RNA4, and RNA5. The ORFs are represented by black bars. The proteins encoded by each ORF are shown as boxes, indicating the predicted molecular mass of the protein. (C) Two-dimensional plot representing the percentage of nucleotide sequence identity in RNA1, RNA2, RNA3, RNA4, and RNA5 of wheat white spike virus (WWSV) to those members of the genus Tenuivirus. Accession numbers are as follows: RNA1: WWSV, MZ_703097; MeCSV, NC_040450; RGSV, NC_002323; RHBV, NC_036597; RSV, NC_003755; EWSMV, MN_160329; RmSV, KR_094115; FSaV, MW_678790. RNA2: WWSV, MZ_703098; IWSV, NC_038748; MSpV, NC_038751; MeCSV, NC_040451; RGSV, NC_002324; RHBV, NC_036598; RSV, NC_003754; EWSMV, MN_160345; RmSV, KR_094116; FSaV, MW_678791. RNA3: WWSV, MZ_703099; EHBV, NC_038934; IWSV, NC_038750; MSpV, NC_038754; MeCSV, NC_040448; RGSV, NC_002325; RHBV, NC_036602; RSV, NC_003776; UHBV, NC_038757; EWSMV, MN_160360; RmSV, KR_698381; FSaV, MW_678792. RNA4: WWSV, MZ_703100; EHBV, NC_038935; MSpV, NC_038752; MeCSV, NC_040454; RGSV, NC_002326; RHBV, NC_036599; RSV, NC_003753; UHBV, NC_038758; IWSV, NC_038749; EWSMV, MN_160376; RmSV, KR_094117; FSaV, MW678793. RNA5: WWSV, MZ_703101; EHBV, NC_038936; MSpV, NC_038753; MeCSV, NC_040449; RGSV, NC_002327; RmSV, KR_094118
HTS sequencing reads were generated from a complementary DNA library (cDNA) using an Illumina HiSeq X Ten platform with paired-end 150-bp sequencing. The cDNA library quality was checked using FastQC software, and low-quality reads and adapters were removed using Trimmomatic [11].
For comparative analysis, the nucleotide (nt) sequences of the tenuiviruses echinochloa hoja blanca virus (EHBV), Iranian wheat stripe virus (IWSV), maize stripe virus (MSpV), melon chlorotic spot virus (MeCSV), rice grassy stunt virus (RGSV), rice hoja blanca virus (RHBV), urochloa hoja blanca virus (UHBV), rice stripe virus (RSV), and European wheat striate mosaic virus (EWSMV) as well as two tentative tenuiviruses, ramu stunt virus (RmSV) and festuca stripe-associated virus (FSaV), were used. Coding regions (ORFs) in the viral genome were identified using ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/) and compared using ClustalW, available in SDT version 1.2.
The alignments used for the phylogenetic analysis were performed using the MUSCLE tool available in the MEGA X program [12]. Phylogenetic trees were built by the maximum-likelihood method implemented in the MEGA X program, using the Tamura 3-paramenter and gamma distribution (G) model, with 9,000 and 5,000 bootstrap replications for RNA1 and other RNAs, respectively. Recombination analysis was performed using Recombination Detection Program version 5 (RDP5) [13]. Only recombination events detected by at least three of the methods available in the program with a p-value less than 0.05 were considered reliable. For detailed information on the methods used, see Supplementary Material. The sequences have been deposited in the NCBI database with the accession numbers MZ703097 (RNA1), MZ703098 (RNA2), MZ703099 (RNA3), MZ703100 (RNA4), and MZ703101 (RNA5). The sequence of the 5’ and 3’ ends of the RNA1 and RNA3 segments were determined using a Rapid Amplification of Complementary DNA (cDNA) Ends (RACE) Kit (Invitrogen) according to the manufacturer’s protocol, using the primers listed in Supplementary Table S1. The other segments may have a few more nucleotides, which is why we refer to the sequence as nearly complete. Fifty-five million reads were generated for the analyzed sample. After processing the reads, 39 contigs were identified as viral sequences. From these contigs, the nearly complete sequence corresponding to the viral genome was assembled.
The viral genome has five segments of single-stranded ribonucleic acid (RNA), which is characteristic of members of the order Bunyavirales [14]. RNAs 1–5 have 8,984, 3,602, 2,217, 1,864, and 1,447 nt, respectively. The genome has eight ORFs. Based on comparisons with other tenuiviruses, RNA1 and RNA5 have negative polarity, while RNA2, RNA3, and RNA4 are ambisense RNAs; that is, they encode proteins on the viral (5′–3′) and complementary (3′–5′) strands (Fig. 1B) [6].
Comparisons with members of the genus Tenuivirus showed differences in sequence similarity among the RNA segments. The RNA1 and RNA2 sequences are 73.5% and 71.7% identical to those of RHBV. The RNA3 sequence is 71% identical to those of EHBV, IWSV, and RHBV. RNA4 and RNA5 share 74.5% and 87% identity with IWSV and EHBV, respectively (Fig. 1C). The non-coding intergenic regions have nucleotide sequence identity values close to 60%, whereas the intergenic regions of RNA2, RNA3, and RNA4 have 63%, 60%, and 57.8% identity to the corresponding segments of MSpV, UHBV, and IWSV, respectively (Supplementary Fig. S1).
RNA1 encodes a 2,921-amino-acid (aa) protein with a possible replicase function. The protein contains the RNA-dependent RNA polymerase (RdRP) domain and has a predicted molecular mass of 336 kDa. Comparative analysis revealed that this protein shares 82% and 80% amino acid sequence identity with the RdRPs of FSaV and RHBV, respectively. RHBV was described in 1983, infecting rice in Colombia; and FSaV was described in 2021, infecting festuca in Germany [15,16,17]. The putative proteins encoded by RNA2 share 74% and 71% identity with the viral proteins pv2 of IWSV and pc2, encoded in the complementary sense of FSaV, respectively. The pv2 and pc2 proteins exhibit homology to glycoprotein precursors that interact with the endoplasmic reticulum [18]. RNA3 encodes two proteins, one with 80% identity to the NS3 protein of IWSV, which acts as a silencing suppressor in RHBV and RSV [19, 20], and one with 74% identity to the NSvc3 of RHBV, which, in the case of RSV, is involved in the virus-insect interactions [21]. RNA4 encodes a protein with 78% identity to the virus-encoded NS4 protein of IWSV and 80% identity to the viral complementary-strand-encoded pc4 protein. The NS4 protein is known to be involved in viral movement in plants [19, 20]. The pc4 protein is multifunctional and has been implicated in cell-to-cell movement, long-distance movement, and induction of leaf necrosis [22]. The protein encoded by RNA5 shares 84% identity with a hydrophilic protein encoded on the complementary-sense strand of EHBV RNA5, which could play a role in cell stability, replication, and movement, as suggested by De Miranda et al. [23] (Supplementary Fig. S2 and Table 1).
Phylogenetic analysis corroborated the sequence comparison data, in which RNA1–5 of the viral isolate characterized in this study showed close relationships to RHBV, IWSV, and EHBV (Supplementary Fig. S3). Intriguingly, based on the sequences of RNA1–5, the Brazilian isolates characterized in this study grouped with different viruses, depending on the RNA segment analyzed, suggesting that the novel virus characterized in this study has RNA segments of different origins, indicating a reassortment event. Reassortment is a shared feature of all segmented RNA viruses [24]. In addition, recombination analysis using the complete RNAs sequences detected a total of seven events involving members of the genus Tenuivirus. There is no evidence of recombination with the novel virus characterized in this study. WWSV was identified as a minor parent of EWSMV and RmSV (Supplementary Table S2). These results suggest that reassortment and recombination are both important mechanisms by which variability is introduced in members of the genus Tenuivirus.
The close relationship between RHBV and EHBV is consistent with their close geographic proximity, since RHBV and EHBV have been reported in Colombia and Costa Rica, infecting rice and Echinochloa colona, respectively [15, 23]. On the other hand, IWSV shares the same host with the novel virus characterized in this study. In addition, WWSV shows a close relationship to FSaV, both of which infect plants of the family Poaceae. Determination of a larger number of sequences of these viruses from different regions would provide precise information regarding their relationships and origins.
Genomic RNAs of members of the genus Tenuivirus range in size from about 9.0 kb to 1.3 kb. Altogether, these RNAs yield a total genome size of about 18–19 kb [25]. Viruses of the genus Tenuivirus have genomes with four or more segments; however, some members of the genus have not had all of their segments sequenced, such as UHBV, which has only two known sequences. The RNA1 of members of this genus is the largest segment, and it has only one coding region; however, for RNA2, RNA3, and RNA4, two coding regions have been described, except in MeCSV and RmSV, which have only one coding region each for both RNA4 and RNA5 [6, 26] (Supplementary Table S3). The virus characterized in this study has a genomic organization similar to that of members of the genus Tenuivirus, showing the most sequence similarity and closer phylogenetic relationships to members of this genus, which are associated with plants of the family Poaceae.
The current criteria for demarcating species in the genus are vector specificity, different ability to infect key plant species, different sizes and/or numbers of RNA segments, <85% aa sequence identity between any corresponding gene products, and <60% nt sequence identity between corresponding non-coding intergenic regions [6, 26]. Based on its genomic organization and comparative sequence analysis, the virus described in this study is most similar to RHBV, IWSV, FSaV, and EHBV; however, it has distinct molecular characteristics that would allow it to be considered a member of a new species in the genus Tenuivirus, and we propose the name “wheat white spike virus” in acknowledgement of the first published work referring to the disease caused by this virus in Brazil. To the best of our knowledge, this is the first report of the genome sequence of this virus, which is the first tenuivirus molecularly characterized in Brazil. The molecular data presented in this study are vital for the development of diagnostic tools and studies on the biology of the virus. More studies are needed to identify its vector and to estimate its prevalence and potential agronomic impact on wheat and other members of the family Poaceae in Brazil.
References
Deslandes JA (1949) Virus diseases in cereals in Pelotas. Agross 2:88–92
Caetano VR, Kitajima EW, Costa AS (1970) White spike disease of wheat, a possible virus disease. Bragantia 29:41–44
Caetano VR (1982) Wheat white spike virus. In: Osório EA (ed) Wheat in Brazil. Cargill Foundation, Campinas, pp 570–573
Silva AR, Kitajima WE, Caetano VR (1979) Ocorrência de espiga branca do trigo no Distrito Federal. Fitopatol Bras 4:152–153
Kitajima EW, Caetano VR, Costa AS (1971) Intracellular inclusions associated with the white spike of wheat. Bragantia 30:101–108
International Committee on Taxonomy of Viruses – ICTV (2021) https://talk.ictvonline.org/taxonomy/. Accessed 23 Jun 2021
Kitajima EW (2020) An annotated list of plant viruses and viroids described in Brazil (1926–2018). Biota Neotrop. https://doi.org/10.1590/1676-0611-bn-2019-0932
Costa AS, Caetano VR, Kitajima EW, Costa CL (1973) Transmission of the white ear by leafhoppers (Sogatella). In: VI Annual Meeting of the Brazilian Society of Plant Pathology, pp 1–2
Valverde RA, Nameth ST, Jordan RL (1990) Analysis of double-stranded RNA for plant virus diagnosis. Plant Dis 74:255–258
Valente JB, Pereira FS, Stempkowski LA et al (2019) A novel putative member of the family Benyviridae is associated with soilborne wheat mosaic disease in Brazil. Plant Pathol 68:588–600
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinf 30:2114–2120
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol and Evol 35:1547–1549
Martin DP, Varsani A, Roumagnac P et al (2021) RDP5: a computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol 7:veaa087
Maes P, Adkins S, Alkhovsky SV et al (2019) Taxonomia da ordem Bunyavirales: update 2019. Arch Virol 164:927–941
Morales FJ, Jennings PR (2010) Rice hoja blanca: a complex plantvirus-vector pathosystem. CAB Rev 5:1–16
Jimenez J, Carvajal-Yepes M, Leiva AM, Cruz M, Romero LE, Bolaños CA, Lozano I, Cuellar WJ (2018) Complete genome sequence of Rice hoja blanca virus isolated from a susceptible rice cultivar in Colombia. Genome Announc 6:e01490-e1517
Gaafar YZ, Rabenstein F, Zia A, Gaafar AR, Ziebell H (2021) Molecular characterisation of a new tenuivirus from Festuca sp. Virus Res. https://doi.org/10.1016/j.virusres.2021.198509
Zheng L, Du Z, Mao Q, Wu K, Wu J, Wei T, Wu Z, Xie L (2015) Rice stripe tenuivirus p2 may recruit or manipulate nucleolar functions through an interaction with fibrillarin to promote virus systemic movement. Mol Plant Pathol 16:921–930
Sõmera M, Kvarnheden A, Desbiez C et al (2020) Sixty years after the first description: genome sequence and biological characterization of European wheat striate mosaic Virus infecting cereal crops. Phytopathol 110:68–79
Xiong R, Wu J, Zhou Y, Zhou X (2009) Characterization and subcellular localization of an RNA silencing suppressor encoded by rice stripe tenuivirus. Virol 387:29–40
Liu W, Gray S, Huo Y, Li L, Wei T, Wang X (2015) Proteomic analysis of interaction between a plant virus and its vector insect reveals new functions of hemipteran cuticular protein. Moll Cell Proteomics 14:2229–2242
Zhang C, Pei X, Wang Z, Jia S, Guo S, Zhang Y, Li W (2012) The Rice stripe virus pc4 functions in movement and foliar necrosis expression in Nicotiana benthamiana. Virol 425:113–121
De Miranda JR, Muñoz M, Wu R, Espinoza AM (1996) Sequence of echinochloa hoja blanca tenuivirus RNA-5. Virus Genes 12:131–134
McDonald SM, Nelson MI, Turner PE, Patton JT (2016) Reassortment in segmented RNA viruses: mechanisms and outcomes. Nat Rev Microbiol 14:448–460
Falk BW, Tsai JH (1998) Biology and molecular biology of viruses in the genus Tenuivirus. Ann Rev Phytopathol 36:139–163
Walker PJ, Siddell SG, Lefkowitz EJ et al (2019) Changes to virus taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses. Arch Virol 164:2417–2429
Acknowledgements
The authors would like to thank Professor Murilo Zerbini for providing reagents and support in RACE analysis. This work was supported by EMBRAPA (project 32.16.04.037.00.00: Viral population analysis and management strategies for the common mosaic in wheat in Brazil), Fundação de Amparo à Pesquisa e Inovação do Estado de Santa Catarina (FAPESC – PAP) (Grant number: PAP2018), and UDESC. F.S.P. and S.C.N are Brazilian Federation for Support and Evaluation of Graduate Education (CAPES) fellows.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None of the authors have a conflict of interest. This article does not contain any studies with animals performed by any of the authors.
Additional information
Handling Editor: Massimo Turina.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pereira, F.S., Stempkowski, L.A., Fajardo, T.V.M. et al. A novel tenuivirus infecting wheat in Brazil. Arch Virol 167, 989–993 (2022). https://doi.org/10.1007/s00705-022-05361-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-022-05361-6