
Abstract
Lytic Proteus phage PM16, isolated from human faeces, is a novel virus that is specific for Proteus mirabilis cells. Bacteriophage PM16 is characterized by high stability, a short latency period, large burst size and the occurrence of low phage resistance. Phage PM16 was classified as a member of the genus Phikmvvirus on the basis of genome organization, gene synteny, and protein sequences similarities. Within the genus Phikmvvirus, phage PM16 is grouped with Vibrio phage VP93, Pantoea phage LIMElight, Acinetobacter phage Petty, Enterobacter phage phiKDA1, and KP34-like bacteriophages. An investigation of the phage-cell interaction demonstrated that phage PM16 attached to the cell surface, not to the bacterial flagella. The study of P. mirabilis mutant cells obtained during the phage-resistant bacterial cell assay that were resistant to phage PM16 re-infection revealed a non-swarming phenotype, changes in membrane characteristics, and the absence of flagella. Presumably, the resistance of non-swarming P. mirabilis cells to phage PM16 re-infection is determined by changes in membrane macromolecular composition and is associated with the absence of flagella and a non-swarming phenotype.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Proteus mirabilis is a Gram-negative, motile, non-sporulating bacterium belonging to the genus Proteus, family Enterobacteriaceae. Members of the genus Proteus are widespread in nature and are found in soil and sewage and in the intestinal tract of humans and animals. An interesting feature of P. mirabilis, as well as other members of the genus Proteus, is their ability to differentiate into elongated mobile multinucleoid cells (60-80 μm in length) that expose hundreds of flagella on their surface, so-called swarming cells [4, 35, 41, 45]. It is believed that the appearance of swarming cells is triggered when P. mirabilis cells and their flagella come into contact with a solid surface [8]. At the time of swarming cell formation, septum formation stops, the cells elongate, become multinucleoid, and synthesize many new flagella. Hundreds of newly formed flagella promote rapid simultaneous swarming cell migration on the surface [35]. After the phase of migration, swarming cells are divided into cells that are 1.5-2.0 μm in length bearing 6-10 peritrichous flagella. These cells live in this form for some time, after which the process of swarming and colonization is repeated. As a result of this cyclic process, a colony of P. mirabilis on agar-containing medium resembles a bull’s eye pattern.
P. mirabilis bacteria, along with Proteus vulgaris and Proteus penneri, are pathogenic for humans, with approximately 90 % of infections caused by P. mirabilis. P. mirabilis is the etiological agent of intestinal infections and urinary tract infections and can cause chronic renal inflammation, infect surgical wounds and contribute to diabetic foot ulcers. P. mirabilis is one of the main causes of catheter-associated urinary tract infections (CAUTI), along with Escherichia coli (E. coli) [35]. Clinical isolates of P. mirabilis often have multiple antibiotic resistance [13, 15, 52], hence the need for alternative therapeutics for the treatment Proteus infections. One such alternative approach could be the use of lytic bacteriophages. However, studies on bacteriophages specific for Proteus spp. are rather limited.
Early studies of Proteus bacteriophages were mostly related to the need to differentiate isolates of P. mirabilis in clinical practice. To do this, phage typing of Proteus isolates was used, along with biochemical and serological identification [23, 48–50]. In recent years, several studies have been published describing the efficacy and safety of using Proteus bacteriophages and cocktails that are currently being used in phage therapy in Poland, Georgia, and the Russian Federation. Long-term clinical results of phage therapy conducted in Poland are presented in a review by Miedzyborski et al. [40]. The ability of the coli-proteus phage cocktail produced by NPO “Microgen” (http://www.microgen.ru) to prevent the formation of biofilms on catheters was studied by Carson et al. [10]. Later, a metagenomic analysis of this phage cocktail was conducted, but linkage was not determined between genomes and particular Proteus phages from this cocktail [38]. Recently, several Proteus phage genome sequences were deposited in the GenBank database (KM819694, KM819695, KM819696, KP890822, KP063118, KP890822, KP890823); however, detailed studies of their characteristics (biological properties, proteome, host range, etc.) were not published.
This study describes biological properties of a novel lytic bacteriophage, PM16, which is specific for P. mirabilis, its genome, structural proteome, and taxonomic classification, and some aspects of its interaction with the host bacterium.
Materials and methods
Bacterial host strain isolation and culture conditions
P. mirabilis strain CEMTC 73 was obtained from a stool sample from a patient after a bariatric surgical operation for the management of severe obesity. The research was approved by the Local Ethical Committee of the Center of New Medical Technology, Novosibirsk, and informed consent from the patient was obtained. We suspended 0.3 g of stool sample in 10 volumes of sterile phosphate-buffered saline (PBS, 5.84 g of NaCl, 4.72 g of Na2HPO4, and 2.64 g of NaH2PO4 × 2H2O per litre, pH 7.5) and clarified by centrifugation at 4,000 g for 5 min at 4 °C. Tenfold dilutions of the supernatant were spread on cystine lactose electrolyte-deficient (CLED) agar (Conda Pronadisa, Spain) plates to prevent the swarming motility of Proteus spp. Plates were incubated overnight at 37 °C. Grown individual colonies were passaged three times under the same conditions. P. mirabilis was identified by sequencing a 1308-bp fragment of 16S rRNA gene using primers 8F 5′-AGRGTTTGATCCTGGCTCA-3′ and 1350R 5′-GACGGGCGGTGTGTACAAG-3′ as described earlier [55]. The sequence of the 16S rRNA gene of the selected P. mirabilis CEMTC 73 strain was deposited in the GenBank database under accession number KF240720. Additionally, P. mirabilis-specific PCR was performed for the CEMTC 73 strain to confirm P. mirabilis phylotyping according to Mansy et al. [36].
Phage isolation and propagation
Bacteriophage PM16 was isolated from the same stool sample that was used to obtain host strain P. mirabilis CEMTC 73. To select bacteriophages, 0.5 g of stool sample was suspended in 2 ml of sterile SM buffer (50 ml of 1 M Tris-HCl, 5.8 g of NaCl, and 2 g of MgSO4 × 6H2O per litre, pH 7.5). The suspension was then clarified by centrifugation at 10,000 g for 15 min and sterilized by filtration through a 0.22-µm filter (Millipore, USA). The filtrate was screened for bacteriophages by spotting 10 µl onto a fresh layer of P. mirabilis CEMTC 73 in the top agar. The plates were incubated for 6 and 18 h at 37 °C, and each plaque was suspended in sterile PBS to extract phage particles. Tenfold dilutions of phage suspensions were spotted on the fresh layer of P. mirabilis CEMTC 73 to obtain single phage plaques for subsequent phage extraction. The cycle of phage dilution and extraction was repeated three times.
Phage PM16 was propagated by infecting 100 ml of a culture of P. mirabilis CEMTC 73 (OD600 = 0.6) with phages at a multiplicity of infection (MOI, i.e., the ratio of phage to bacterium) of 0.1. Phage particles were purified from phage lysate by polyethylene glycol (PEG) precipitation as described earlier [5].
Phage plaque morphology and phage particle stability
The morphology of plaques formed by phage PM16 on a layer of sensitive P. mirabilis CEMTC 73 culture was determined using the double agar overlay method [27]. Plaques were examined after 18 h of incubation.
The sensitivity of phage PM16 to temperature, chloroform, pH and freezing-thawing were tested in parallel using a bacteriophage preparation with an initial titre of 107 plaque-forming units per ml (pfu/ml) in SM buffer. All experiments were carried out twice, three times in each repeat. The final titre in a sample was determined at the end of each experiment using a small-drop plaque assay [37]. For the temperature sensitivity test, the aliquots of phage preparation were incubated at 55 °C for 24 h and at 80 °C for 1 h. For the chloroform susceptibility test, the aliquots of phage preparation containing 5 % (v/v) chloroform were incubated for eight days at room temperature (RT). The sensitivity of PM16 at different pH values was tested by exposing aliquots of phage suspension in SM buffer (range pH 5.0–9.0) for seven days at RT. The ability of PM16 to withstand freezing and thawing was investigated by exposing the bacteriophage sample to eight consecutive freeze-thaw cycles (−20 °C/RT for 30 min in each cycle).
Biological properties of PM16 bacteriophage and host range study
All experiments on the biological properties of phage PM16 were performed twice, three times in each repeat. Phage adsorption experiments were performed according to Kropinski [28]. A one-step growth curve and burst size experiments were carried out as described earlier [43] with slight modifications. Ten millilitres of a mid-exponential-phase culture of P. mirabilis CEMTC 73 was pelleted by centrifugation, and the pellet was re-suspended in 0.5 ml of Luria-Bertani (LB) medium. PM16 bacteriophage with an MOI of 0.001 was added to the cell suspension, and the mixture was incubated for 5 min at 37 °C for phage adsorption. Then, the cells were pelleted by centrifugation and resuspended in 10 ml of LB medium. Incubation was continued for 1 h at 37 °C. Culture aliquots were collected every 5 min, and the phage titre was determined.
A lytic activity assay of phage PM16 was performed as described earlier [22] with our modifications. An exponentially growing culture of P. mirabilis CEMTC 73 (107 cfu/ml) was mixed with phage PM16 (MOI of 0.001). The mixture was then incubated with shaking at 37 °C. Every 30 min, aliquots were taken, and the appropriate dilutions were spread on CLED agar plates, and incubated overnight at 37 °C. The next day, colonies were counted, and these data were used to generate a multistep bacterial killing curve for PM16.
The frequency of the occurrence of phage-resistant mutant bacterial cells was determined by the method described earlier with our modifications [39]. Bacteriophage PM16 with an MOI of 100 was added to a growing culture P. mirabilis CEMTC 73 (108 cfu/ml). The suspension was then mixed and incubated for 10 min at 37 °C. Thereafter, aliquots of the mixture were spread in parallel on CLED agar plates to count colonies, and on LB plates to estimate swarming motility. Plates were incubated overnight at 37 °C, and the next day, the frequency of occurrence of phage-resistant bacteria was determined. A part of each phage-resistant colony from the LB plate was suspended in sterile water and heated at 95 °C for 5 min, and cellular debris was precipitated by centrifugation. The supernatants were then used as templates for further PCR and sequencing. The other parts of colonies used for PCR were passaged three times to assure that the culture was phage-free and then checked for resistance to reinfection by phage PM16 using the small-drop plaque assay [37].
To confirm that each phage-resistant mutant was P. mirabilis, P. mirabilis-specific PCR according to Mansy et al. [36] was performed. In addition, these colonies were identified by sequencing of the 16S rRNA gene fragment using primers 8F 5′-AGRGTTTGATCCTGGCTCA-3′ and 1350R 5′-GACGGGCGGTGTGTACAAG-3′, as described earlier [55].
The absence of PM16 DNA in phage-resistant P. mirabilis-passaged colonies was confirmed using a PCR method to detect three different regions of the phage PM16 genome. Three pairs of primers specific for PM16 DNA polymerase, DNA-dependent RNA polymerase, and internal core protein gene sequences were used for this assay: P16_8585 5′-AACGTGGTGGTAGGGTGTGGTG-3′ and P16_9009 5′-TTCATCGGGTAAGTCATCAGGCA-3′; P16_16880 5′-GGGTGGACGAACTGCTGACCTG-3′ and P16_17277 5′-TCCAAGGCTCATGCTCAAGGTG-3′; P16_30166 5′-GCTAGCAAAGGTGCTCCAGCTAC-3′ and P16_30715 5′-AGCACCAATCACAGCAGTAACAGC-3′; respectively. Touchdown PCR was performed, and the thermal cycling conditions were as follows: 94 °C for 2 min followed by 10 cycles of 94 °C for 30 s; annealing temperature stepdown every cycle of 1 °C (from 60 °C to 50 °C); 72 °C for 1 min. The annealing temperature for the final 20 cycles was 50 °C with denaturation and extension phases as above.
P. mirabilis swarming motility was investigated as described earlier [24]. Bacterial cells were inoculated by picking on the surface of an LB plate with an agar concentration of 0.8 % and then incubated for 16 h at 37 °C.
Lytic activity of phage PM16 was defined for ATCC 25933 P. mirabilis and 11 clinical strains of P. mirabilis collected in Railway Clinical Hospital (Novosibirsk, Russia). The host range was determined by spotting of serial phage dilutions onto freshly prepared lawns of bacteria on agar plates as described earlier [29].
Electron microscopy
A drop of phage PM16 suspension (109 pfu/ml) was adsorbed for 1 min on a copper grid covered with formvar film. The excess liquid was then removed, and the grid was contrasted on a drop of 1 % uranyl acetate for 5-7 s.
To examine the interaction between the phage and the cell, suspensions of P. mirabilis cells (108 cfu/ml) and phage PM16 (109 pfu/ml) were mixed in a droplet on Parafilm and incubated for 30 s. The suspension was adsorbed on a grid and stained with uranyl acetate as described above. In addition, to investigate the interaction of phage PM16 with swarming and non-swarming P. mirabilis cells, ultrathin sectioning was applied. Suspensions of phage PM16 (170 µl with the titre of 109 pfu/ml) and P. mirabilis cells (0.5 ml with 108 cfu/ml) were mixed, incubated for 5 and 15 min, fixed by adding 8 % paraformaldehyde (200 µl), and then pelleted by centrifugation for 10 min at 103 g. The pellets were post-fixed in 1 % osmium tetroxide, routinely processed and embedded in an epon-araldit mixture. Hard blocks were cut using a diamond knife (Diatome, Switzerland) on an EM UC7 (Leica, Germany) ultramicrotome. The sections were routinely contrasted with uranyl acetate and lead citrate.
All of the samples were examined with a JEM 1400 transmission electron microscope (JEOL, Japan), and digital images were collected using a side-mounted Veleta digital camera (Olympus SIS, Germany).
PM16 DNA purification and complete genome sequencing
Phage DNA was extracted from the phage preparation as described earlier [42]. RNase and DNase (Thermo Fisher Scientific, USA) were added to the phage preparation to a final concentration 5 µg/ml, and the mixture was incubated for 1 h at 37 °C. Then, the phage suspension was supplemented with EDTA, proteinase K (Thermo Fisher Scientific, USA) and SDS to final concentrations of 20 mM, 100–200 µg/ml, and 0.5 %, respectively, and the mixture was incubated for 3 h at 55 °C. After that, phage DNA was purified by phenol/chloroform extraction and subsequent ethanol precipitation.
A paired-end library of bacteriophage PM16 was made using a Nextera DNA Sample Preparation Kit (Illumina, Inc, San Diego, USA). Sequencing was carried out using a MiSeq Benchtop Sequencer (SB RAS Genomics Core Facility, ICBFM SB RAS, Novosibirsk, Russia) and a MiSeq Reagent Kit v.1 (2 × 150 base reads). The genome was assembled de novo using CLC Genomics Workbench software v.6.0.1 and resulted in one genomic contig with an average coverage of 304.
To identify DNA fragments containing 5′- and 3′-end genome sequences, phage PM16 DNA was digested with endonuclease EcoRI. Putative 5′- and 3′-end genome fragments were sequenced by the Sanger method using primers 16_Start_L26 5′-TACAGCACCAATAACAGCACTAAGCA-3′ and 16_Fin_U26 5′-CAGACTCAAGGGATGTCCTAGATGGT-3′ designed on the basis of genome sequencing. The PM16 phage genome sequence was deposited in the GenBank database with the accession number KF319020.
Genome analysis
To compare the PM16 complete genome and other phiKMV-like phage genomes from the GenBank database (http://www.ncbi.nlm.nih.gov), Nucleotide BLAST and MAFFT software [26] were used. Putative ORFs in the PM16 phage genome were determined using BioEdit and Vector NTI software [33, 54] with a 150-bp minimum size for each putative ORF. To compare the products encoded by the predicted ORFs with the sequences deposited in the GenBank database, BLASTX and DELTA-BLAST algorithms were used. The predicted ORFs encoding hypothetical proteins identical to hypothetical phage proteins and ORFs without homology to the sequences deposited in the GenBank database were analysed using InterProScan and HHPred software [21, 46]. In addition, the PM16 phage genome was analysed for the presence of potential phage promoter and terminator sequences using the MEME and PHIRE programs [6, 32] and the ERPIN and RNAMotif software [19, 34], respectively. The similarity of the PM16 genome and other phiKMV-like virus genomes was investigated by the cluster analysis method (CLANS), which allows the visualization of the degree of nucleotide sequence resemblance in 2 or 3 dimensions [18].
Phylogenetic analysis
Similar protein sequences were obtained from GenBank using the BLASTP algorithm. Sequences were aligned using MAFFT software. Phylogenetic analysis was performed by the maximum-likelihood method in PhyML software [20]. The amino acid substitution models LG + I + Γ + F as proposed by the ProtTest program 2.4 [1] were used. Edge support was assessed by the Bayes branch supports.
Bacteriophage structural proteins analysis by SDS-PAGE and MALDI-TOF mass spectrometry
Phage particles were purified from phage preparations by CsCl gradient ultracentrifugation [5]. Proteins from purified phage PM16 particles were separated in a 15 % (w/v) SDS-PAGE gel and visualized by Coomassie R250 staining. Gel fragments containing protein bands were cut, out and trypsin digestion was carried out as described earlier [51]. Peptides were extracted from the gel, purified using Zip Tip pipette tips (Millipore, USA), and spotted onto a MALDI-TOF target. Protein identification was performed in a MALDI-TOF Autoflex Speed (Bruker Daltonics, Germany). Tandem mass spectra were obtained in a data-dependent pattern by collecting one full MS scan (m/z range = 700-4500) followed by MS/MS spectra of the most abundant precursor ions. The MS/MS spectra were processed and analysed using FlexAnalysis 3.3 and BioTools 3.2 software (Bruker Daltonics, Germany). Database searches were performed against the PM16 protein database generated from its genome (this study) using Mascot version 2.3 software (Matrix Science, UK).
Results and discussion
Phage PM16 plaque morphology and phage particle stability
Bacteriophage PM16 was obtained from a sample of stool using P. mirabilis strain CEMTC 73 isolated from the same stool sample. Phage PM16 forms large clear plaques with a diameter of approximately 3 mm surrounded with a translucent halo on a bacterial lawn of P. mirabilis CEMTC 73.
Examination of phage PM16 particle stability revealed that phage PM16 titre remains unchanged after incubation of phage PM16 suspension for 24 h at 55 °C, but the phage titre quickly decreased from an initial titre of 107 pfu/ml to 102 pfu/ml after incubation for 30 min at 80 °C and was completely lost after incubation for 1 h at the same temperature. Storage in a buffered solution with pH 5.0–9.0 for a week and incubation in a chloroform-containing suspension (5 % v/v) for eight days did not affect phage viability. Bacteriophage PM16 was stably stored at -20 °C for six months, and exposure to at least eight cycles of freezing and thawing had no effect on its infectivity. Therefore, PM16 appeared to be a stable phage capable of withstanding harsh environmental conditions.
Electron microscopy of bacteriophage PM16 and its interaction with host cells
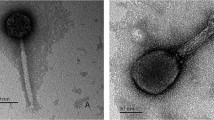
Negative staining of phage PM16 revealed icosahedral heads with a diameter of 50-55 nm connected with a short tail of approximately 10-12 nm in length (Fig. 1A). The morphology and size of the phage particles corresponded to those of Podoviridae family members [2].

Phage PM16 interaction with P. mirabilis CEMTC 73 cells. (A) Icosahedral bacteriophage with a short tail composed of subunits. (B) Phage PM16 particles on the surface of P. mirabilis cells after 30 s of incubation of bacteria with phage. The particle attached to the cell surface is shown in a frame. Negative staining with 1 % uranyl acetate, transmission electron microscopy
After mixing P. mirabilis CEMTC 73 cells with the phage PM16 suspension, the majority of the phage particles were found attached to the cell surface, not on the bacterial flagella (Fig. 1B). Single phage particles were found in close vicinity to the flagella of a few cells; however, these findings were too rare to suggest that sliding on the flagella could be the mechanism to reach the cell surface.
Bacteriophage PM16 biological properties and host specificity
The adsorption rate constant of bacteriophage PM16 to the P. mirabilis CEMTC 73 cells was calculated as 1.6 × 10−8 ml/min (Fig. 2A). A one-step growth curve for phage PM16 revealed a duration of the latent period of 15 min with a burst size of ~100 phage particles per infected cell (Fig. 2B). The multistep bacterial killing curve of the phage PM16 life cycle is shown in Fig. 2C. The number of living bacteria decreased dramatically in the 1.5 h after infection, and afterwards started to increase slowly. The frequency of occurrence of phage-insensitive mutants (BIMs) was (6.3 ± 0.4) × 10−6. Thus, the data demonstrated the high lytic activity of phage PM16 against the sensitive strain P. mirabilis CEMTC 73. Phage PM16 was able to infect only clinical strain CEMTC 73 and none of the 12 tested P. mirabilis strains that were screened in the host range assay. Presumably, PM16 has a narrow host range, but this should be checked using other bacterial strains.

Biological properties of bacteriophage PM16. (A) Adsorption curve of phage PM16 to bacterial cells. Po, initial number of phages; P, number of non-adsorbed phages. (B) One-step growth curve of phage PM16. LP, latent period; BS, burst size. (C) Multistep bacterial killing curve in the phage life cycle of phage PM16. Intact growing P. mirabilis CEMTC 73 cells were used as a control
Complete phage PM16 genome sequencing and analysis
Phage PM16 complete genome sequencing was carried out using a MiSeq Benchtop Sequencer. The assembled nucleotide sequence appeared to be circular, and restriction analysis of phage DNA was performed to estimate the positions of the 5’ and 3’ ends. The exact positions of the 5’ and 3’ ends were determined by the Sanger sequencing method. The size of the PM16 genome was 41,268 bp with terminal repeats 450 bp in length. The GC content of the PM16 genome was 41.4 %, which is close to that in the microbial host genome (GC content, 38.88 %) [44]. This fact indirectly testifies to the long coexistence between phage PM16 and host cells.
The PM16 genome nucleotide sequence was compared with other phage sequences deposited in the GenBank database using algorithms of Nucleotide BLAST, and similarity to the genome of the unclassified Proteus phage PM 75 (NC_027363) was revealed. In addition, the BLASTN algorithm demonstrated low similarity with genome fragments of Vibrio phage VP93 (FJ896200) and Klebsiella phage KP34 (NC_013649), which were earlier classified as phiKMV-like phages [7, 16]. The level of sequence identity to the complete genome sequences of Proteus phage PM16, Proteus phage PM75, Vibrio phage VP93, and Klebsiella phage KP34 was determined using MAFFT software, and it turned out to be 80.7 % for PM16/PM75, 37 % for PM16/VP93, and 40.6 % for PM16/KP34.
Analysis of predicted ORFs
Forty-four predicted ORFs were identified in the phage PM16 genome using the BioEdit Sequence Alignment Editor and Vector NTI Suite 8.0 Software (Table 1). Two more ORFs (32 and 41) were identified by mass spectrometry methods. The coding part of the phage PM16 genome contains 36,847 bp (89 % of the genome). All predicted ORFs are co-directional (Fig. 3). Most of the adjacent ORFs are separated by sequences containing from 1 to 256 bp; seventeen pairs of adjacent ORFs overlapped. ATG is a start codon for 44 ORFs, and only two ORFs contain GTG as a start codon (Table 1).

A schematic map of the Proteus phage PM16 genome. Arrows and black circles denote promoters and terminators, respectively
Protein sequences encoded by the predicted ORFs of the PM16 genome were examined using the BLASTX algorithm. Briefly, products of 20 ORFs shared maximum similarity with proteins with known functions encoded by the genomes of phiKMV-like phages (Table 1). Six amino acid sequences were similar to the hypothetical phage proteins, mostly of the genus Phikmvvirus. For the eighteen putative proteins, no homology was found in the GenBank Database. At the 5’ and 3’ ends of the PM16 genome, three ORFs were found to encode proteins that are similar, with varying degrees of reliability, to the proteins of enterobacteria-specific phages of the families Myoviridae and Siphoviridae. In addition, analysis of all hypothetical ORFs with InterProScan and HHPred software revealed functions for three more ORFs (Table 1).
ORFs of phage PM16 are organized in three functional clusters (Fig. 3). The first of these includes eleven predicted ORFs (Table 1). Presumably, they are early genes expressed at the beginning of infection that provide metabolic changes in cells necessary for the subsequent development of phage infection [47]. The second cluster includes DNA metabolism genes with the DNA-dependent RNA polymerase gene (ORF 26) being the last of them. The third cluster is associated with the assembly of phage capsids, DNA maturation and the outburst of the mature phage particles (Table 1, Fig. 3). The precise determination of functional clusters is difficult, because the PM16 phage genome contains some potential ORFs without homology to known sequences.
The similarity of the putative proteins of PM16 phage to proteins of the phiKMV-like bacteriophages, the presence of the long terminal repeats (450 bp) and the position of the DNA-dependent RNA polymerase gene at the end of the DNA metabolism gene cluster (Table 1, Fig. 3) allow us to classify Proteus phage PM16 as a member of the genus Phikmvvirus, subfamily Autographivirinae [11, 31].
The lysis cassette of phage PM16
The classic mechanism of bacterial host cell destruction during lytic phage infection is mediated by the proteins of the phage lysis cassette. In the Pseudomonas phage phiKMV genome, the prototype representative of the genus Phikmvvirus, the lysis cassette includes the genes encoding the SAR-endolysin, pinholin, Rz, and Rz1 proteins. These genes were identified for phiKMV phage in silico and later confirmed experimentally [9]. Similar lysis cassettes were found during genome analysis of some other Pseudomonas phages from this genus [11, 30]. However, only ORFs encoding putative endolysins had been identified in the genomes of some non-Pseudomonas phiKMV-like phages [3] due to the great variety of bacteriophage lytic protein sequences. In particular, putative genes of lysis cassettes were found in the genomes of the group of KP34-like phages. Their lysis gene cluster differs from the prototype cluster of phage phiKMV and includes spanin, pinholin, and SAR-endolysin [17]. In the genome of PM16, we were able to determine that ORF 45 putatively encodes endolysin, and ORF 43 presumably encodes a protein similar to the Rz1 protein (YP_008059735) of Shigella phage pSf-1 (Table 1).
The regulatory sequences of the PM16 genome
Six potential promoters were found at the 5’ end of the PM16 genome, upstream from the ORF 1 (Table 2, Fig. 3). The presence of few host promoters at the beginning of the genome is a common feature for phiKMV-like phages and their relatives, T7-like phages [11, 16, 31]. No additional potential promoters similar to previously predicted promoters of members of the genera T7virus and Phikmvvirus and specific for phage-encoded DNA-dependent RNA-polymerase were found in the PM16 genome [11, 25]. It is likely that sequences providing late-gene expression are highly specific for the DNA-dependent RNA-polymerase of PM16 phage and differ from other known phage promoter sequences.
Four potential ρ-independent terminators were identified using the RNAMotif and ERPIN software. Two of them, t1 and t2, are located downstream of ORF 1 and ORF 2, respectively, while two other terminators, t3 and t4, are downstream of ORF 31 and ORF 36, respectively (Table 1, 2, Fig. 3). The presence of the terminator downstream of ORF 31, encoding the capsid protein, is typical for members of the genera T7virus and Phikmvvirus [11]. It is known that a similar terminator of T7 bacteriophage operates with an efficiency of less than 100 %, so it is possible that terminator t3 of PM16 allows the structural genes, which are located downstream, to be expressed.
Cluster analysis of phiKMV-like phage genome sequences
Comparative analysis of the PM16 genome and 32 complete genome sequences of other phages of the genus Phikmvvirus was carried out using the CLANS software package [18]. Earlier, this method was used by Drulis-Kawa et al. [16] to compare the Klebsiella phage KP34 genome with the genomes of other bacteriophages of the subfamily Autographivirinae.
PhiKMV-like phage genomes were downloaded from the GenBank database, and phylogenetic information was obtained from GenBank annotations. The list of phiKMV-like phage genomes used in the analysis is shown in Table S1 (Supplementary). It was revealed that the PM16 phage genome lies at a significant distance from other members of this genus and groups with genomes of Vibrio phage VP93 (NC_012662), Klebsiella KP34-like phages (NC_013649, KP708985, KP708986, NC_023567, NC_025418), Pantoea phage LIMElight (NC_019454), Acinetobacter phage Petty (NC_023570), and Enterobacter phage phiKDA1(JQ267518) (Fig. 4).

Graphic layout by the CLANS software for the genome sequences of 33 phiKMV-like viruses using BLASTN searches. The analysed sequences are shown as vertices connected by the edges reflecting attractive forces proportional to the negative logarithm of the HSP P value. The intensity of grayness for connections is proportional to these forces
Phylogenetic analysis of PM16 structural proteins
Phylogenetic analysis was carried out for three structural proteins of phage PM16 encoded by ORF 31, ORF 33, and ORF 34 (Table 1), as well as for the most similar phage protein sequences downloaded from the GenBank database. It was shown that these PM16 structural protein sequences have maximal similarity with the corresponding protein sequences of VP93, KP34-like, phiKDA1 and LIMElight phages (Fig. 5). These data confirmed the result of the comparative genome analysis.

Phylogenetic analysis for three structural proteins of phage PM16. (A) Tail tubular protein A (ORF 33). (B) Tail tubular protein B (ORF 34). (C) Capsid protein (ORF 31). GenBank identifiers (gi) for the sequences are in parentheses. Bayes branch support values above 90 % are given at nodes. The Proteus phage PM16 sequences are indicated by black circles
Mass-spectrometry analysis of phage PM16 structural proteins
Eleven protein bands were revealed by SDS-PAGE (Fig. 6). As a result of peptide mass fingerprinting analysis [53], proteins encoded by ORF 29, ORF 31, and ORF 33–ORF 38 were confirmed to be structural. In addition, MS/MS analysis revealed structural proteins encoded by ORF 28 and ORF 45 (Table 1, Fig. 6) and allowed the identification of two new small ORFs (ORF 32 and ORF 41) (Table 1, Fig. 6). Peptide coverage of protein sequences is shown in detail in Table S2 (Supplementary). It should be noted that the electrophoretic motility of proteins encoded by ORF 35 and ORF 38 was found to be lower than predicted (Table 1, Fig. 6). However, MS/MS analysis confirmed the identification of ORF 35 and ORF 38 (Table S2, Supplementary).

SDS-PAGE of purified bacteriophage PM16 particles, followed by staining with Coomassie brilliant blue R250. Lane M is unstained protein standards SM0431 (Thermo Fisher Scientific, USA)
The influence of phage PM16 infection on P. mirabilis swarming motility
P. mirabilis cells obtained during the phage-resistant bacterial cell assay were additionally investigated. Sixty randomly picked phage-resistant colonies were designated as P. mirabilis CEMTC 73 BIMs 1–60 and were selected for further analysis. As these colonies showed a non-swarming phenotype compared to the original P. mirabilis CEMTC 73 strain (Fig. 7A and B), confirmation that BIMs were indeed derived from CEMTC 73 was required. P. mirabilis CEMTC 73 BIMs 1–60 were tested by P. mirabilis-specific PCR as described earlier [36], and sequencing of the 16S rRNA gene fragment was carried out. P. mirabilis-specific PCR-fragments were found in all tested samples, and all 16S rRNA sequences were identical to the original sequence KF240720. To investigate whether the absence of swarming motility is temporary, the same P. mirabilis CEMTC 73 BIMs 1–60, for which sequencing of the 16S rRNA gene fragment was carried out previously, were passaged five times on LB agar plates with an agar content of 0.8 %. It was demonstrated that the absence of swarming motility remained after five passages (Fig. 7C and D).

Photographs of LB plates containing bacterial colonies of P. mirabilis. (A) Original P. mirabilis CEMTC 73. (B) P. mirabilis mutants obtained during the phage-resistant bacterial cell assay. (C) Swarming colony of original P. mirabilis CEMTC 73. (D) Non-swarming phage-resistant colony of P. mirabilis after five passages on LB plates
The influence of phages on bacterial swarming motility was demonstrated previously with the infection of P. aeruginosa with temperate phages DMS3 and MP22 [14, 55]. It was shown that the genomes of these bacteriophages could be integrated into particular regions of the bacterial host genome and thus change the expression of some host cell genes [14, 56]. In our study, bacteriophage PM16 was determined to be a lytic phage on the basis of its genome organization (Table 1, Fig. 3) and the results of the lytic activity studies (Fig. 2). To confirm that BIMs do not contain phage PM16 DNA, P. mirabilis CEMTC 73 BIMs 1–60 after three passages on LB plates were tested using PCR with primers specific for genes located in different regions of the PM16 genome that are essential for phage viability: PM16 DNA-polymerase, internal core protein, and DNA-dependent RNA polymerase genes. None of the 60 lysates of phage-resistant colonies contained these genes (data not shown).
Electron microscopy of negatively stained phage-resistant cells that had lost their swarming motility revealed prominent changes in their morphology compared to the original P. mirabilis cells (Fig. 8A and B). The original P. mirabilis cells had a rod shape (0.6-0.9 × 2-8 µm) and contained a cytoplasm with high electron density and numerous flagella on the surface. In contrast, phage-resistant cells became roundish (0.8 × 1.3 µm), and they were completely devoid of flagella.

Morphology of phage-sensitive (left column) and phage-resistant (right column) P. mirabilis cells. (A) Rod-shaped cell with flagella. (B) Roundish cells devoid of flagella. (C) Interaction of phage PM16 particles with phage-sensitive cells. Phage-cell interaction is shown in a frame with high magnification. (D) Absence of phages close to phage-resistant cells. (A), (B) - negative staining with 1 % uranyl acetate. Ultrathin sections of the cells are shown in the frames. (C), (D) Ultrathin sections of preparations of bacterial cells co-incubated with phage PM16 for 5 min. Transmission electron microscopy
The study of ultrathin sections of phage-susceptible and phage-resistant bacterial cells revealed more morphological differences between two types of bacterial cells. Phage-susceptible rod-shaped bacteria showed the typical structure of Gram-negative cells with a wavy outer membrane and a distinct periplasmic space. In contrast, roundish phage-resistant cells had a smooth outer membrane, waviness was absent, and the periplasmic space was not visible (Fig. 8). Clear differences in the virus-cell interaction were observed on ultrathin sections of phage-susceptible and phage-resistant cells incubated with PM16 phage. Numerous phage particles were seen around susceptible cells and on their surface after 5 and 15 minutes of incubation, while phages were absent in sections of phage-resistant cells (Fig. 8C and D).
Morphological differences between phage-susceptible and phage-resistant cells indicate some changes in membrane characteristics. The phage-resistant cells showed signs of swelling, presumably related to damage of water-ion transport and membrane charge. It is likely that these differences are associated with a change in membrane macromolecular composition that makes adsorption of phage particles on the cell surface impossible. Taking into consideration that phage PM16 particles are able to attach to the Proteus cell surface but do not use flagella to reach cells (Fig. 1B), we can propose that the resistance of non-swarming P. mirabilis cells to phage PM16 re-infection is connected to the absence of the appropriate receptors on the surface of these cells. The presence of these receptors is possibly associated with the existence of flagella and swarming motility of P. mirabilis cells. This phenomenon needs further investigation.
Conclusion
The genus Phikmvvirus consists of a well-known group of Pseudomonas bacteriophages with a high degree of nucleotide sequence similarity (80–97 %) [11, 12] and bacteriophages specific mostly for γ-proteobacteria [e.g., 3, 7, 17]. Lytic Proteus phage PM16 shows low sequence similarity to the genomes of other phiKMV-like phages and was classified as a member of the genus Phikmvvirus on the basis of cluster organization of its genome, gene synteny, and protein sequence similarities. Phage PM16 is grouped with Vibrio phage VP93, Pantoea phage LIMElight, Acinetobacter phage Petty, Enterobacter phage phiKDA1, and KP34-like bacteriophages. The low nucleotide sequence similarity of phage PM16 to even the most closely related bacteriophages might indicate early divergence of this phage from other phiKMV-like phages.
Bacteriophage PM16 is characterized by high stability, a short latency period, a large burst size, and the occurrence of low phage resistance. This bacteriophage could be included in a Proteus phage cocktail in the future. Notably, Proteus cells resistant to phage PM16 reinfection have reduced swarming motility and a presumably decreased ability to spread infection. Taking into consideration that lytic bacteriophage PM16 and its host swarming strain P. mirabilis CEMTC 73 were found in the same clinical sample, further investigations of PM16-host cell interaction are required.
References
Abascal F, Zardoya R, Posada D (2005) ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105
Ackermann HW (2009) Phage classification and characterization. In: Clokie MRJ (ed), Kropinski AM (ed) Bacteriophages: methods and protocols, vol 1. Humana Press, New York, pp 127–140. doi:10.1007/978-1-60327-164-613
Adriaenssens EM, Ceyssens PJ, Dunon V, Ackermann HW, Van Vaerenbergh J et al (2011) Bacteriophages LIMElight and LIMEzero of Pantoea agglomerans, belonging to the “phiKMV-like viruses”. Appl Environ Microbiol 77:3443–3450
Armbruster CE, Mobley HLT (2012) Merging mythology and morphology: the multifaceted lifestyle of Proteus mirabilis. Nat Rev Microbiol 10:743–754. doi:10.1038/nrmicro2890
Bacteriophage λ and its vectors (2001) In: Sambrook J, Russell D (eds) Molecular cloning. vol. 1. Cold Spring Harbour Laboratory Press, pp 187–303
Bailey TL, Elkan C (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2:28–36
Bastías R, Higuera G, Sierralta W, Espejo RT (2010) A new group of cosmopolitan bacteriophages induce a carrier state in the pandemic strain of Vibrio parahaemolyticus. Environ Microbiol 12(4):990–1000
Belas R, Suvanasuthi R (2005) The ability of Proteus mirabilis to sense surfaces and regulate virulence gene expression involves FliL, a flagellar basal body protein. J Bacteriol 187(19):6789–6803
Briers Y, Peeters LM, Volckaert G, Lavigne R (2011) The lysis cassette of bacteriophage ϕKMV encodes a signal-arrest-release endolysin and a pinholin. Bacteriophage 1:25–30
Carson L, Gorman SP, Gilmore BF (2010) The use of lytic bacteriophages in the prevention and eradication of biofilms of Proteus mirabilis and Escherichia coli. FEMS Immunol Med Microbiol 59:447–455
Ceyssens PJ, Lavigne R, Mattheus W, Chibeu A, Hertveldt K et al (2006) Genome analysis of Pseudomonas aeruginosa phages LKD16 and LKA1: establishment of the phiKMV subgroup within the T7 supergroup. J Bacteriol 188:6924–6931
Ceyssens PJ, Glonti T, Kropinski NM, Lavigne R, Chanishvili N et al (2011) Phenotypic and genotypic variations within a single bacteriophage species. Virol J 8:134
Chong Y, Shimoda S, Yakushiji H, Ito Y, Miyamoto T, Kamimura T, Shimono N, Akashi KJ (2013) Community spread of extended-spectrum β-lactamase-producing Escherichia coli, Klebsiella pneumoniae and Proteus mirabilis: a long-term study in Japan. J Med Microbiol 62(Pt 7):1038–1043. doi:10.1099/jmm.0.059279-0
Chung IY, Sim N, Cho YH (2012) Antibacterial efficacy of temperate phage-mediated inhibition of bacterial group motilities. Antimicrob Agents Chemother 56(11):5612–5617
De Vecchi E, Sitia S, Romanò CL, Ricci C, Mattina R, Drago L (2013) Aetiology and antibiotic resistance patterns of urinary tract infections in the elderly: a 6-month study. J Med Microbiol 62(Pt 6):859–863. doi:10.1099/jmm.0.056945-0
Drulis-Kawa Z, Mackiewicz P, Kęsik-Szeloch A, Maciaszczyk-Dziubinska E, Weber-Dąbrowska B et al (2011) Isolation and characterisation of KP34—a novel phiKMV-like bacteriophage for Klebsiella pneumoniae. Appl Microbiol Biotechnol 90:1333–1345
Eriksson H, Maciejewska B, Latka A, Majkowska-Skrobek G, Hellstrand M, Melefors Ö, Wang JT, Kropinski AM, Drulis-Kawa Z, Nilsson AS (2015) A suggested new bacteriophage genus, “Kp34likevirus”, within the Autographivirinae subfamily of Podoviridae. Viruses 7(4):1804–1822
Frickey T, Lupas AN (2004) CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics 20:3702–3704
Gautheret D, Lambert A (2001) Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J Mol Biol 313:1003–1011
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Jones P, Binns D, Chang H-Yu, Fraser M, Li W, McAnulla C, McWilliam H et al (2014) InterProScan 5: genome-scale protein function classification. Bioinformatics. doi:10.1093/bioinformatics/btu031
Heo YJ, Lee YuR, Jung HH, Lee J, Ko G, Cho YH (2009) Antibacterial Efficacy of phages against Pseudomonas aeruginosa infections in mice and Drosophila melanogaster. Antimicrob Agents Chemother 53(6):2469–2474
Hickman FW, Farmer JJ 3rd (1976) Differentiation of Proteus mirabilis by bacteriophage typing and the Dienes reaction. J Clin Microbiol 3(3):350–358
Hola V, Peroutkova T, Ruzicka F (2012) Virulence factors in Proteus bacteria from biofilm communities of catheter-associated urinary tract infections. FEMS Immunol Med Microbiol 65:343–349
Kassavetis GA, Chamberlin MJ (1979) Mapping of class II promoter sites utilized in vitro by T7-specific RNA polymerase on bacteriophage T7 DNA. J Virol 29(1):196–208
Katoh K, Kuma K, Toh H, Miyata T (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 33:511–518
Kropinski AM, Mazzocco A, Waddell TE, Lingohr E, Johnson RP (2009) Enumeration of bacteriophages by double agar overlay plaque assay. In: Clokie MRJ, Kropinski AM (eds) Bacteriophages: methods and protocols. Humana Press, New York, pp 69–76
Kropinski AM (2009) Measurement of the rate of attachment of bacteriophage to cells. In: Clokie MRJ, Kropinski AM (eds) Bacteriophages: methods and protocols. Humana Press, New York, pp 151–155
Kutter E (2009) Phage host range and efficiency of plating. In: Clokie MRJ, Kropinski AM (eds) Bacteriophages: methods and protocols. Humana Press, New York, pp 141–149
Lammens E, Ceyssens PJ, Voet M, Hertveldt K, Lavigne R, Volckaert G (2009) Representational difference analysis (RDA) of bacteriophage genomes. J Microbiol Methods 77(2):207–213
Lavigne R, Burkal’tseva MV, Robben J, Sykilinda NN, Kurochkina LP et al (2003) The genome of bacteriophage phiKMV, a T7-like virus infecting Pseudomonas aeruginosa. Virology 312:49–59
Lavigne R, Sun WD, Volckaert G (2004) PHIRE, a deterministic approach to reveal regulatory elements in bacteriophage genomes. Bioinformatics 20:629–635
Lu G, Moriyama EN (2004) Vector NTI, a balanced all-in-one sequence analysis suite. Brief Bioinform 5:378–388
Macke TJ, Ecker DJ, Gutell RR, Gautheret D, Case DA, Sampath R (2001) RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res 29:4724–4735
Manos J, Belas R (2006) The genera Proteus, Providencia, and Morganella. In: Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E (eds) Prokaryotes. Springer, Berlin, pp 245–260
Mansy MS, Fadl AA, Ashour MS, Khan MI (1999) Amplification of Proteus mirabilis chromosomal DNA using the polymerase chain reaction. Mol Cell Probes 13:133–140
Mazzocco A, Waddell TE, Lingohr E, Johnson RP (2009) Enumeration of bacteriophages using the small drop plaque assay system. In: Clokie MRJ, Kropinski AM (eds) Bacteriophages: methods and protocols. Humana Press, New York, pp 81–85
McCallin S, Alam Sarker S, Barretto C, Sultana S, Berger B, Huq S, Krause L, Bibiloni R, Schmitt B, Reuteler G, Brüssow H (2013) Safety analysis of a Russian phage cocktail: from metagenomic analysis to oral application in healthy human subjects. Virology 443(2):187–196. doi:10.1016/j.virol.2013.05.022
Merabishvili M, Vandenheuvel D, Kropinski AM, Mast J, De Vos D, Verbeken G, Noben JP, Lavigne R, Vaneechoutte M, Pirnay JP (2014) Characterization of newly isolated lytic bacteriophages active against Acinetobacter baumannii. PLoS One 9(8):e104853. doi:10.1371/journal.pone.0104853
Międzybrodzki R, Borysowski J, Weber-Dąbrowska B, Fortuna W, Letkiewicz S, Szufnarowski K, Pawełczyk Z, Rogóż P, Kłak M, Wojtasik E, Górski A (2012) Clinical aspects of phage therapy. Adv Virus Res 83:73–121. doi:10.1016/B978-0-12-394438-2.00003-7
Morgenstein RM, Szostek B, Rather PN (2010) Regulation of gene expression during swarmer cell differentiation in Proteus mirabilis. FEMS Microbiol Rev 34(5):753–763. doi:10.1111/j.1574-6976.2010.00229.x
O’Flaherty S, Coffey A, Edwards R, Meaney W, Fitzgerald GF, Ross RP (2004) Genome of staphylococcal phage K: a new lineage of Myoviridae infecting gram-positive bacteria with a low GC content. J Bacteriol 186:2862–2871
Pajunen M, Kiljunen S, Skurnik M (2000) Bacteriophage phiYeO3-12, specific for Yersinia enterocolitica serotype O:3, is related to coliphages T3 and T7. J Bacteriol 182(18):5114–5120
Pearson MM, Sebaihia M, Churcher C, Quail MA, Seshasayee AS et al (2008) Complete genome sequence of uropathogenic Proteus mirabilis, a master of both adherence and motility. J Bacteriol 190:4027–4037
Rather PN (2005) Swarmer cell differentiation in Proteus mirabilis. Environ Microbiol 7(8):1065–1073. doi:10.1111/j.1462-2920.2005.00806.x
Remmert M, Biegert A, Hauser A, Söding J (2011) HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods 9(2):173–175. doi:10.1038/nmeth.1818
Roucourt B, Lavigne R (2009) The role of interactions between phage and bacterial proteins within the infected cell: a diverse and puzzling interactome. Environ Microbiol 11:2789–2805
Schmidt WC, Jeffries CD (1974) Bacteriophage typing of Proteus mirabilis, Proteus vulgaris, and Proteus morganii. Appl Microbiol 27(1):47–53
Sekaninová G, Hofer M, Rychlík I, Pillich J, Kolárová M et al (1994) A new phage typing scheme for Proteus mirabilis and Proteus vulgaris strains. Morphological analysis. Folia Microbiol 39:381–386
Sekaninová G, Rychlík I, Kolárová M, Pillich J, Seménka J et al (1998) A new bacteriophage typing scheme for Proteus mirabilis and Proteus vulgaris strains. Analysis of lytic properties. Folia Microbiol 43:136–140
Shevchenko A, Jensen ON, Podtelejnikov AV, Sagliocco F, Wilm M, Vorm O, Mortensen P, Shevchenko A, Boucherie H, Mann M (1996) Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels. Proc Natl Acad Sci 93(25):14440–14445
Siebor E, Neuwirth C (2013) Emergence of Salmonella genomic island 1 (SGI1) among Proteus mirabilis clinical isolates in Dijon, France. J Antimicrob Chemother 68(8):1750–1756. doi:10.1093/jac/dkt100
Thiede B, Höhenwarter W, Krah A, Mattow J, Schmid M, Schmidt F, Jungblut PR (2005) Peptide mass fingerprinting. Methods 35(3):237–247
Tippmann HF (2004) Analysis for free: comparing programs for sequence analysis. Brief Bioinform 5(1):82–87
Wang Y, Qian PY (2009) Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS One 4(10):e7401. doi:10.1371/journal.pone.0007401
Zegans ME, Wagner JC, Cady KC, Murphy DM, Hammond JH, O’Toole GA (2009) Interaction between bacteriophage DMS3 and host CRISPR region inhibits group behaviors of Pseudomonas aeruginosa. J Bacteriol 191(1):210–219
Acknowledgments
Funding: This research was financially supported by the Program of Presidium of the Russian Academy of Sciences “Basic researches for development of medical technologies” (Grant No. 2014-155) and by Ministry of Education and Science of the Russian Federation (Grant No. VI.55.1.1 “Genomics of bacterial and viral communities”).
The authors wish to thank I.V. Saranina, G.B. Kaverina and E.P. Panferova for excellent technical support. They also thank doctors V.V. Anishenko and S.A. Semenov from Railway Clinical Hospital (Novosibirsk, Russia) for providing clinical samples.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors have seen and agree with the contents of the manuscript, and there is no financial interest to report. All co-authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The research was approved by the Local Ethical Committee of the Center of New Medical Technology, Novosibirsk and informed consent from the patient was obtained.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Morozova, V., Kozlova, Y., Shedko, E. et al. Lytic bacteriophage PM16 specific for Proteus mirabilis: a novel member of the genus Phikmvvirus . Arch Virol 161, 2457–2472 (2016). https://doi.org/10.1007/s00705-016-2944-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-2944-2