
Abstract
African swine fever (ASF), caused by African swine fever virus (ASFV), was first reported in Kenya in 1921, but an effective vaccine or antiviral drug is still not available for ASFV control. Rapid and effective diagnostics are key steps in managing ASF. We generated two monoclonal antibodies (MAbs) against the ASFV phosphoprotein P30 and designated these as 3H7A7 and 6H9A10. Epitope mapping revealed that MAb 3H7A7 and 6H9A10 recognized aa 144-154 and aa 12-18 of P30, respectively. A signal-amplified sandwich colloidal gold test strip for rapid detection of ASFV was developed based using these MAbs. Sensitivity and specificity analysis showed that the detection limit of the strip was 2.16 ng of P30. The strip only reacted with ASFV and did not react with other common porcine viruses. In detection tests using 153 clinical field samples including sera, plasma, anticoagulant-treated blood, and tissue, the strip had 95.42% concordance with real-time PCR. The new MAbs specific for P30 and the rapid colloidal gold test strip helped to reveal novel B cell epitopes in P30 and provide an efficient diagnostic test for on-site clinical detection of ASF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
African swine fever (ASF) is a hemorrhagic febrile disease caused by African swine fever virus (ASFV). The mortality rate in domestic pigs and wild boars infected with virulent strains of ASFV can reach 100% [1,2,3]. ASFV was introduced into Georgia in 2007 and spread quickly to neighboring countries [4,5,6]. ASFV transmission is now expanding from Eastern European to Middle European countries [7,8,9,10]. In August 2018, ASF was first reported in Liaoning [11, 12], and the disease then spread to other provinces in China. There is no available vaccine or antiviral drug available to control ASF [13, 14], so the ASF epidemic in China has resulted in severe economic losses.
To control ASF, rapid and accurate diagnosis, followed by effective measures such as restriction of animals and vehicle traffic, delimitation of epidemic areas, and treatment of infected pigs, is needed in epidemic countries. The diagnostic methods for ASFV currently recommended by the World Organization for Animal Health (OIE) include virus isolation as well as detection of viral antigen by immunofluorescence assay (IFA), DNA by polymerase chain reaction (PCR), and antibodies by enzyme-linked immunosorbent assay (ELISA) [15]. Although these diagnostic approaches are feasible, a rapid and pen-side approach for detecting viral antigen would be more practical and could identify pigs suspected of being infected with ASFV. Sastre et al. developed a lateral flow method for detecting the P72 protein of ASFV in blood, and a result was obtained 10 min after adding the sample. The sensitivity of the strip was somewhat better than that of the commercial antigen detection kit, the Double Antibody Sandwich (DAS) ELISA test manufactured by INGENASA, but there were many false negatives compared to real-time PCR. This was true even for samples for which the Ct value was less than 25, which were easily detected by standard PCR [16]. P30, encoded by ORF CP204L of ASFV, is an important and relatively invariant capsid protein [17, 18]. In virions, the amount of P30 is lower than that of VP72, the major viral capsid protein. However, P30 is abundant in the cytoplasm of infected cells [19]. Therefore, P30 is a good target for detecting the presence of an ASFV infection.
In the present study, two novel monoclonal antibodies, recognizing different epitopes of P30, were developed and used to produce an efficient colloidal gold test strip for detection of ASFV.
Materials and methods
Ethics statement
Six-week-old female BALB/c mice were purchased from the Institute of Comparative Medicine, Yangzhou University (Yangzhou, China). Three-month-old healthy pigs were purchased from Jiangsu Academy of Agricultural Sciences (Nanjing, China). All applicable national and institutional guidelines for the care and use of animals were followed to minimize suffering. Animal procedures were approved by the Animal Care and Use Committee of Yangzhou University.
Viruses and clinical samples
Viruses, including pseudorabies virus (PRV) Bartha-K61, classical swine fever virus (CSFV), reproductive and respiratory syndrome virus (PRRSV), senecavirus type A (SVA), porcine circovirus type 2 (PCV-2), porcine epidemic diarrhea virus (PEDV), porcine transmissible gastroenteritis virus (TGEV), and porcine parvovirus (PPV), were preserved in our laboratory. Clinical samples from domestic pigs (for details, see Table 1) and ASFV strain SY18 were provided by the China Animal Health & Epidemiology Center (Qingdao, China).
Production of monoclonal antibodies against P30
Based on the genome sequence of ASFV strain BA71V (GenBank accession no. U18466.1), a codon-optimized p30 gene designed using the JAVA Codon Adaption Tool was synthesized and cloned into the plasmid pET-30a. The recombinant P30 with a 6×His tag was expressed in E. coli BLR (DE3) cells, induced by adding isopropyl-β-d-thiogalactopyranoside (IPTG, Sangon Biotech, China) at a concentration of 1 mM and purified using Ni-NTA agarose (QIAGEN, Germany). After verification by Western blot, using an anti-His-Tag MAb, 50 μg of the purified recombinant P30 protein was mixed with an equal volume of complete Freund’s adjuvant and incomplete Freund’s adjuvant (Sigma, St. Louis, MO, USA) and inoculated into 6-week-old BALB/c mice at 10-day intervals. On day 3 after the immunization, the mice were euthanized, and the splenocytes were collected, fused with mouse myeloma cell line Sp2/0 (Mingzhou Biotech, China), and cultured in 96-well plates in HAT selection medium. At 9 days post-fusion, the supernatants of fused cells were screened by indirect ELISA. The hybridoma cells in positive wells were subcloned twice to isolate individual clones and verified again by indirect ELISA. For preparation of ascites, female BALB/c mice were injected intraperitoneally with individual hybridoma cell lines (106 cells/mouse) at 7 days after pretreatment with liquid paraffin. Five days later, the ascites were collected and centrifuged for 5 min at 10,000 g. Before use, the MAbs were purified by ammonium sulfate precipitation and dialyzed three times in PBS as described previously [20].
Indirect ELISA
Indirect ELISA was performed as described previously [21]. Briefly, 10 ng of the recombinant P30 per mL in carbonate buffered saline was coated onto the wells of high-binding 96-well assay plates (Corning, USA) overnight at 4°C. The plates were washed twice with 0.01 M PBS, pH 7.4, containing 0.05% Tween 20 (PBST) and blocked with 200 μL/well of PBST with 5% nonfat dry milk for 1 h at 37°C, then washed twice with PBST. Supernatants (100 μL/well) were added and incubated for 1 h at 37°C, followed by three washes with PBST. A 1/5,000 dilution of HRP-conjugated goat anti-mouse IgG antibody (Abcam, USA) in blocking solution was dispensed into the plates at 100 μL/well and incubated for 1 h at 37°C. The plates were washed three times with PBST, 100 μL of 3,3,5,5-tetramethyl benzidine substrate (Sangon Biotech, China) was added to each well, and the reaction was stopped with 50 μL of 2 M sulfuric acid per well. The antibody level was measured 10 min later using a microplate reader (Bio Tek, USA).
Immunofluorescence assay
The immunofluorescence assay was performed as described previously [22]. Briefly, the p30 gene was subcloned into the plasmid pcDNA3.0 (Invitrogen, USA) to construct recombinant plasmid pcDNA-p30. PK15 cells (ATCC® CCL-33™) were transfected with pcDNA3-p30 and cultured for 48 h after transfection. Porcine alveolar macrophages (PAMs) were collected from the lungs of healthy pigs and cultured as described previously [23]. The PAMs were infected with ASFV strain SY18 (Genotype II) at an MOI of 1.0 and cultured for 36 h. The transfected and infected cells were fixed with 4% paraformaldehyde (Sangon Biotech, China) for 10 min, and then washed with PBS. A 0.2% (v/v) Triton X-100 (Sigma, USA) solution diluted in PBS was then applied to each well for 10 min. After washing three times with PBS, the cells were overlaid with PBS containing 2.5% (w/v) bovine serum albumin (Sangon Biotech, China) and incubated for 30 min at 37 °C. The supernatants of hybridomas were used as the primary antibody. After 45 min of incubation at 37 °C, the cell monolayers were washed three times with PBS. The cells were then stained using diluted goat anti-mouse IgG conjugated with FITC (KPL, America) and incubated for an additional 45 min at 37 °C. The stained cells were rinsed with PBS and examined under a fluorescence microscope (Nikon, Japan).
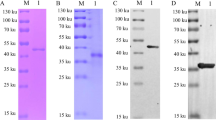
Mapping of epitopes by Western blot analysis
The different lengths of DNA fragments of the p30 gene were subcloned into pET-30a, and the truncated recombinant P30 proteins listed in Fig. 1A were expressed in E. coli BLR (DE3) cells induced with IPTG. The cells were lysed in SDS loading buffer at 95°C for 10 min before being run on 5%–12% Bis-Tris mini gels. The denatured proteins were then transferred onto nitrocellulose membranes (Amersham, USA), treated with blocking solution for 1 h at 37°C, and washed three times with PBST. The membranes were the incubated with the supernatants from the hybridomas for 1 h at 37°C, washed three times with PBST, and immersed in diluted DyLight 800–conjugated goat anti-mouse IgG antibody (KPL, USA) for 1 h at 37°C. After washing four times with PBST, the blots were scanned using an Odyssey infrared imaging system (LI-COR, Lincoln, NE, USA).

Detection of P30 expressed in cells by IFA. (A and B) Expressed P30 reacted with MAbs 6H9A10 and 3H7A7 in PK15 cells transfected with pcDNA-p30. The p30 gene was from Ba71V, genotype I of ASFV. (D and E) Expressed P30 reacted with MAbs 6H9A10 and 3H7A7 in PAM cells infected with SY18 strain (genotype II of ASFV). (C and F) Negative controls of PK15 cells and PAM cells
Synthesis of colloidal gold
A 750-μL amount of 10% HAuCl4 (Aladdin, China) solution was added to 500 mL of ultrapure water in a clear siliconized beaker on a magnetic stirrer and boiled for 3 min. A 900-μL amount of 10% trisodium citrate (Aladdin, China) solution was dropped into the boiled solution, stirred, and heated continuously for 5 min until the color changed to red-purple. The resulting colloidal gold solution was stirred continuously and cooled in ice water.
MAb biotinylation
A 2-mg quantity of NHS-biotin was dissolved in 590 μL of dimethyl sulfoxide (DMSO) to make a NHS-biotin solution. One mL of the purified MAb at a concentration of 2 mg/mL and 15 μL of NHS-biotin solution were mixed, vortexed, incubated at room temperature for 30 min, and dialyzed three times in PBS at 4 °C.
Preparation of the colloidal gold–biotinylated MAb conjugate
A 100-mL quantity of colloidal gold solution was added to a clear siliconized beaker on a magnetic stirrer, and stirred until the solution reached room temperature, using an appropriate volume of 0.2 M K2CO3 to adjust the pH to 8.5. Then, 1 mL of biotinylated MAb was added to the colloidal gold solution with gentle stirring for 30 min. Five mL of 10% bovine serum albumin (BSA, Solarbio, China) was added to the solution, which was gently stirred for an additional 30 min. The solution was centrifuged at 10,000 rpm for 20 min at 4 °C, the precipitate was resuspended in 10 mL of re-dissolved solution (0.23% Tris, 0.25% casein, 2% sucrose, 1% trehalose, and 0.2% Triton X-100, pH 8.0), and stored at 4 °C.
Preparation of the conjugate pad, sample pad, and nitrocellulose membrane
The colloidal gold–biotinylated MAb conjugate was added to a glass fiber (JieYi Biotech, China) with a volume of 100 μL/cm as a conjugate pad and dried at 37 °C for 12 h. The glass fiber used as the sample pad was incubated with the solution (0.6% Tris, 0.5% casein, 0.5% PVP30, 1% Triton X-100, 0.5% Tetronic 1307 M, 0.25% NaCl, 0.5% sucrose, and 3% streptavidin, pH 8.0) and dried at 45°C for 12 h. Another MAb at a concentration of 1.2 mg/mL and goat anti-mouse IgG antibody at a concentration of 1.5 mg/mL (Huaan Biotechnology, China) were dispensed onto the NC membrane as the test line (T line) and control line (C line) using a ZX 1000 BioDot dispenser system (BioDot, USA). The two lines were approximately 0.5 cm apart.
Assembly of the colloid gold test strip
The colloid gold immunochromatographic strip was composed of a gold–biotinylated MAb conjugate pad, a sample pad, an absorbent pad, and an NC membrane. All of these were pasted onto an adhesive plastic backing using previously described methods with slight modifications [24]. Briefly, the NC membrane was pasted in the center of the backing plate, the conjugate pad was pasted by overlapping 1 mm on the bottom of the NC membrane, the sample pad was pasted by overlapping 2 mm on the bottom of the conjugate pad, and the absorbent pad was pasted by overlapping 1 mm on the upper position of the NC membrane. The whole assembled strip was cut lengthwise into 3-mm-wide strips using a guillotine cutter, and then installed on the test card. The strip was stored with a desiccant in a sealed foil pouch.
Colloid gold strip test procedure
The field tissue samples (diluted 1:10 in PBS) listed in Table 1, including lymph node, spleen, and liver, were homogenized thoroughly using a tissue homogenizer. The homogenates were centrifuged at 10,000 rpm for 1 min, and the supernatants were used for detection. Anticoagulated blood was diluted 1:10 with PBS before testing, but serum and plasma were tested directly without extra treatment. Before loading specimens, the pouch was allowed to reach temperature before opening, and the test strip was taken from the foil pouch. A 30-μL processed specimen was transferred into the specimen well (S), and 60 μL of loading buffer (0.01 M PBS, pH 7.4, and 1% Triton X-100) was added to the specimen well. The results were checked 5–7 minutes after dispensing the specimen. A positive sample was recognized by the appearance of two red lines, one in the test region (T) and one in the control region (C), whereas negative samples only had a red line in the control region (C) and lacked a red line in the test region (T).
Evaluation of the immunochromatographic strip
To test the sensitivity of the strip, purified recombinant P30 protein was diluted to different concentrations (72 μg/mL, 7.2 μg/mL, 720 ng/mL, 72 ng/mL, and 7.2 ng/mL) as a standard antigen to determine the limit of detection; PBS was used as a negative control. For specificity analysis, different swine viruses (> 105.5 TCID50/mL) including PRV, CSFV, PRRSV, SVA, PCV-2, PEDV, TGEV and PPV were applied to the strip. For the stability assay, the strips were stored for 12 months at room temperature (25°C) and at 4°C and were then used to test 30 μL of recombinant P30 at a concentration of 72 ng/ml (minimum limit of detection); PBS was the negative control.
Testing of clinical samples
A total of 153 field samples, including serum, plasma, anticoagulated blood, and tissues collected from pigs with or without clinical symptoms, were tested using the strips, following the procedure described above. These samples were also tested using a commercial ASFV real-time PCR detection kit (Lijian Diagnostic Technology Development Center, China) for comparison.
Results
Production of two novel MAbs against P30 of ASFV
To generate MAbs against the P30 protein of ASFV, mice were immunized with purified recombinant P30, the splenocytes were fused with Sp2/0 myeloma cells, and two lines of hybridoma cells secreting antibodies against P30 were detected by indirect ELISA. The MAbs secreted by the hybridomas were designated as 3H7A7 and 6H9A10, respectively. An immunofluorescence assay was used to verify the reactivity of the Mabs. Bright green fluorescence in the cytoplasm of PK15 cells transfected with p30 and PAM cells infected with ASFV demonstrated that these two Mabs not only reacted with P30 of ASFV BA71V but also with P30 of the prevalent ASFV strain (Fig. 1).
Antigenic epitopes recognized by the MAbs
Epitope mapping was performed by Western blot using truncated recombinant P30 proteins expressed in E. coli. The results (Fig. 2) indicated that MAb 3H7A7 and 6H9A10 could efficiently recognize aa 144-154 (TVQHIEQYGKA) and aa 12-18 (EVIFKTD) of P30, respectively. Sequence analysis by MegAlign in DNAstar 8.0 revealed that the two linear epitopes recognized by MAb 3H7A7 and 6H9A10 were highly invariant between ASFV genotype I (BA71V; U18466.2) and genotype II (SY18; MH766894.1). The characteristics of the two MAbs against P30 allow development of efficient diagnostics for ASFV.

Epitope identification using monoclonal antibodies. (A) The truncated fragments of P30 expressed in E. coli used for epitope mapping. (B and C) Epitope regions (dotted box and black box in A) of P30 recognized by MAb 3H7A7 and 3H9A10 using Western blot
Establishment of a colloidal gold test strip for ASFV
To develop a rapid and efficient diagnostic test for ASFV, a novel colloidal gold test strip was generated using MAb 3H7A7 and Mab 6H9A10. In the test strip, MAb 6H9A10 was biotinylated and labeled with colloid gold. MAb 3H7A7 was dispensed on the NC membrane to capture the P30 antigen. When the sample was added to the sample pad, free P30 or P30 released from infected cells by treatment with the nonionic detergent Triton X-100 migrated into the conjugate pad and was captured by the biotinylated 6H9A10-colloidal gold conjugate. The binding of streptavidin from the sample pad at a ratio of 1:4 to the biotin coupled to MAb 6H9A10 amplified the signal of the colloidal gold. The complex on the NC membrane, captured by MAb 3H7A7, was the T line, and the excess colloid gold compound captured by the anti-mouse IgG antibody was the C line.
Sensitivity of the colloidal gold strip
To evaluate the sensitivity of the strip, the purified recombinant P30 was serially diluted in tenfold steps. The strip was able to detect as little as 2.16 ng of P30 (Fig. 3)

Minimum detection limit of the colloidal gold test strip. Recombinant P30 concentrations of 2.16 μg/mL, 216 ng/mL, 21.6 ng/mL, 2.16 ng/mL and 216 pg/mL were tested using colloid gold test strips. PBS was the control.
Specificity of the colloidal gold strip
Common swine viruses, including PRV, CSFV, PRRSV, SVA, PCV-2, PEDV, TGEV and PPV present in the supernatants of cultured cells and viral proteins in lysates of infected cells were tested using the strip. The strip did not react with any of the common viruses or viral products, indicating that the test is highly specific (Fig. 4).

Specificity of the colloidal gold test strip. (A) Supernatants containing virions of PRV, CSFV, PRRSV, SVA, PCV-2, PEDV, TGEV and PPV tested using the colloid gold test strips. (B) Lysed cells infected with PRV, CSFV, PRRSV, SVA, PCV-2, PEDV, TGEV and PPV tested using the strips.
Stability of the colloidal gold strip
To evaluate the stability of the colloidal gold strip, strips from the same batch were stored at room temperature (25 °C) or 4 °C for 12 months. Thirty μL of recombinant protein P30 at a concentration of 72 ng/mL was tested using strips that had been kept at the above temperatures. The strips were found to be stable for at least 1 year at room temperature or 4 °C, which makes them suitable for practical applications (Fig. 5).

Stability of the colloidal gold test strip. (1 and 2) 30 μL of the recombinant P30 at a concentration of 72 ng/mL and PBS tested using strips stored at 4°C for 12 months. (3 and 4) 30 μL of the recombinant P30 and PBS tested using strips stored at room temperature (25 °C) for 12 months
Applicability of the colloidal gold test strip for clinical samples
To evaluate the applicability of the strip for testing clinical samples, 100 sera and 20 lymph nodes collected in 2012 before the outbreak of ASF in China and 33 samples including serum, plasma, anticoagulated blood, spleens, lymph nodes and livers collected in 2018–2019 after the outbreak of ASF in China were tested using the new procedure. Four sera collected in 2012 gave false positive results, and three sera collected in 2018–2019 gave false negative results in the strip test compared with real-time PCR (Table 1). Further analysis showed that 119 out of 123 negative samples were negative using the strip, resulting in a specificity of 96.75%. Of the 30 samples that were positive by real-time PCR, 27 were positive in the strip test. These results indicate a sensitivity of 90%. The three samples that gave a false negative result in the strip test had real-time PCR values higher than 32.45. Out of all the clinical samples, the results for 146 samples were the same in both tests, indicating 95.42% concordance between the strip and real-time PCR. These data demonstrate that the colloidal gold test strip can be used efficiently for detecting the P30 antigen of ASFV in clinical samples.
Discussion
No commercial vaccine or antiviral drug is available to efficiently prevent or treat ASF. Rapid and efficient diagnostics are critical for identifying infected pigs, monitoring the infection, and tracking the spread of the disease. Although real-time PCR has been widely used in diagnosing ASF in the laboratory [25], it is poorly accepted by small farms due to its equipment expense, operator training requirements, and frequent aerosol contamination. For these reasons, development of an easier diagnostic device is helpful for early identification of ASF. The p30 gene of ASFV is transcribed at an early stage of virus proliferation [26]. Some of the expressed products of p30 are packed into viral particles, but most remain in the cytoplasm of infected monocytes and macrophages. P30 can be released from infected cells to the blood during infection [17]. Therefore, P30 is an appropriate target in the development of diagnostic methods for ASFV. In this study, two MAbs against P30 (3H7A7 and 6H9A10) were generated, and the linear epitope regions (aa 144-154 and aa 12-18) recognized by these MAbs were different from a linear epitope (aa 61-93) and a conformational epitope (aa 120-204) reported previously [27]. A novel immunochromatography colloidal gold test strip was established for detection of ASFV based on MAbs 3H7A7 and 6H9A10. The experimental data showed that the efficiency of the strip is comparable to real-time PCR. The strip has high specificity and sensitivity for testing different kinds of clinical samples, including serum, plasma, anticoagulated blood, and tissue homogenates, and all of the positive samples with Ct values ≤ 32.45 in PCR were detected correctly. A previously reported lateral flow assay can only be used to detect P72 of ASFV in anticoagulated blood, and the CT values of 10 of 17 false-negative field samples were less than 30 [16]. These data indicate that P30 may be a more suitable antigen for diagnosis of ASF.
During the detection experiments, we attempted to estimate the ratio of P30 and copies of ASFV. We found that a plasma sample with a Ct value of 30.42 gave a weak detection signal on the strip, but serum and anticoagulated blood with Ct values of 32.45 and 32.34 gave strong signals on the strip. These results demonstrate that there is no obvious linear relationship between the amount of P30 released from cells and the number of free viral particles. However, it was better to assay fresh or frozen samples than samples kept at 4°C for more than 48 h. This might relate to the potential degradation of P30 in infected cells by an activated lysosome or ubiquitin system.
The strip did not efficiently identify pigs with low levels of viremia (Ct values in real-time PCR > 32.45), but dead or diseased pigs with clinical signs could be diagnosed efficiently (100%). Its high specificity (96.75%) and sensitivity (90%) relative to real-time PCR makes the strip suitable for pen-side detection, especially for distinguishing ASF from other common swine diseases. The positive diagnostic results rapidly provided by the strip can allow producers to implement immediate ASF control measures.
References
Sanchez-Vizcaino JM, Mur L, Gomez-Villamandos JC, Carrasco L (2015) An update on the epidemiology and pathology of African swine fever. J Comp Pathol 152(1):9–21. https://doi.org/10.1016/j.jcpa.2014.09.003
Sanchez-Cordon PJ, Montoya M, Reis AL, Dixon LK (2018) African swine fever: a re-emerging viral disease threatening the global pig industry. Vet J 233:41–48. https://doi.org/10.1016/j.tvjl.2017.12.025
Dixon LK, Sun H, Roberts H (2019) African swine fever. Antivir Res 165:34–41. https://doi.org/10.1016/j.antiviral.2019.02.018
Rowlands RJ, Michaud V, Heath L, Hutchings G, Oura C, Vosloo W, Dwarka R, Onashvili T, Albina E, Dixon LK (2008) African swine fever virus isolate, Georgia, 2007. Emerg Infect Dis 14(12):1870–1874. https://doi.org/10.3201/eid1412.080591
Gogin A, Gerasimov V, Malogolovkin A, Kolbasov D (2013) African swine fever in the North Caucasus region and the Russian Federation in years 2007–2012. Virus Res 173(1):198–203. https://doi.org/10.1016/j.virusres.2012.12.007
Kolbasov D, Titov I, Tsybanov S, Gogin A, Malogolovkin A (2018) African Swine fever virus, Siberia, Russia, 2017. Emerg Infect Dis 24(4):796–798. https://doi.org/10.3201/eid2404.171238
Costard S, Mur L, Lubroth J, Sanchez-Vizcaino JM, Pfeiffer DU (2013) Epidemiology of African swine fever virus. Virus Res 173(1):191–197. https://doi.org/10.1016/j.virusres.2012.10.030
Gallardo C, Fernandez-Pinero J, Pelayo V, Gazaev I, Markowska-Daniel I, Pridotkas G, Nieto R, Fernandez-Pacheco P, Bokhan S, Nevolko O, Drozhzhe Z, Perez C, Soler A, Kolvasov D, Arias M (2014) Genetic variation among African Swine fever genotype II viruses, Eastern and Central Europe. Emerg Infect Dis 20(9):1544–1547. https://doi.org/10.3201/eid2009.140554
Boklund A, Cay B, Depner K, Földi Z, Guberti V, Masiulis M, Miteva A, More S, Olsevskis E, Šatrán P, Spiridon M, Stahl K, Thulke HH, Viltrop A, Wozniakowski G, Broglia A, Cortinas Abrahantes J, Dhollander S, Gogin A, Verdonck F, Amato L, Papanikolaou A, Gortázar C (2018) Epidemiological analyses of African swine fever in the European Union (November 2017 until November 2018). EFSA J. https://doi.org/10.2903/j.efsa.2018.5494
Cwynar P, Stojkov J, Wlazlak K (2019) African Swine fever status in Europe. Viruses. https://doi.org/10.3390/v11040310
Ge S, Li J, Fan X, Liu F, Li L, Wang Q, Ren W, Bao J, Liu C, Wang H, Liu Y, Zhang Y, Xu T, Wu X, Wang Z (2018) Molecular characterization of African Swine Fever virus, China, 2018. Emerg Infect Dis 24(11):2131–2133. https://doi.org/10.3201/eid2411.181274
Zhou X, Li N, Luo Y, Liu Y, Miao F, Chen T, Zhang S, Cao P, Li X, Tian K, Qiu HJ, Hu R (2018) Emergence of African Swine Fever in China, 2018. Transbound Emerg Dis 65(6):1482–1484. https://doi.org/10.1111/tbed.12989
Sanchez EG, Perez-Nunez D, Revilla Y (2019) Development of vaccines against African swine fever virus. Virus Res 265:150–155. https://doi.org/10.1016/j.virusres.2019.03.022
Teklue T, Sun Y, Abid M, Luo Y, Qiu HJ (2020) Current status and evolving approaches to African swine fever vaccine development. Transbound Emerg Dis 67(2):529–542. https://doi.org/10.1111/tbed.13364
OIE (2020) World Organisation for Animals Health. Terrestrial Manual Chapter 3.8.1: African swine fever (Infection with African swine fever virus). https://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/3.08.01_ASF.pdf. Accessed 12 Jun 2020
Sastre P, Gallardo C, Monedero A, Ruiz T, Arias M, Sanz A, Rueda P (2016) Development of a novel lateral flow assay for detection of African swine fever in blood. BMC Vet Res 12:206. https://doi.org/10.1186/s12917-016-0831-4
Afonso CL, Alcaraz C, Brun A, Sussman MD, Onisk DV, Escribano JM, Rock DL (1992) Characterization of p30, a highly antigenic membrane and secreted protein of African swine fever virus. Virology 189(1):368–373. https://doi.org/10.1016/0042-6822(92)90718-5
Dixon LK, Chapman DA, Netherton CL, Upton C (2013) African swine fever virus replication and genomics. Virus Res 173(1):3–14. https://doi.org/10.1016/j.virusres.2012.10.020
Alejo A, Matamoros T, Guerra M, Andres G (2018) A proteomic Atlas of the African swine fever virus particle. J Virol. https://doi.org/10.1128/JVI.01293-18
Kohler G, Milstein C (1976) Derivation of specific antibody-producing tissue culture and tumor lines by cell fusion. Eur J Immunol 6(7):511–519. https://doi.org/10.1002/eji.1830060713
Myint O, Yoshida A, Sekiguchi S, Van Diep N, Fuke N, Izzati UZ, Hirai T, Yamaguchi R (2019) Development of indirect enzyme-linked immunosorbent assay for detection of porcine epidemic diarrhea virus specific antibodies (IgG) in serum of naturally infected pigs. BMC Vet Res 15(1):409. https://doi.org/10.1186/s12917-019-2123-2
Zhang X, Ren D, Li T, Zhou H, Liu X, Wang X, Lu H, Gao W, Wang Y, Zou X, Sun H, Ye J (2018) An emerging novel goose astrovirus associated with gosling gout disease, China. Emerg Microb Infect 7(1):152. https://doi.org/10.1038/s41426-018-0153-7
Carrascosa AL, Bustos MJ, de Leon P (2011) Methods for growing and titrating African swine fever virus: field and laboratory samples. Curr Protoc Cell Biol. https://doi.org/10.1002/0471143030.cb2614s53
Yu X, Wei L, Chen H, Niu X, Dou Y, Yang J, Wang Z, Tang Y, Diao Y (2018) Development of colloidal gold-based immunochromatographic assay for rapid detection of goose parvovirus. Front Microbiol 9:953. https://doi.org/10.3389/fmicb.2018.00953
Tignon M, Gallardo C, Iscaro C, Hutet E, Van der Stede Y, Kolbasov D, De Mia GM, Le Potier MF, Bishop RP, Arias M, Koenen F (2011) Development and inter-laboratory validation study of an improved new real-time PCR assay with internal control for detection and laboratory diagnosis of African swine fever virus. J Virol Methods 178(1–2):161–170. https://doi.org/10.1016/j.jviromet.2011.09.007
Prados FJ, Vinuela E, Alcami A (1993) Sequence and characterization of the major early phosphoprotein p32 of African swine fever virus. J Virol 67(5):2475–2485
Petrovan V, Yuan F, Li Y, Shang P, Murgia MV, Misra S, Rowland RRR, Fang Y (2019) Development and characterization of monoclonal antibodies against p30 protein of African swine fever virus. Virus Res 269:197632. https://doi.org/10.1016/j.virusres.2019.05.010
Funding
This research was funded by the National Project for Prevention and Control of Transboundary Animal Diseases (Grant no. 2017YFD0501805), the National Key R&D Program for the 13th Five-Year Plan, and the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Contributions
XZ and XL performed most of the work; XW provided the field samples, the ASFV SY18 strain, and workspace in the biosafety level III laboratory; WR and YZ were responsible for testing field samples; XX helped in material preparation; and HS gave experimental instruction.
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that they have no competing interests regarding the publication of the data from this study.
Additional information
Handling Editor: Patricia Aguilar.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, X., Liu, X., Wu, X. et al. A colloidal gold test strip assay for the detection of African swine fever virus based on two monoclonal antibodies against P30. Arch Virol 166, 871–879 (2021). https://doi.org/10.1007/s00705-020-04915-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-020-04915-w