
Abstract
Macrophomina phaseolina is a pathogenic fungus of the family Botryosphaeriaceae that causes stem rot or leaf blight in many economically important plants. Mycoviruses exist widely in fungi, but there are only a limited number of reports on mycovirus infection in M. phaseolina. A novel dsRNA virus, tentatively named “Macrophomina phaseolina fusagravirus 1” (MpFV1), was isolated from strain 2012-19 of M. phaseolina, and its molecular features were examined. The full-length cDNA of MpFV1 comprises 9,289 nucleotides with a predicted GC content of 48.1% and two discontinuous open reading frames (ORF 1 and 2). A-1 frameshift region with two typical factors, including a shifty heptamer (GGAAAAC) and an H-type pseudoknot, was predicted in the junction region of ORF1 and ORF2. The protein encoded by ORF1 shows significant similarity to a hypothetical protein, whereas ORF2 encodes an RNA-dependent RNA polymerase (RdRp) via a ribosomal frameshifting mechanism. Homology searches and phylogenetic analysis based on the RdRp sequence suggested that MpFV1 is a new member of the proposed family “Fusagraviridae”.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mycoviruses infect fungi and replicate in fungal cells. They are ubiquitous in various types of fungi, including endophytic, medical, entomopathogenic, and phytopathogenic fungi [1,2,3,4,5]. Most mycoviruses have a double-stranded RNA (dsRNA), positive-stranded RNA (+ssRNA), or negative-stranded RNA (-ssRNA) genome [6,7,8], but a single-stranded circular DNA virus has been reported in filamentous fungi [9]. Mycoviruses with a dsRNA genome are classified into six recognized families whose members have different numbers of genome segments, including Reoviridae (11–12 segments), Chrysoviridae (4–5 segments), Quadriviridae (4 segments), Megabirnaviridae (2 segments), Partitiviridae (2 segments), and Totiviridae (1 segment) [10]. Along with the development of viral metatranscriptomics techniques, the number of studies on virology, especially those on discovering novel viruses is increasing [11,12,13,14]. Newly discovered mycoviruses have been shown to have unique molecular and biological properties, and new families such as “Botytiviridae” and “Fusagraviridae” have been proposed to accommodate these unassigned mycoviruses, which share molecular features with, but are significantly different from, other known mycoviruses [15,16,17,18].
Macrophomina phaseolina, belonging to the ascomycete family Botryosphaeriaceae, is an important destructive necrotrophic fungus [19] that is capable of infecting over 500 plant species worldwide, including the important oil crops sesame (Sesamum indicum) and soybean (Glycine max) [20]. The typical symptom of sesame plants infected by M. phaseolina is charcoal rot; the fungus forms spindle-shaped lesions with a dark border and light-gray center that are covered with black pinhead-sized pycnidia and microsclerotia. This severe disease can kill plants and cause huge losses in annual yield [21]. Compared with other phytopathogenic fungi such as Sclerotinia sclerotiorum and Fusarium graminearum, mycoviruses or dsRNA elements have been rarely reported in M. phaseolina. Previous reports have suggested that approximately 21.7% of M. phaseolina strains isolated from diseased Cyamopsis tetragonoloba plants harbor dsRNA elements, but sequence data for these putative viruses have not been obtained [22]. A high-throughput-sequencing-based metatranscriptomics approach has recently been utilized to identify virus-related sequences in M. phaseolina strains isolated from diseased soybean plants. Eleven novel viruses that were temporarily assigned to five distinct families, including Hypoviridae, Narnaviridae, Virgaviridae, Tombusviridae, and Chrysoviridae, and unsigned -ssRNA viruses belonging to the order Bunyavirales have been discovered [12].
Here, we report the isolation of a new mycovirus, Macrophomina phaseolina fusagravirus 1 (MpFV1), isolated from the strain 2012-19 of M. phaseolina. Genomic characterizations of MpFV1 allowed the elucidation of its tentative taxonomic position. MpFV1 is suggested to belong to the proposed family “Fusagraviridae”, along with other newly reported similar mycoviruses found in Fusarium poae [15], Botrytis cinerea [16], and Rosellinia necatrix [17]. Our results further expand our knowledge regarding the genomic diversity and host range of fusagraviruses and provide insights into genome evolution.
Provenance and sequencing of strains
The strain 2012-19 of M. phaseolina was isolated from the diseased stem of a sesame plant with typical symptoms of charcoal rot, which was collected from Fuyang county, Anhui province, China, in 2012. Strain 2012-19 has a hypovirulent phenotype, characterized by abnormal colony morphology, slow growth on potato dextrose agar (PDA), and lower virulence on sesame plants (unpublished data). Mycelia agar plugs of strain 2012-19 were inoculated on PDA plates covered with cellophane membranes and cultured in the dark at 28-30°C for 2-4 days. Mycelia were harvested using a medicine spoon and stored at -80 °C until further use. New mycovirus discovery by next-generation sequencing was conducted based on a previously reported protocol with modifications [23]. Total RNA was extracted from 1.0 g of mycelia using an RNAiso Kit (TaKaRa, Dalian, China), and rRNA was depleted using a Ribo-Zero™ rRNA Removal Kit (Illumina, CA, USA). Paired-end sequencing libraries was prepared and sequenced on an Illumina HiSeq 2500 platform at Shanghai Bohao Biotechnology Co., Ltd. To obtain clean sequences, the raw reads from deep sequencing were processed to remove the adaptor sequences, and low-quality reads were discarded. These clean transcripts were then matched against genome sequences of M. phaseolina using Bowtie (1.0) software. The unmatched RNAs were next assembled into longer contiguous sequences (contigs), using Velvet software, and used to search the non-redundant protein sequences (nr) of GenBank database (http://www.ncbi.nlm.nih.gov/). Contigs that were identical or complementary to the viral genomic sequence were extracted and identified as potential viral sequences.
Synthesis cDNA from strain 2012-19 was conducted according to the manufacturer’s instructions for the PrimeScriptTM II 1st Strand cDNA Synthesis Kit (TaKaRa) (TaKaRa, Dalian, China). MpFV1 was identified using specific primers (F1, 5’-CCTGTTGAGCATACCG-3’; R1, 5’-ATTGTCCTGCCGAGCCT-3’) based on the putative viral sequence. To complete the 5’- and 3’-terminal genomic sequences, rapid amplification of cDNA ends (RACE) was performed using a SMARTer RACE cDNA Amplification Kit (Clontech, CA) with the help of gene-specific primers (GSPs). GSP-R1 (5’-CGCCAGCGATGTAAGCGATGACCG-3’) and GSP-R2 (5’-GGCGTGAACTTTTGACGATCC-3’) were used for the 5’-RACE reaction. GSP-F1 (5’-GACGATTACCAGTTATTCGC-3’) and GSP-F2 (5’-CGGAAAAGACCTAATCAAGAATGA-3’) were used for the 3’-RACE reaction. The procedures were performed according to the user manual provided with the kit. All PCR products were purified and cloned into the pMD19-T vector (Sangon Biotech, Zhengzhou, China) and introduced into Escherichia coli Trelief 5α (TSINGKE Biotech, Zhengzhou, China) by transformation. At least three recombinant clones were sent to Sangon Biotech for sequencing.
Prediction of putative open reading frames (ORFs) was performed using ORF Finder at NCBI. A protein domain search was conducted using the Conserved Domain Database (CDD). Multiple sequence alignments of the protein sequences were performed using DNAMAN software (version 9) and the CLUSTALX program (version 2.1) [24]. A phylogenetic tree was constructed by the maximum-likelihood (ML) method in the MEGA (version 7) program with 1,000 bootstrap replicates [25].
Sequence properties
From the assembled dataset remaining after discarding genomic sequences of M. phaseolina, we identified a long contig containing 9161 nucleotides (nt) that encoded two putative proteins related to proteins encoded by fusagraviruses or related mycoviruses. Thus, this contig was believed to be a partial sequence of a mycovirus, and this mycovirus was temporarily designated as MpFV1. Finally, the full-length sequence of MpFV1 was obtained and submitted to the GenBank database under the accession number MK780821.
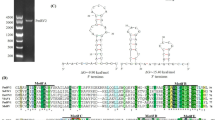
The full genome of MpFV1 comprises a double-stranded RNA with a length of 9289 nt and containing two disconnected ORFs, namely ORF1 (nt 891-5168) and ORF2 (nt 5354-9166). The length of the 5’-UTR and 3’-UTR was 890 nt and 123 nt, respectively (Fig. 1A). The sequences at the 5’ terminus and 3’ terminus of the positive strand of MpFV1 were predicted to form stem-loop structures, and their initial ∆G values were -14.60 kcal/mol and -9.50 kcal/mol, respectively (Fig. 1D).

Schematic representation of the genomic organization and translational strategy of MpFV1. (A) Schematic diagram of the genomic organization of MpFV1. MpFV1 shows the presence of two ORFs (ORF1 and ORF2). The dotted-lined box indicates a possible extension of ORF2 via a-1 translational frameshift mechanism. OFR1 encodes a putative hypothetical protein (P1), whereas ORF2 encodes a putative protein containing two conserved domains, i.e., the RdRp and S7 domains. (B) Schematic representation of the predicted H-type RNA pseudoknot, which is located 1357 nt directly upstream of the RdRp_4 motif in MpFV1, and the proposed mechanism underlying -1 ribosomal frameshifting. (C) P predicted secondary structures for the 5’-UTR and 3’-UTR of the coding strand of MpFV1, constructed using RNA mfold online (http://unafold.rna.albany.edu/?q=mfold/RNA-Folding-Form). (D) Predicted secondary structure of the H-type RNA pseudoknot. The RNA secondary structure was predicted using the KnotSeeker program
The 5’-proximal ORF1 of MpFV1 encodes a hypothetical protein comprising 1425 amino acids (aa), with a predicted molecular mass of 159.3 kDa and a predicted isoelectric point of 7.04. A multiple alignment of the sequence of the protein encoded by ORF1 showed that it shared the highest sequence identity (58%) with a hypothetical protein of Macrophomina phaseolina RNA virus 2 (MpRV2), but relatively low identity (26%–38%) with the hypothetical proteins of eleven other unclassified dsRNA viruses (Table 1). Moreover, a search using the CDD showed that the ORF1-encoded protein does not contain any conserved domains.
MpFV1 has a putative shifty heptamer sequence (5159GGAAAAC5165) located immediately upstream of the stop codon of ORF1 (Fig. 1A). Moreover, a candidate recoding stimulatory element (RSE) structure (H-type RNA pseudoknot located at nt 5219 to 5257) was predicted to lie just downstream of the slippery site (Fig. 1B). These two functional elements in MpFV1 are the cis-acting frameshift signals of the -1 ribosomal frameshifting mechanism, suggesting that the ORF2-encoded protein may be translated by ribosomal frameshifting. The 3’-proximal ORF2 of MpFV1 encodes an RNA-dependent RNA polymerase (RdRp) comprising 1270 amino acids (aa), with a predicted molecular mass of 143.2 kDa and predicted isoelectric point of 8.84. A BLASTp search of the protein encoded by ORF2 showed that it shared the highest sequence identity (61%) with the RdRp of MpRV2 but relatively low identity (31%–38%) with the RdRps of eleven other unclassified dsRNA viruses (Table 1). A search of the CDD and multiple protein sequence alignment indicated that the ORF2-encoded protein contains two conserved domains: a conserved RdRp domain (RdRp_4; pfam02123) with eight conserved motifs (I–VIII) that are characteristic of the RdRps of dsRNA viruses [26] (Supplementary Figure S1) and a conserved phytoreovirus S7 domain (S7; pfam07236) (Supplementary Figure S2). The MpFV1 S7 conserved domain is 115 aa long (aa 820 to 935), and polypeptides homologous to S7 were also identified in other fusagraviruses and related mycoviruses (Table 1).
To examine the relationship between MpFV1 and other mycoviruses, we performed phylogenetic analysis using protein alignments of the conserved RdRp domains from MpFV1 and 20 other selected RNA viruses (Fig. 2). The results showed that MpFV1 clustered with the previously reported MpRV2 and 15 other unclassified dsRNA mycoviruses, forming a distinct clade, indicating a close evolutionary relationship. However, MpFV1 and its related mycoviruses are distantly related to the clade including members of the families Totiviridae and Partitiviridae. Furthermore, a new family, “Fusagraviridae”, was recently proposed to accommodate these MpFV1-related but unassigned dsRNA viruses identified in filamentous fungi [15]. Moreover, MpFV1 has a shorter intervening sequence between ORF1 and ORF2, a shorter 5’-UTR, and a longer 3’-UTR than MpRV2 at the corresponding locations. Thus, MpFV1 is a potential new member of the proposed family “Fusagraviridae”.

Phylogenetic analysis of MpFV1 (marked with a red dot) and other related RNA viruses. The phylogenetic tree was generated by the maximum-likelihood method (1000 bootstrap replicates) based on the amino acid sequences of the putative RdRp domains using MEGA 7. Two partitiviruses (PsVF and PsVS) and two totiviruses (ScV L-A and ScV L-B) were included as outgroups. The scale bar is equivalent to a genetic distance of 1.0 amino acid substitutions per site
References
Rosseto P, Costa AT, Polonio JC, da Silva AA, Pamphile JA, Azevedo JL (2016) Investigation of mycoviruses in endophytic and phytopathogenic strains of Colletotrichum from different hosts. Genet Mol Res. 15(1):15017651
Li W, Zhang T, Sun H, Deng Y, Zhang A, Chen H, Wang K (2014) Complete genome sequence of a novel endornavirus in the wheat sharp eyespot pathogen Rhizoctonia cerealis. Arch Virol 159:1213–1216
Liu H, Fu Y, Jiang D, Li G, Xie J, Peng Y, Yi X, Ghabrial SA (2009) A novel mycovirus that is related to the human pathogen hepatitis E virus and rubi-like viruses. J Virol 83:1981–1991
Chiba S, Salaipeth L, Lin YH, Sasaki A, Kanematsu S, Suzuki N (2009) A novel bipartite double-stranded RNA mycovirus from the white root rot fungus Rosellinia necatrix: molecular and biological characterization, taxonomic considerations, and potential for biological control. J Virol 83:12801–12812
Xie J, Jiang D (2014) New insights into mycoviruses and exploration for the biological control of crop fungal diseases. Annu Rev Phytopathol 52:45–68
Son M, Yu J, Kim KH (2015) Five questions about mycoviruses. PLoS Pathog 11:e1005172
Liu L, Xie J, Cheng J, Fu Y, Li G, Yi X, Jiang D (2014) Fungal negative-stranded RNA virus that is related to bornaviruses and nyaviruses. Proc Natl Acad Sci USA 111:12205–12210
Donaire L, Pagán I, Ayllón MA (2016) Characterization of Botrytis cinerea negative-stranded RNA virus 1, a new mycovirus related to plant viruses, and a reconstruction of host pattern evolution in negative-sense ssrna viruses. Virology 499:212–218
Yu X, Li B, Fu Y, Jiang D, Ghabrial SA, Li G, Peng Y, Xie J, Cheng J, Huang J, Yi X (2010) A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. Proc Natl Acad Sci USA 107:8387–8392
Ghabrial SA, Castón JR, Jiang D, Nibert ML, Suzuki N (2015) 50-plus years of fungal viruses. Virology 479–480:356–368
Mu F, Xie J, Cheng S, You M, Barbetti MJ, Wang Q, Cheng J, Fu Y, Chen T, Jiang D (2018) Virome characterizatiodn of a collection of S. sclerotium from Australia. Front Microbiol 8:2540
Marzano S-YL, Nelson BD, Ajayi-Oyetunde O, Bradley CA, Hughes TJ, Hartman GL, Eastburn DM, Domier LL (2016) Identification of diverse mycoviruses through metatranscriptomics characterization of the viromes of five major fungal plant pathogens. J Virol 90:6846–6863
Bartholomäus A, Wibberg D, Winkler A, Pühler A, Schlüter A, Varrelmann M (2016) Deep sequencing analysis reveals the mycoviral diversity of the virome of an avirulent isolate of Rhizoctonia solani AG-2-2 IV. PLoS One 11(11):e0165965
Shi M, Lin X, Tian J, Chen L, Chen X, Li C, Qin X, Li J, Cao J, Eden J, Buchmann J, Wang W, Xu J, Holmes EC, Zhang Y (2016) Redefining the invertebrate RNA virosphere. Nature 540:539–543
Wang L, Zhang J, Zhang H, Qiu D, Guo L (2016) Two novel relative double-stranded RNA mycoviruses infecting Fusarium poae strain SX63. Int J Mol Sci 17:641–654
Arjona-Lopez JM, Telengech P, Jamal A, Hisano S, Kondo H, Yelin MD et al (2018) Novel, diverse RNA viruses from mediterranean isolates of the phytopathogenic fungus, Rosellinia necatrix: insights into evolutionary biology of fungal viruses. Environ Microbiol 20(4):1464–1483
Hao F, Ding T, Wu M, Zhang J, Yang L, Chen W et al (2018) Two novel hypovirulence-associated mycoviruses in the phytopathogenic fungus Botrytis cinerea: molecular characterization and suppression of infection cushion formation. Viruses 10(5):254
Wu M, Jin F, Zhang J, Yang L, Jiang D, Li G (2012) Characterization of a novel bipartite double-stranded RNA mycovirus conferring hypovirulence in the pathogenic fungus Botrytis porri. J Virol 86:6605–6619
Islam MS, Haque MS, Islam MM, Emdad EM, Halim A et al (2012) Tools to kill: genome of one of the most destructive plant pathogenic fungi Macrophomina phaseolina. BMC Genom 13(1):493
Wyllie TD (1988) Charcoal rot of soybean-current status. In: Wyllie TD, Scott DH (eds) Soybean diseases of the north central region. APS Press, St. Paul, MN, p 106–113
Li L (1993) Progress in research on sesame diseases in the world. Oil Crop Sci 2:75–77
Arora P, Dilbaghi N, Chaudhury A (2012) Detection of double stranded RNA in phytopathogenic Macrophomina phaseolina causing charcoal rot in Cyamopsis tetragonoloba. Mol Biol Rep 39:3047–3054
Nerva L, Varese GC, Turina M (2018) Different approaches to discover mycovirus associated to marine organisms. Methods Mol Biol 1746:97–114
Thompson JD, Gibson J, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: fexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33(7):1870–1874
Bruenn JA (1993) A closely related group of RNA-dependent RNA polymerases from double-stranded RNA viruses. Nucleic Acids Res 21:5667–5669
Acknowledgements
We are extremely grateful to Dr. Huiquan Liu (Northwest A&F University) and Dr. Mingde Wu (Huazhong Agricultural University) for their help with sequence analysis.
Funding
This research was supported by the China Agriculture Research System (CARS-14) and National Key R&D Program of China (2018YFD0201006). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare that they have no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Massimo Turina.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, J., xiao, Y., Zhao, H. et al. A novel double-stranded RNA mycovirus that infects Macrophomina phaseolina. Arch Virol 164, 2411–2416 (2019). https://doi.org/10.1007/s00705-019-04334-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-019-04334-6