
Abstract
In this study, we present the characterization and genomic data of three Achromobacter phages belonging to the family Siphoviridae. Phages 83-24, JWX and JWF were isolated from sewage samples in Paris and Braunschweig, respectively, and infect Achromobacter xylosoxidans, an emerging nosocomial pathogen in cystic fibrosis patients. Analysis of morphology and growth parameters revealed that phages 83-24 and JWX have similar properties, both have nearly the same head and tail measurements, and both have a burst size between 85 and 100 pfu/cell. In regard to morphological properties, JWF had a much longer and more flexible tail compared to other phages. The linear double-stranded DNAs of all three phages are terminally redundant and not circularly permutated. The complete nucleotide sequences consist of 81,541 bp for JWF, 49,714 bp for JWX and 48,216 bp for 83-24. Analysis of the genome sequences showed again that phages JWX and 83-24 are quite similar. Comparison to the GenBank database via BLASTN revealed partial similarities to Roseobacter phage RDJL phi1 and Burkholderia phage BcepGomr. In contrast, BLASTN analysis of the genome sequence of phage JWF revealed only few similarities to non-annotated prophage regions in different strains of Burkholderia and Mesorhizobium.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Achromobacter xylosoxidans is a motile Gram-negative rod-shaped bacterium [1] that is widely distributed in natural environments [2, 3]. During the last decade, it has also been recognized as an emerging nosocomial pathogen in cystic fibrosis [4, 5], but it also causes different human infections, e.g., endocarditis [6, 7], bacteremia [8, 9], ocular infections [10, 11] and urinary tract infections [12]. It has recently been shown that Achromobacter strains [13] are resistant to a large number of antimicrobial substances, such as penicillins, lincosamides, and cephalosporines, which emphasizes the urgent need to develop alternative strategies to combat this opportunistic pathogen. The use of bacteriophages as therapeutic agents to overcome bacterial infections is well established, and phages have already been applied for the treatment of infections in humans [14], animals [15] and plants [14, 16]. Bacteriophages against Achromobacter xylosoxidans were nearly unknown for a long time; only some older publications existed [17, 18]. Recently, Wittmann et al. [13] isolated bacteriophages against Achromobacter from soil and sewage samples. Most of the described bacteriophages belonged to the family Siphoviridae, and had long and flexible tails. Two members of the family Podoviridae, phages JWAlpha and JWDelta, were described in more detail. According to their genomic organization and sequence similarity, they could be grouped into the highly conserved N4 family [19]. So far, few genomic sequences of Achromobacter phages have been determined [20–22]. In this study, we present further genomic data for Achromobacter phages that belong to the family Siphoviridae and are different from those already known.
Materials and methods
Genome sequence accession numbers
Genome sequences of JWF, JWX and phage 83-24 were deposited at NCBI GenBank under accession numbers KP202969-KP202971.
Bacterial strains and growth
Achromobacter xylosoxidans strains LMG 3465 (JWX), CCUG 48386 (JWF) and HER 83-190 (83-24) were used for phage propagation. All strains were cultured in TBY medium (10 g of tryptone, 5 g of yeast extract, and 5 g of NaCl per liter, pH 7.5) or on Petri dishes of TBY medium supplemented with 1.5% agar (w/v) for approximately 16 h (overnight) at 28 °C.
Purification of phages and phage DNA
Phages were purified by CsCl gradient centrifugation and their DNA was isolated by phenol extraction as described previously [23].
Sample preparation and sequencing
SMRTbell™ template libraries were prepared according to instructions from Pacific Biosciences, Menlo Park, CA, USA, following the Procedure & Checklist for “Low-Input 10 kb Template Preparation and Sequencing” using C2 Chemistry as described previously [20].
Bioinformatic analysis
Phage genome sequence assembly was performed using the “RS_HGAP_Assembly.3” protocol included in SMRTPortal version 2.2.0., applying standard parameters. For all phages, one final contig could be obtained, which was linearized due to recognition of distinct start and end points in the phage assemblies where the majority of sequences ended abruptly. A quality check of the final phage genomes regarding overall coverage as well as SNPs was performed using SMRT View and IGV [24]. All phage genomes were annotated using PROKKA 1.8 (PMID 24642063) with subsequent manual curation in Artemis [25]. The intergenic genome regions of the phage were searched for transcriptional regulation elements. A search for tRNA genes was done with the tRNAscan-SE program v1.2.1 [26] and ARAGORN v1.2.36 [27]. Homology assignments were based on amino acid sequence alignment searches (BlastP) and were accepted only if the statistical significance of the sequence similarities (E value) was less than 1×10−5, the percentage query cover was ≥60%, and the percentage identity between the aligned sequences was ≥35%.
One-step growth curve analysis
The classical method of Ellis and Delbruck for one-step growth curve analysis was performed according to the modifications described [28]. A 50-ml logarithmic culture grown at 28°C in TBY medium was centrifuged at a titer of 3×108/ml. Cells were resuspended in 5 ml of TBY. Phage lysates were adjusted to a titer of 3×109 plaque-forming units (pfu) per ml. Infection was done at a multiplicity of infection (moi) of 0.01 (1.5×108 phages = 50 µl). After 8 min at room temperature to allow adsorption, the mixture was centrifuged to remove the excess phage. The supernatant was discarded, and the cell pellets were resuspended in 50 ml of fresh TBY medium and incubated with shaking at 28 °C. Two samples were collected at the intervals shown in Fig. 2. One set of samples was treated with a final concentration of 1% chloroform to determine the total amount of viable phages (intracellular and extracellular). All samples were immediately serially diluted in tenfold steps and plated for phage titration. All experiments were done in triplicate.
SDS-PAGE analysis of phage proteins
Treatment of phage samples for SDS-PAGE analysis and SDS-PAGE analysis itself were performed as described previously [29].
MALDI-TOF analysis
MALDI-TOF analysis of peptides was performed as described previously [20]. Coomassie-blue-stained protein bands were excised from the gel and washed twice in water for 5 min. After dehydration by adding 200 μl of 50% acetonitrile for 5 min, the bands were then washed in 200 μl of 0.1 M NH4HCO3 for 15 min at room temperature. The washing solution was discarded, and the slices were dehydrated by adding acetonitrile again. Dehydrated gel pieces were completely dried in a Speed Vac concentrator for 10 min. For tryptic digestion of the proteins, the gel slices were reswelled by adding 50 μl of digestion solution containing trypsin and incubated overnight at 37°C. For extraction of peptides, 50 μl of acetonitrile was added, followed by incubation with vigorous shaking for 15 min at 37°C. Supernatants were removed, and gel slices were treated with 50 μl of 5% formic acid with vigorous shaking for 15 min at 37°C. After that, 50 μl of acetonitrile was added, followed again by incubation with vigorous shaking for 15 min at 37°C. Supernatants were combined with the removed supernatants and concentrated using a Speed Vac concentrator. Forty μl of 32% methanol/0.25% HCOOH was added, followed by ultrasound treatment for 3 min and analysis by MALDI Ultraflex-TOF/TOF. Results were compared with a database of predicted phage proteins using Mascot (in-house system).
Negative staining of phages
Thin carbon support films were prepared by sublimation of a carbon thread onto a freshly cleaved mica surface. Phages were negatively stained with 2% (w/v) aqueous uranyl acetate, pH 5.0, according to the method of Valentine et al. [30]. Samples were examined in a TEM 910 transmission electron microscope (Carl Zeiss, Oberkochen) at an acceleration voltage of 80 kV. Magnification was calibrated with a cross-lined carbon replica. At least 10 phage particles were measured, and the standard error was determined.
Results and discussion
General phage properties
Morphological characterization
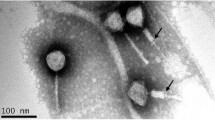
Phage 83-24 was obtained from the Félix d’Hérelle Reference Center for Bacterial Viruses at the University of Laval; it was originally isolated from sewage by Vieu and Binette in Paris in 1983. Phages JWX and JWF were isolated from sewage in Braunschweig in 2012 with host strains LMG 3465 and CCUG 48386, respectively. Morphological characterization via transmission electron microscopy revealed that they all belong to the family Siphoviridae [13], but whereas phage JWF has a quite long and flexible tail with a length of 277 nm +/- 17 nm, the tails of phages 83-24 and JWX are much shorter, with 126 nm +/- 6 nm and 148 nm +/- 9 nm, respectively. In addition, phage 83-24 had three to four tail fibers, which could not be identified in JWX (Fig. 1).

Electron micrographs of Achromobacter phages 83-24 (A), JWX (B) and JWF (C). Negative staining (2% [w/v] uranyl acetate, pH 5.0). Bars represent 100 nm
One-step growth curve
In order to characterize the infection cycle of the three phages, we determined the burst size and the eclipse and latent period – three characteristic parameters of phage life cycle – by one- step growth experiments as described previously [28]. These experiments revealed that phages JWX and 83-24 have an eclipse period of 36 +/- 1 min and 47 +/- 3 min, respectively- and latent periods of 55 +/- 2 min, and 76 +/- 4 min, respectively, while the eclipse period of JWF takes 97 +/- 4 min and the latent period is 133 +/- 4 min. Phage JWX has a burst size of 78 +/- 8, and phage 83-24 has a burst size of 78 +/- 16 (Fig. 2), whereas JWF has a significantly lower burst size of 20 +/- 1 new phages per infected cell. Fig. 1 shows that JWF had been readsorbed onto cell debris, resulting in clumps and empty heads. Thus, the real number of released phages may be higher.

One-step growth curve analysis of phages JWX, 83-24 and JWF. Experiments were conducted in triplicate, and average values are shown in the graph
General genomic properties
All three phages contained linear, double-stranded DNA, which was confirmed by digestion with different restriction endonucleases as described [13]. Whole-genome, long-read sequencing was performed using the PacBio RSII system, which resulted in genome assembly lengths of 81,541 bp (JWF; G+C content, 60.1%), 49,714 bp (JWX; G+C content, 55.4%) and 48,216 bp (83-24; G+C content, 54.8%). These phages had a lower G+C content than their host species (65-67% G+C [31, 32]), a phenomenon that has been described before for other phages and is quite common [33]. Looking for repeat regions in the genome sequences of the three phages, we identified terminal redundancies of 3358 bp (JWX), 2642 bp (83-24), and 1465 bp (JWF), which was confirmed via long-read sequencing. These redundancies were determined by analyzing high-coverage regions with distinct start and end points. For the identification of protein-coding sequences, open reading frames (ORFs) were identified automatically using Prokka [34] and then curated manually in Artemis [25], resulting in 118 ORFs for JWF (113 ATG [Met], 4 GTG [Met], 1 TTG [Met]; coding percentage, 93.7%; Supplemental File S1), 68 ORFs for JWX (64 ATG [Met], 2 GTG [Met], 2 TTG [Met]; coding percentage, 90%; Supplemental File S2), and 62 ORFs for phage 83-24 (56 ATG [Met], 3 GTG [Met], 3 TTG [Met]; coding percentage, 90.6%; Supplemental File S3). Genome annotation using BLASTP, Prokka, and PHAST [35] led to the identification of different functional clusters, including DNA packaging, head and tail morphogenesis, replication, and lysis. Further sequence analysis using tRNAscan-SE v.1.21 [26] revealed a proline tRNA gene in the genomes of JWX and 83-24 (both anti-codon TGG) and a lysine tRNA gene (anti-codon CTT) in JWF.
Genomic analysis of phages JWX and 83-24
The morphology, gene content, and genomic organization of phages JWX and 83-24 are quite similar (Fig. 3A) despite the fact that they were isolated from different geographic origins more than 30 years apart. Analysis via BLASTN revealed that they exhibit 76% sequence identity at the DNA level over 86% of their genome length. Thus, their sequence divergence is quite high, and they cannot be considered isolates of one phage species (species cutoff, 94 or 95% average nucleotide sequence identity) [19].

Genomic organization of phages 83-24, JWX and JWF in comparison with related phages. Functional clusters are colored: DNA packaging genes in orange, structural genes in red, replication genes in light blue, genes for host lysis in yellow, and putative integrase\primpol genes in green. Genes for tRNAs are indicated by blue triangles. The figure was generated using Easyfig with amino acid sequence comparison. The amino acid identity range is indicated by the gradient scale (color figure online)
The genome sequences gave no indication of prophage functions, such as a repressor gene or an integrase gene of the serine or tyrosine type. However, in both genomes, a gene with a primase-polymerase (primpol) domain is located at the right end (JWAP_00058 in 83-24 and JWX_00062 in JWX). Homologs of these genes have already been described in a Pseudomonas phage [36] and some Vibrio phages and annotated as integrases [37]. This type of putative integrase differs significantly from the typical tyrosine and serine site-specific recombinases [38]. These proteins contain a primpol domain, which is unusual for integrases, and they are longer than the tyrosine integrases and more acidic. The predicted primpol proteins of both Achromobacter phages are 846 amino acids long, and their isoelectric points pI are 5.34 (JWX) and 5.44 (83-24), respectively. However, further experiments are needed to verify that the primpol proteins of the phages really act as integrases.
Gene cluster for DNA packaging and structural proteins
Both phage genomes carry clearly identifiable genes with a conserved domain (Terminase_6, pfam03237; E-value, 6.11e-12) in their deduced amino acid sequence for the small and large subunits of the terminase that are required for the packaging of the phage genome into the capsid. Based on a set of amino acid sequences of phages with known packaging mechanisms [39–41], we conducted a phylogenetic analysis of the large terminase subunits of these Achromobacter phages to compare them to others with a known mechanism of DNA packaging (Fig. 4). We found that the terminases of JWX and 83-24 could not be grouped together with other terminases with known packaging mechanisms; however, together with terminases from Pseudomonas and Burkholderia phages, they formed a cluster with bootstrap support in the phylogenetic tree. The functions of these proteins have not been tested experimentally, and the DNA packaging mechanism of these phages remains unclear.

Phylogenetic analysis of terminase large subunits of JWX, JWF and phage 83-24 compared to those with known DNA packaging strategies. The neighbor-joining tree was constructed based on ClustalW alignment of terminase subunit amino acid sequences with 1000 bootstrap replicates (MEGA5) [42]
Analysis of the gene clusters for structural proteins of JWX and 83-24 revealed that both phages share all genes in this cluster, including genes for the portal and scaffold protein, capsid proteins, minor and major tail proteins, a tail fiber, and a tail tape measure protein.
To verify the correct annotation of these genes, phage particles were denatured and separated by SDS-PAGE (Fig. 5). Afterwards, different protein bands were extracted and identified by peptide mass fingerprinting. We identified the portal proteins (JWX_00009 in JWX and JWAP_00007 in phage 83-24), the putative major tail proteins (JWX_00017 in JWX and JWAP_00015 in phage 83-24), and the major coat proteins (JWX_00011 in JWX and JWAP_00009 in phage 83-24) which formed the most prominent bands in their protein profiles.

Identification of structural proteins of phages JWF, JWX and 83-24 by SDS-PAGE analysis and mass peptide fingerprinting. Proteins were separated by 17.5% SDS-PAGE. M, SeeBlue®Pre-Stained Standard. After extraction of gel slices from the corresponding bands and tryptic digestion, samples were analyzed via peptide mass fingerprinting. Identified gene products are shown
Although these two phages seem to be quite similar with regard to their genomic properties, there was a high degree of variation in their host spectra [13]. BLASTP analysis of the deduced amino acid sequences of two genes (JWX_00027 in JWX [pfam09327, 3.56e-21] and JWAP_00025 in 83-24 [pfam09327, E-value 6.88e-19]) revealed conserved domains that might be responsible for host recognition and therefore also for host specificity. Comparison of theses sequences showed that they shared at least 80% amino acid sequence identity. When we compared the whole genomes of these phages at the amino acid level (Fig. 3A), we found two regions in the structural cluster with less similarity than was observed in the rest of this cluster. Gp22 (JWAP_00022) in 83-24 and Gp24 (JWX_00024) in JWX differed in length and were only weakly similar in sequence. However, according to BLASTP analysis, both genes showed similarities to tail fiber proteins that are also involved in host recognition. Furthermore, both phages possess two additional genes at the end of this cluster (JWX_00028/JWX_00029 and JWAP_00026/JWAP_00027) that differ from each other and do not show any similarities to known phage genes.
Lysis cluster
dsDNA phages use a holin-endolysin lysis system to disrupt the peptidoglycan layer in a precisely timed manner and to lyse the bacterial host cell to release new phage progeny. Downstream from the gene cluster for structural proteins, we identified a possible cluster for host cell lysis that consists of three genes in 83-24 (JWAP_00031-JWAP_00033) and of four genes in JWX (JWX_00032-JWX_00035). Although holins often share little similarity, they can be identified by their secondary structure. Analysis with TMHMM Server v. 2.0 [43] predicted that the deduced amino acid sequences of the first two genes in 83-24 and three genes in JWX all contained one transmembrane domain, either in the middle or in the N-terminal part of the protein, which led to the assumption that these proteins might function as class III holins [44]. Endolysins might have different enzymatic activities, e.g., N-acetyl muramidase, amidase or endopeptidase activity. The gene that is located downstream from the holin genes might code for an endolysin (JWAP_00033 in 83-14 and JWX_00035 in JWX) showing similarities to M15 peptidases from Burkholderia and Pseudomonas phages, e.g., endolysin gp23 from Burkholderia phage KS9 (YP_003090199) or a peptidase from Pseudomonas phage PaBG (YP_008433519).
Replication cluster
Immediately after the cluster for host cell lysis, the genes were found to be in the opposite transcriptional orientation. Apart from several genes with unknown function that might play a role in host shutoff, nearly the whole cluster coded for proteins that are putatively involved in replication of the genome. Downstream from the gene for a tRNA with an anticodon (TGG) for the amino acid proline, a gene with a highly conserved domain for a DNA polymerase (JWX_00048 in JWX and JWAP_00046 in 83-24) was identified, followed by genes necessary for replication and nucleotide metabolism, e.g., genes for a helicase, an NTPase, a thymidylate synthase, and a dCTPase.
Genomic characterization of phage JWF
Host interaction and DNA packaging
Though isolation of JWF lysogenic cells and reinduction of the prophage was successful previously [13], analysis of the JWF genome did not reveal the presence of a gene for a putative integrase. Instead, the 5’ end of the genome contains two genes (PJWF_00003 and PJWF_00004) with conserved domains for putative HNH endonucleases, followed by a gene with a highly conserved domain for a bifunctional DNA primase/polymerase (PJWF_00005), as is found in phages JWX and 83-24. With regard to genes involved in DNA packaging, we could only identify one gene (PJWF_00007) with a conserved domain Terminase_GpA (pfam05876; E-value, 1.78e-20) for the large subunit of a terminase, but no gene for a small subunit. Phylogenetic analysis of this gene revealed that it did not form a branch in a phylogenetic tree based on sequences of terminases with known packaging mechanisms (Fig. 4). However, the JWF terminase showed similarities to the terminase of Bacillus phage SPO1 at the amino acid level. The SPO1 genome has terminal redundancies [45], as has been shown for JWF, and thus, these phages might have a similar packaging mechanism.
Head and tail cluster of JWF
Downstream of the terminase gene, we identified a cluster of genes that showed no similarities to any phage genes, but did show similarity to genes of Burkholderia sp. MSh1 [46] (23 of 26 genes, 31-74% identity at the aa level) and Mesorhizobium sp. STM 4661 (14 of 26 genes, 22-53% identity at the aa level) (Fig. 3C). The results of the peptide mass fingerprinting analysis (Fig. 5) confirmed that genes located in this region encode structural proteins for the head and tail of the phage. Whereas Gp11 represented the major capsid protein as the most prominent band in the protein profile of JWF, Gp20 to Gp31 (PJWF_00020-PJWF_00031) seem to be proteins for the phage tail, including a tail tape measure protein (tape_meas_TP901 [TIGR01760]), a minor tail protein (phage_NlpC_pfam TIGR02219; E-value, 5.12e-13), a putative tail assembly protein (COG4723) and a putative host specificity protein (COG4733, Phage-tail_3, pfam13550; E-value, 1.25e-09) based on conserved domains identified after BLASTP analysis. Further downstream, we found two additional genes with similarities to tail fibers and other phage tail proteins (PJWF_00036 and PJWF_00037).
Lysis cluster of JWF
Putative genes involved in the lysis of the bacterial host cell could be identified embedded between the genes for structural proteins. The amino acid sequence of Gp32 (PJWF_00032) contained a conserved domain (COG3179) for a putative lysozyme, whereas analysis of Gp33 and Gp34 with TMHMM Server v. 2.0 predicted the presence of two and four transmembrane domains, respectively, making them candidates for holin-like proteins. Downstream from these two genes, we identified a gene (PJWF_00035) for a protein with a conserved domain (Phage_lysis, pfam03245; E-value, 3.49e-04) that is often found in Rz-like proteins, which are also involved in lysis by many phages of Gram-negative hosts [47].
Cluster for replication and nucleotide metabolism
Downstream from the cluster for lysis and head and tail morphogenesis, the sequence analysis revealed that the direction of transcription changes, and all further hypothetical genes were identified on the other strand. Whereas gp38 (PJWF_00038) coded for a putative SGNH hydrolase, the following genes all encoded proteins involved either in nucleotide metabolism – gp39 (PJWF_00039) for a deoxynucleoside monophosphate kinase and gp40 (PJWF_00040) for a thymidylate synthase – or replication of the phage genome. This cluster contained genes for an NTPase (PJWF_00041), an ATPase (PJWF_00043), endo- and exonucleases (PJWF_00044-PJWF_00046), a DNA ligase (PJWF_00049), a DNA polymerase (PJWF_00056) and a helicase (PJWF_00064). Upstream of the replication cluster, BLASTP analysis revealed only a few similarities to phage genes or any known genes in general. Generally, so-called early genes, which are used for host shutdown, protection of phage DNA after injection, or modification of bacterial RNA polymerases, for example, are poorly examined and annotated in databases.
Comparison with phages of different bacterial hosts
One aim of this study was to get insight into the relationship and similarities of Achromobacter phages of the family Siphoviridae to other phages with regard to gene content and genome organization, as there was previously no information on genomic properties, apart from Achromobacter phage phiAxp-1 [21], which showed similarities at the amino acid level in the cluster for head and tail structure. In general, most of the examined genes of phages 83-24 and JWX showed similarities to genes from Burkholderia, Roseobacter and Pseudomonas phages. Interestingly, the positive strands of both phage genomes, which contain the clusters for DNA packaging, head and tail structure, and host lysis closely resemble the genomic organization of Burkholderia phage BcepGomr (NCBI reference sequence NC_009447), whereas the negative strands with the replication clusters showed a high degree of similarity to Roseobacter phage RDJL Phi 1. Whereas 83-24 and JWX exhibited a high level of DNA sequence identity, analysis of JWX with BLASTN, for example, revealed little similarity to Burkholderia phage BcepGomR (72% sequence identity over 2% coverage) and Roseobacter RDJL Phi 1 (66% sequence identity over 4% coverage), with no relevance at the DNA level.
In support of these findings, further analysis of the genomes at the amino acid level, using CoreGenes3.0, revealed that the two Achromobacter phages shared 51 homologous genes with each other, but a smaller number with BcepGomr and RDJL Phi 1, respectively (Table 1).
According to previous studies [49], JWX and 83-24 could be grouped into the same phage genus, as they share more than 40% homologous genes. Apart from the gene for a putative integrase from phage RDJL Phi1, no significant similarities to genes of BcepGomr (NC_009447.1) or RDJL Phi 1 (HM151342.1) could be identified on the opposite strand (Fig. 3B). This finding is another example of genomic mosaicism in bacteriophage genomes [50, 51], putatively caused by horizontal exchange and recombination. In contrast to phages 83-24 and JWX, the genome of JWF is similar to only a small extent to other described phages. Analysis of deduced amino acid sequences of JWF with BLASTP revealed little similarity to known phages, but high similarity in the morphogenetic gene cluster to unannotated putative prophage regions from Burkholderia sp. MSh1 and Mesorhizobium sp. STM 4661 [52] (Fig. 3C). This supports the suggestion that phage JWF might be related to prophages that have the ability to be integrated into the host genome [13], although these genes are not annotated as a prophage region in the bacterial genome. Furthermore, in contrast to JWX and 83-24, there are not only one or two dominating phages with regard to the identified similarities, but a few similarities to many phages from different host species could be determined.
Conclusion
So far, only few genome sequences of Achromobacter phages have been determined. These phages mainly belong to the family Podoviridae, and their genomic structure and main characteristics are highly conserved and indicate a relationship to N4-like phages. Although members of the N4 family are widespread among different host genera, but have a highly conserved genomic organization, no close relatives or putative characteristics of Achromobacter phages could be determined on the basis of these sequences. Thus, we were interested in the analysis of the genomic structures of different siphoviruses of Achromobacter. In general, all three phages are further examples of mosaicism in the genomic organization of tailed phages with partial relationship to other phages with hosts that are also wide spread in the environment, e.g., Pseudomonas and Burkholderia.
For therapeutic applications, temperate phages need to be excluded. After infection with the phages examined in this study at an moi of 5 [13], very few cells survived, suggesting that these phage are virulent. However, all three phages contain putative genes that encode a primpol integrase, which could be a characteristic of this group of phages. Genes with this combined primase/polymerase motif were also identified in the genomes of Pseudomonas phage YuA [36] and several Vibrio phages [37]. In these studies, it was speculated that the gene products are integrases. The failure to isolate stable lysogens of phages with a predicted primpol integrase has already been described by Ceyssens et al. [36] for phage YuA and its host Pseudomonas aeruginosa PAO1, and in other publications describing studies with φJL001 [53], which infects a sponge-associated bacterium, and Vibrio haemolyticus phages VP16T and VP16C [37]. The authors assume that, as these putative integrases differ from well-studied serine and tyrosine recombinases, they might show a distinct behavior that is dependent on specific physiological conditions of the host that have not been determined yet, causing them to exhibit a lytic behavior under specific experimental conditions. It will be necessary to clarify the function of this group of enzymes experimentally to determine whether these phages are temperate and, if so, under which conditions they can lysogenize their hosts.
References
Yabuuchi E, Oyama A (1971) Achromobacter xylosoxidans n. sp. from human ear discharge. Jpn J Microbiol 15(5):477–481
Busse HJ, Auling G (2005) Genus II. Achromobacter Yabuuchi and Yano 1981, 477VP emend. Yabuuchi, Kawamura, Kosako and Ezaki 1998a, 1083. In: Brenner DJ, Krieg NR, Staley JT (eds) Bergey’s manual of systematic bacteriology. Springer, New York, pp 658–662
Spear JB, Fuhrer J, Kirby BD (1988) Achromobacter xylosoxidans (Alcaligenes xylosoxidans subsp. xylosoxidans) bacteremia associated with a well-water source: case report and review of the literature. J Clin Microbiol 26(3):598–599
Mahenthiralingam E (2014) Emerging cystic fibrosis pathogens and the microbiome. Paediatr Respir Rev 15(Suppl 1):13–15. doi:10.1016/j.prrv.2014.04.006
Trancassini M, Iebba V, Citera N, Tuccio V, Magni A, Varesi P, De Biase RV, Totino V, Santangelo F, Gagliardi A, Schippa S (2014) Outbreak of Achromobacter xylosoxidans in an Italian Cystic fibrosis center: genome variability, biofilm production, antibiotic resistance, and motility in isolated strains. Front Microbiol 5:138. doi:10.3389/fmicb.2014.00138
Ahmed MS, Nistal C, Jayan R, Kuduvalli M, Anijeet HK (2009) Achromobacter xylosoxidans, an emerging pathogen in catheter-related infection in dialysis population causing prosthetic valve endocarditis: a case report and review of literature. Clin Nephrol 71(3):350–354
van Hal S, Stark D, Marriott D, Harkness J (2008) Achromobacter xylosoxidans subsp. xylosoxidans prosthetic aortic valve infective endocarditis and aortic root abscesses. J Med Microbiol 57(Pt 4):525–527. doi:10.1099/jmm.0.47496-0
Behrens-Muller B, Conway J, Yoder J, Conover CS (2012) Investigation and control of an outbreak of Achromobacter xylosoxidans bacteremia. Infect Control Hosp Epidemiol 33(2):180–184. doi:10.1086/663710
Tena D, Carranza R, Barbera JR, Valdezate S, Garrancho JM, Arranz M, Saez-Nieto JA (2005) Outbreak of long-term intravascular catheter-related bacteremia due to Achromobacter xylosoxidans subspecies xylosoxidans in a hemodialysis unit. Eur J Clin Microbiol Infect Dis 24(11):727–732. doi:10.1007/s10096-005-0028-4
Park JH, Song NH, Koh JW (2012) Achromobacter xylosoxidans keratitis after contact lens usage. Korean J Ophthalmol 26(1):49–53. doi:10.3341/kjo.2012.26.1.49
Reddy AK, Garg P, Shah V, Gopinathan U (2009) Clinical, microbiological profile and treatment outcome of ocular infections caused by Achromobacter xylosoxidans. Cornea 28(10):1100–1103. doi:10.1097/ICO.0b013e3181a1658f
Tena D, Gonzalez-Praetorius A, Perez-Balsalobre M, Sancho O, Bisquert J (2008) Urinary tract infection due to Achromobacter xylosoxidans: report of 9 cases. Scand J Infect Dis 40(2):84–87. doi:10.1080/00365540701558714
Wittmann J, Dreiseikelmann B, Rohde C, Rohde M, Sikorski J (2014) Isolation and characterization of numerous novel phages targeting diverse strains of the ubiquitous and opportunistic pathogen Achromobacter xylosoxidans. PLoS One 9(1):e86935. doi:10.1371/journal.pone.0086935
Abedon ST, Kuhl SJ, Blasdel BG, Kutter EM (2011) Phage treatment of human infections. Bacteriophage 1(2):66–85. doi:10.4161/bact.1.2.15845
Johnson RP, Gyles CL, Huff WE, Ojha S, Huff GR, Rath NC, Donoghue AM (2008) Bacteriophages for prophylaxis and therapy in cattle, poultry and pigs. Anim Health Res Rev 9(2):201–215. doi:10.1017/s1466252308001576
Zaczek M, Weber-Dabrowska B, Gorski A (2014) Phages in the global fruit and vegetable industry. J Appl Microbiol. doi:10.1111/jam.12700
Jones PT, Pretorius GHJ (1981) Achromobacter sp. 2 Phage a3: a physical characterization. J Gen Virol 55:275–281
Thomson JA, Woods DR (1974) Bacteriophages and cryptic lysogeny in Achromobacter. J Gen Virol 22(1):153–157
Wittmann J, Klumpp J, Moreno Switt AI, Yagubi A, Ackermann HW, Wiedmann M, Svircev A, Nash JH, Kropinski AM (2015) Taxonomic reassessment of N4-like viruses using comparative genomics and proteomics suggests a new subfamily—”Enquartavirinae”. Arch Virol 160(12):3053–3062. doi:10.1007/s00705-015-2609-6
Wittmann J, Dreiseikelmann B, Rohde M, Meier-Kolthoff JP, Bunk B, Rohde C (2014) First genome sequences of Achromobacter phages reveal new members of the N4 family. Virol J 11:14. doi:10.1186/1743-422x-11-14
Li E, Zhao J, Ma Y, Wei X, Li H, Lin W, Wang X, Li C, Shen Z, Zhao R, Jiang A, Yang H, Yuan J, Zhao X (2016) Characterization of a novel Achromobacter xylosoxidans specific siphoviruse: phiAxp-1. Sci Rep 6:21943. doi:10.1038/srep21943
Ma Y, Li E, Qi Z, Li H, Wei X, Lin W, Zhao R, Jiang A, Yang H, Yin Z, Yuan J, Zhao X (2016) Isolation and molecular characterisation of Achromobacter phage phiAxp-3, an N4-like bacteriophage. Sci Rep 6:24776. doi:10.1038/srep24776
Beilstein F, Dreiseikelmann B (2006) Bacteriophages of freshwater Brevundimonas vesicularis isolates. Res Microbiol 157(3):213–219. doi:10.1016/j.resmic.2005.07.005
Thorvaldsdottir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14(2):178–192. doi:10.1093/bib/bbs017
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B (2000) Artemis: sequence visualization and annotation. Bioinformatics 16(10):944–945
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25(5):955–964
Laslett D, Canback B (2004) ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res 32(1):11–16. doi:10.1093/nar/gkh152
Park M, Lee JH, Shin H, Kim M, Choi J, Kang DH, Heu S, Ryu S (2012) Characterization and comparative genomic analysis of a novel bacteriophage, SFP10, simultaneously inhibiting both Salmonella enterica and Escherichia coli O157:H7. Appl Environ Microbiol 78(1):58–69. doi:10.1128/aem.06231-11
Wittmann J, Gartemann KH, Eichenlaub R, Dreiseikelmann B (2011) Genomic and molecular analysis of phage CMP1 from Clavibacter michiganensis subspecies michiganensis. Bacteriophage 1(1):6–14. doi:10.4161/bact.1.1.13873
Valentine RC, Shapiro BM, Stadtman ER (1968) Regulation of glutamine synthetase. XII. Electron microscopy of the enzyme from Escherichia coli. Biochemistry 7(6):2143–2152
Jakobsen TH, Hansen MA, Jensen PO, Hansen L, Riber L, Cockburn A, Kolpen M, Ronne Hansen C, Ridderberg W, Eickhardt S, Hansen M, Kerpedjiev P, Alhede M, Qvortrup K, Burmolle M, Moser C, Kuhl M, Ciofu O, Givskov M, Sorensen SJ, Hoiby N, Bjarnsholt T (2013) Complete genome sequence of the cystic fibrosis pathogen Achromobacter xylosoxidans NH44784-1996 complies with important pathogenic phenotypes. PLoS One 8(7):e68484. doi:10.1371/journal.pone.0068484
Strnad H, Ridl J, Paces J, Kolar M, Vlcek C, Paces V (2011) Complete genome sequence of the haloaromatic acid-degrading bacterium Achromobacter xylosoxidans A8. J Bacteriol 193(3):791–792. doi:10.1128/JB.01299-10
Rocha EP, Danchin A (2002) Base composition bias might result from competition for metabolic resources. Trends Genet 18(6):291–294. doi:10.1016/s0168-9525(02)02690-2
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14):2068–2069. doi:10.1093/bioinformatics/btu153
Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS (2011) PHAST: a fast phage search tool. Nucleic Acids Res 39((Web Server issue)):W347–W352. doi:10.1093/nar/gkr485
Ceyssens PJ, Mesyanzhinov V, Sykilinda N, Briers Y, Roucourt B, Lavigne R, Robben J, Domashin A, Miroshnikov K, Volckaert G, Hertveldt K (2008) The genome and structural proteome of YuA, a new Pseudomonas aeruginosa phage resembling M6. J Bacteriol 190(4):1429–1435. doi:10.1128/jb.01441-07
Seguritan V, Feng IW, Rohwer F, Swift M, Segall AM (2003) Genome sequences of two closely related Vibrio parahaemolyticus phages, VP16T and VP16C. J Bacteriol 185(21):6434–6447
Groth AC, Calos MP (2004) Phage integrases: biology and applications. J Mol Biol 335(3):667–678
Casjens SR, Gilcrease EB, Winn-Stapley DA, Schicklmaier P, Schmieger H, Pedulla ML, Ford ME, Houtz JM, Hatfull GF, Hendrix RW (2005) The generalized transducing Salmonella bacteriophage ES18: complete genome sequence and DNA packaging strategy. J Bacteriol 187(3):1091–1104. doi:10.1128/jb.187.3.1091-1104.2005
Fouts DE, Klumpp J, Bishop-Lilly KA, Rajavel M, Willner KM, Butani A, Henry M, Biswas B, Li M, Albert MJ, Loessner MJ, Calendar R, Sozhamannan S (2013) Whole genome sequencing and comparative genomic analyses of two Vibrio cholerae O139 Bengal-specific Podoviruses to other N4-like phages reveal extensive genetic diversity. Virol J 10:165. doi:10.1186/1743-422x-10-165
Merrill BD, Grose JH, Breakwell DP, Burnett SH (2014) Characterization of Paenibacillus larvae bacteriophages and their genomic relationships to firmicute bacteriophages. BMC Genom 15:745. doi:10.1186/1471-2164-15-745
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739. doi:10.1093/molbev/msr121
Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305(3):567–580. doi:10.1006/jmbi.2000.4315
Young R (2002) Bacteriophage holins: deadly diversity. J Mol Microbiol Biotechnol 4(1):21–36
Stewart CR, Casjens SR, Cresawn SG, Houtz JM, Smith AL, Ford ME, Peebles CL, Hatfull GF, Hendrix RW, Huang WM, Pedulla ML (2009) The genome of Bacillus subtilis bacteriophage SPO1. J Mol Biol 388(1):48–70. doi:10.1016/j.jmb.2009.03.009
Ong KS, Aw YK, Gan HM, Yule CM, Lee SM (2014) Draft genome sequences of two antimicrobial-producing Burkholderia sp. strains, MSh1 and MSh2, isolated from Malaysian Tropical Peat Swamp Forest Soil. Genome Announc. doi:10.1128/genomeA.01032-14
Summer EJ, Berry J, Tran TA, Niu L, Struck DK, Young R (2007) Rz/Rz1 lysis gene equivalents in phages of Gram-negative hosts. J Mol Biol 373(5):1098–1112. doi:10.1016/j.jmb.2007.08.045
Mahadevan P, King JF, Seto D (2009) CGUG: in silico proteome and genome parsing tool for the determination of “core” and unique genes in the analysis of genomes up to ca. 1.9 Mb. BMC Res Notes 2:168. doi:10.1186/1756-0500-2-168
Lavigne R, Seto D, Mahadevan P, Ackermann HW, Kropinski AM (2008) Unifying classical and molecular taxonomic classification: analysis of the Podoviridae using BLASTP-based tools. Res Microbiol 159(5):406–414. doi:10.1016/j.resmic.2008.03.005
Hatfull GF (2008) Bacteriophage genomics. Curr Opin Microbiol 11(5):447–453. doi:10.1016/j.mib.2008.09.004
Casjens SR, Thuman-Commike PA (2011) Evolution of mosaically related tailed bacteriophage genomes seen through the lens of phage P22 virion assembly. Virology 411(2):393–415. doi:10.1016/j.virol.2010.12.046
Maynaud G, Brunel B, Mornico D, Durot M, Severac D, Dubois E, Navarro E, Cleyet-Marel JC, Le Quere A (2013) Genome-wide transcriptional responses of two metal-tolerant symbiotic Mesorhizobium isolates to zinc and cadmium exposure. BMC Genom 14:292. doi:10.1186/1471-2164-14-292
Lohr JE, Chen F, Hill RT (2005) Genomic analysis of bacteriophage PhiJL001: insights into its interaction with a sponge-associated alpha-proteobacterium. Appl Environ Microbiol 71(3):1598–1609. doi:10.1128/aem.71.3.1598-1609.2005
Acknowledgements
We sincerely thank Simone Severitt, Nicole Heyer and Anja Meier for technical assistance. We further thank Professor Ed Moore (CCUG Culture Collection, University of Gothenburg, Sweden) and Dr. Danielle Janssens (BCCM/LMG, Laboratory for Microbiology, University of Gent, Belgium) for providing us with strains of Achromobacter xylosoxidans, and Professor Sylvain Moineau (Félix d`Hérelle Reference Center for Bacterial Viruses, Université Laval, Canada) for phage 83-24.
Author contributions
BD conceived and designed the experiments and wrote the paper, BB and CS performed sequencing and genome analysis, MR performed morphological analysis via TEM microscopy, MN analyzed the peptide mass fingerprinting data, JW conceived and designed the experiments, analyzed data and wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dreiseikelmann, B., Bunk, B., Spröer, C. et al. Characterization and genome comparisons of three Achromobacter phages of the family Siphoviridae . Arch Virol 162, 2191–2201 (2017). https://doi.org/10.1007/s00705-017-3347-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-017-3347-8