
Abstract
Bluetongue virus (BTV) mainly infects sheep but can be transmitted to other domestic and wild ruminants, resulting in a considerable financial burden and trade restriction. Our understanding of the origin, movement, and distribution of BTV has been hindered by the fact that this virus has a segmented genome with the possibility of reassortment, the existence of 27 identified serotypes, and a lack of complete sequences of viruses isolated from different parts of the world. BTV serotype 7 is one of the prevalent BTV serotypes in Asia. Nonetheless, no complete genomic sequence of an Asian isolate of this serotype is available. In an effort to understand the molecular epidemiology of BTV infection in China, for the first time, we report here the complete genome sequence of a BTV serotype 7 strain, GDST008, which was isolated in 2014 in China. This sequence also represents the first complete genome sequence of a BTV serotype 7 from Asia and the third one in the world. Sequence analysis suggests that GDST008 consists of segments from BTV viruses of African lineage as well as those from China. Together, these results improve our understanding of the origin, emergence/re-emergence, and movement of BTV and thus can be applied in the development of vaccines and diagnostics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Bluetongue virus (BTV) is the prototype member of the genus Orbivirus in the family Reoviridae and is the etiological agent of bluetongue (BT), an arthropod (Culicoides)-transmitted disease that affects both ruminant and camelid species. BTV infection poses significant risks to animal health, particularly in native ruminant populations, and restricts international trade in livestock. It is therefore listed as a ‘notifiable disease’ by the Office International des Epizooties (OIE) [13].
BTV is a double-stranded RNA (dsRNA) virus, and the viral genome is composed of 10 linear segments (Seg-1 to Seg-10), which encode 12 distinct viral proteins. Seven of these (VP1 to VP7) are structural proteins that form the double shell of the viral particle, whereas the remaining five are non-structural proteins (NS1, NS2, NS3/3a, and NS4) that are required for viral replication in infected cells but are not incorporated into virions [2, 16]. The available BTV sequences show significant variations that reflect different geographic origins around the world, with a clear division of most genome segments into “eastern” and “western” groups/topotypes [9, 10]. Reassortment of BTV genome segments has been identified in vitro as well as in vivo in both vertebrate and vector hosts [14, 17]. The reassortment process provides BTV with a unique capability to acquire new antigenic and biological phenotypes in an efficient fashion. As more full-genome sequences of BTV field strains become available, it is clear that the occurrence of BTV reassortment among isolates is extensive [1, 10, 12, 18].
To date, 27 distinct BTV serotypes (BTV-1 to BTV-27) have been identified worldwide [4], and seven serotypes (BTV-1, -2, -3, -4, -12, -15 and -16) have been identified in China [6]. BTV-7 has been found in Africa, Australia, and Indonesia and has been detected serologically in India and Pakistan, but it has not been reported in China. There are only two BTV-7 full-genome sequences available in the GenBank database: one from South Africa and the other from Australia [3]. As part of our effort to understand the molecular epidemiology of BTV infections in China, we report here the isolation of a BTV-7 strain in China and characterization of its genetic composition. To our knowledge, this is the first report of the isolation of BTV-7 in China and of the first complete genome sequence of the BTV-7 strain from Asia.
In 2014, blood samples taken from sentinel cattle in the Shantou District of Guangdong Province were sent to Yunnan Tropical and Subtropical Animal Virus Diseases Laboratory (YTSAVDL). The ethylenediaminetetraacetic acid (EDTA) blood sample was positive for BTV RNA by quantitative reverse transcription polymerase chain reaction (qRT-PCR) targeting BTV segment 1 (Seg-1) [19]. Embryonated chicken eggs were subsequently inoculated with heparinized blood. Dead embryos at days 2–5 post-inoculation were collected, and their livers were harvested and homogenized. The liver homogenate was passaged once on C6/36 cells. Extensive cytopathic changes were observed after two passages of BHK-21 cells. A subsequent qRT-PCR assay of cell culture supernatants confirmed the presence of BTV RNA. Virus neutralization tests (VNTs) were performed to determine the serotype of the BTV isolate. The viral infection was significantly blocked when the antiserum raised to the BTV-7 South African reference strain was mixed with the prepared viruses, indicating that this isolate belongs to the BTV-7 serotype. The isolated BTV-7 strain was designated GDST008.
For synthesis of full-length cDNA of BTV genome segments, RNA was extracted from infected BHK-21 monolayers using TRIzol Reagent (Life Technologies). BTV dsRNA was separated using differential precipitation of single-stranded RNA (ssRNA) in the presence of 2 M LiCl, and the dsRNA remaining in the supernatant was precipitated with isopropanol. Full-length amplification of cDNAs (FLAC) as described by Maan et al. [8] was conducted using the purified viral dsRNA. The purified cDNA was further subjected to library construction (200 bp in length) using a TruSeq DNA Sample Prep Kit v2 (Illumina) with multiple indexes. Paired-end sequencing was performed on an Illumina MiSeq instrument. Multiple filtering steps were used to remove low-quality sequences, including reads that contained greater than 50 % of low-quality bases (Q20), and to trim the adaptor sequences. After filtration, we performed de novo assembly using Newbler 2.6 and Velvet [22] to construct a consensus sequence for each segment of the BTV genome.
Sequence alignments were performed with the MAFFT software [5]. Phylogenetic trees were constructed by the neighbor-joining method using distance matrices generated by the p-distance determination algorithm in MEGA 6 [20] with 1000 bootstrap replicates. Sequence relatedness was reported as percentage identity. Our nucleotide sequences were submitted to GenBank under the accession numbers KT002578 to KT002587.
Genome segments 1–10 of GDST008 range in size from 3944 (Seg-1) to 822 base pairs (Seg-10), encoding proteins with 1302 (VP1) to 229/216 (NS3/NS3a) amino acids, respectively. The GDST008 strain is a genetic reassortant virus with segments derived from both western and eastern topotype genomes. Seg-1, -3 and -4 of GDST008 are most closely related to several serotypes of BTV from Africa (90 %–97 %) that genetically resemble the western topotype. However, Seg-5, -7, -8, -9, and -10 are genetically most closely related to other BTV isolates from China (95 %–99 %) corresponding to the eastern topotype. The details of individual genome segments are presented in Table 1.
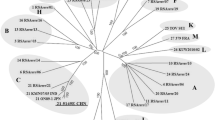
Sequence analysis of GDST008 Seg-2 confirmed the serotype 7 designation determined by VNT, thus eliminating any doubt regarding the serotype classification of this isolate due to possible cross-reaction in serological tests. Phylogenetic analysis of the GDST008 Seg-2 sequence with 27 BTV serotype reference strains and the available BTV-7 full-length Seg-2 sequence confirmed the existing serotype lineages and showed that GDST008 Seg-2 is a new addition to the previously assigned nucleotype F [7] (Fig. 1). The GDST008 Seg-2 sequence grouped with the BTV-7 South African reference strain RSArrrr/7 and Australian strain v6963, showing nt/aa identities of 92.8 %/95.2 % and 92.7 %/94.8 %, respectively. Seg-6 of GDST008 was segregated into the previously assigned nucleotype D [9] along with BTV-7 South African reference strain RSArrrr/7 and Australian strain v6963 and showed nt/aa identities of 94.3 %/99.4 % and 93.9 %/98.7 %, respectively.

Phylogenetic analysis of all available BTV-7 Seg-2 sequences by the neighbor-joining method. The relationships between Seg-2 of GDST008 and 27 BTV serotype reference strains are shown. Twelve nucleotypes (A–L) are indicated. The tree was constructed using distance matrices, using the p-distance determination algorithm in MEGA 6.0 (1000 bootstrap replicates). The bar length is equivalent to 0.05 % sequence divergence. 1 RSArrrr/01-24 represent BTV-1 to -24 serotype reference strains deposited in GenBank (AJ585122-AJ585145); 25 SWI TOV(EU839840), 26 KUW 2010/02 (HM590642), and 27 FRA 379 (KM200718) are the BTV-25, -26, and -27 strains isolated in Switzerland, Kuwait and France, respectively; 7 AUS V6939 (JQ086292), 7 ZAF 1504 (JX272550) and 7 CHN GDST008 (KT002579) are BTV-7 strains isolated in Australia, South Africa, and China, respectively. The new sequence determined in the present study is underlined and in bold
Seg-1, -3 and -4 of GDST008 correspond to the western topotype and are most closely related to BTV strains from Africa (Table 1). Previously, the full genomes of two BTV-1 field strains isolated from Yunnan Province of China were sequenced. All segments of one strain, Y863, isolated in 1979, belong to the eastern topotype, suggesting no reassortment with the viral genomes of western topotype strain [23]. However, all segments of strain SZ97/1, isolated in 1996, were closely related to a South African BTV-1 strain [21], suggesting that the virus of African lineage had entered China probably after 1979. It is worth noting that Seg-1 and -4 of GDST008 also show 96.0 %/99.1 % and 95.3 %/97.8 % nt/aa identity to SZ97/1, indicating that Seg-1 and -4 of this isolate may be derived from the SZ97/1 strain. The genetic evolution of the Chinese BTV strains may have been primarily driven by segment reassortment between the incursive and native strains in China, which may have made the virus more suitable for transmission and survival in the local ecosystem. This suggestion was supported by the fact that Seg-5 of various Indian BTV strains was replaced with the Seg-5 of the invasive western topotype strains [11, 15].
Seg-5, -7, -8, -9 and -10 of GDST008 correspond to the eastern topotype and are genetically most closely related to the BTV-1 Y863 and BTV-4 YTS-4 strains, which were isolated in 1979 and 1996 from China (Table 1), indicating that these segments were likely derived from theY863 or YTS-4 strains. These segments also show higher sequence similarity to various strains isolated in India (93 %–97 %), suggesting that several segments of BTV strains from China and India may share a common origin.
In conclusion, our study provides evidence for the entry of African lineage BTV strains into China and shows that segment reassortment occurred between the introduced and native strains. It is unclear how African strains entered China, and it remains to be determined whether BTV reassortments among member strains of different BTV topotypes resulted in alteration of biological properties, especially virulence. Our data will aid in the development of diagnostic assays, assessments of east-west reassortants, and monitoring of BTV infection in China.
References
Batten CA, Maan S, Shaw AE, Maan NS, Mertens PP (2008) A European field strain of bluetongue virus derived from two parental vaccine strains by genome segment reassortment. Virus Res 137:56–63
Belhouchet M, Mohd Jaafar F, Firth AE, Grimes JM, Mertens PP, Attoui H (2011) Detection of a fourth orbivirus non-structural protein. PLoS One 6:e25697
Boyle DB, Bulach DM, Amos-Ritchie R, Adams MM, Walker PJ, Weir R (2012) Genomic sequences of Australian bluetongue virus prototype serotypes reveal global relationships and possible routes of entry into Australia. J Virol 86:6724–6731
Jenckel M, Breard E, Schulz C, Sailleau C, Viarouge C, Hoffmann B, Hoper D, Beer M, Zientara S (2015) Complete coding genome sequence of putative novel bluetongue virus serotype 27. Genome A 3:e00016-15
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Kirkland PD, Zhang N, Hawkes RA, Li Z, Zhang F, Davis RJ, Sanders DA, Li H, Zhang K, Ben J, He GF, Hornitzky CL, Hunt NT (2002) Studies on the epidemiology of bluetongue virus in China. Epidemiol Infect 128:257–263
Maan S, Maan NS, Samuel AR, Rao S, Attoui H, Mertens PP (2007) Analysis and phylogenetic comparisons of full-length VP2 genes of the 24 bluetongue virus serotypes. J Gen Virol 88:621–630
Maan S, Rao S, Maan NS, Anthony SJ, Attoui H, Samuel AR, Mertens PP (2007) Rapid cDNA synthesis and sequencing techniques for the genetic study of bluetongue and other dsRNA viruses. J Virol Methods 143:132–139
Maan S, Maan NS, Ross-smith N, Batten CA, Shaw AE, Anthony SJ, Samuel AR, Darpel KE, Veronesi E, Oura CA, Singh KP, Nomikou K, Potgieter AC, Attoui H, van Rooij E, van Rijn P, De Clercq K, Vandenbussche F, Zientara S, Breard E, Sailleau C, Beer M, Hoffman B, Mellor PS, Mertens PP (2008) Sequence analysis of bluetongue virus serotype 8 from the Netherlands 2006 and comparison to other European strains. Virology 377:308–318
Maan S, Maan NS, van Rijn PA, van Gennip RG, Sanders A, Wright IM, Batten C, Hoffmann B, Eschbaumer M, Oura CA, Potgieter AC, Nomikou K, Mertens PP (2010) Full genome characterisation of bluetongue virus serotype 6 from the Netherlands 2008 and comparison to other field and vaccine strains. PLoS One 5:e10323
Maan S, Maan NS, Guimera M, Nomikou K, Singh KP, Pullinger G, Belaganahalli MN, Mertens PP (2012) Genome sequence of a reassortant strain of bluetongue virus serotype 23 from western India. J Virol 86:7011–7012
Maan S, Maan NS, Belaganahalli MN, Kumar A, Batra K, Rao PP, Hemadri D, Reddy YN, Putty K, Krishnajyothi Y, Reddy GH, Singh KP, Hegde NR, Nomikou K, Sreenivasulu D, Mertens PP (2015) Genome sequence of bluetongue virus type 2 from India: evidence for reassortment between outer capsid protein genes. Genome A 3:e00045-15
Maclachlan NJ (2011) Bluetongue: history, global epidemiology, and pathogenesis. Prev Vet Med 102:107–111
Ramig RF, Garrison C, Chen D, Bell-Robinson D (1989) Analysis of reassortment and superinfection during mixed infection of Vero cells with bluetongue virus serotypes 10 and 17. J Gen Virol 70(Pt 10):2595–2603
Rao PP, Reddy YV, Hegde NR (2013) Isolation and complete genome sequencing of bluetongue virus serotype 12 from India. Transbound Emerg Dis. doi:10.1111/tbed.12199
Roy P (1992) Bluetongue virus proteins. J Gen Virol 73(Pt 12):3051–3064
Samal SK, Livingston CW Jr, McConnell S, Ramig RF (1987) Analysis of mixed infection of sheep with bluetongue virus serotypes 10 and 17: evidence for genetic reassortment in the vertebrate host. J Virol 61:1086–1091
Shafiq M, Minakshi P, Bhateja A, Ranjan K, Prasad G (2013) Evidence of genetic reassortment between Indian isolate of bluetongue virus serotype 21 (BTV-21) and bluetongue virus serotype 16 (BTV-16). Virus Res 173:336–343
Shaw AE, Monaghan P, Alpar HO, Anthony S, Darpel KE, Batten CA, Guercio A, Alimena G, Vitale M, Bankowska K, Carpenter S, Jones H, Oura CA, King DP, Elliott H, Mellor PS, Mertens PP (2007) Development and initial evaluation of a real-time RT-PCR assay to detect bluetongue virus genome segment 1. J Virol Methods 145:115–126
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Yang T, Liu N, Xu Q, Sun E, Qin Y, Zhao J, Feng Y, Wu D (2012) Complete genomic sequence of bluetongue virus serotype 1 from China. J Virol 86:1288–1289
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829
Zhu J, Yang H, Li H, Xiao L, Wang J, Li N, Zhang N (2013) Full-genome sequence of bluetongue virus serotype 1 (BTV-1) strain Y863, the first BTV-1 isolate of eastern origin found in China. Genome A 1:e00403-13
Acknowledgments
This study was supported by the Special Fund for Agro-Scientific Research in the Public Interest (Project No. 201303035), the National Natural Science Foundation of China (Project No. 31360621) and the Yunnan Applied Basic Research Foundation of China (Project No. 213FZ16).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Heng Yang and Minna Lv contributed equally to this article.
Rights and permissions
About this article

Cite this article
Yang, H., Lv, M., Sun, M. et al. Complete genome sequence of the first bluetongue virus serotype 7 isolate from China: evidence for entry of African-lineage strains and reassortment between the introduced and native strains. Arch Virol 161, 223–227 (2016). https://doi.org/10.1007/s00705-015-2624-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-015-2624-7