
Abstract
Beauveria bassiana is a ubiquitous entomopathogen infecting hundreds of insect species. We have determined the genomic organization and the complete nucleotide sequence of a novel virus isolated from the isolate A24 of B. bassiana. Phylogenetic analysis of the polymerase gene reveals that the virus, tentatively named Beauveria bassiana virus 1, belongs to the family Amalgaviridae and represents a distinct lineage of amalgaviruses infecting fungi.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A growing number of viruses have been detected recently in ascomycetous and, less frequently, basidiomycetous (pathogenic as well as industrially cultivated micro- and macrofungi) fungal species. The taxonomic system for these viruses is continuously growing and being refined. Since 2009, a few new viruses with an undivided, relatively small dsRNA genome separately infecting fungal and plant hosts have been described. Due to similarities in their RdRp sequences with those of members of the families Totiviridae and Partitiviridae but significant differences in genome organization, a family named Amalgaviridae has been newly established [1].
Here, we describe a new RNA virus infecting the ascomycete fungus Beauveria bassiana (Balls.-Criv.) Vuill. The virus has been provisionally named Beauveria bassiana virus 1 (BbV1-A24). B. bassiana is a ubiquitous entomopathogen infecting hundreds of insect species. It is commercially used for microbial control as a mycoinsecticide, and its isolates are used for producing a large number of biologically active secondary metabolites [2]. To date, two viruses have been reported to infect B. bassiana cultures. Both of these are members of the family Totiviridae [3, 4].
A series of fungal isolates belonging to Beauveria bassiana sensu lato obtained from the collection of the Faculty of Agriculture at the University of South Bohemia were screened for the presence of dsRNA elements. Selective extraction of dsRNA was carried out as described by Castillo et al. [5]. The isolate A24 was found to harbor a dsRNA element with an approximate molecular size of 3 kbp and was further investigated. A random cDNA library was prepared from the excised band using a Maxima First Strand cDNA Synthesis Kit for RT-qPCR (Fermentas, Lithuania) with 0.5 pmol of dN6 random primer 5′-CCTGAATTCGGATCCTCCNNNNNN-3′ (synthesized by Generi Biotech, Czech Republic) according to a protocol described by Darissa et al. [6]. PCR reactions were carried out using 2x PPP Master Mix (Top-Bio, Czech Republic) in total volume 30 μL with 0.5 μM of 5′-CCTGAATTCGGATCCTCC-3′ SPA primer (Generi Biotech, Czech Republic). The products obtained were resolved on a 2 % agarose gel and stained with SYBR Green I dye. The PCR products were visualized as a smear with sizes of 100 bp to ~1 kbp. Fragments larger than 500 bp were excised from the gel, purified using a GeneJET Gel Extraction Kit (Fermentas, Lithuania) and ligated into pGEM-T Easy Vector (Promega, USA). The clones that were obtained were screened for the presence of inserts of the appropriate size. Selected clones were sequenced using standard primers (GATC Biotech, Germany). After trimming the raw sequence data to remove the vector backbone and the SPA primer, the processed sequences were assembled (Geneious, Biomatters) into one large contig of around 2.4 kbp. The 3′ and 5′ ends were amplified using a Mint-2 cDNA synthesis kit (Evrogen) with appropriate sequence-specific primers and then sequenced.
The identity of the fungal isolate was confirmed by internal transcribed spacer sequencing using primers ITS1 and ITS4 [7] and was deposited in the GenBank database under accession number KP027410.
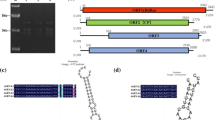
The final contig assembly of BbV1-A24 resulted in a sequence of 3191 bp, which was deposited in the GenBank database under accession number KM233415. Analysis using ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) detected two open reading frames (ORFs) (Fig. 1).

Schematic representation of the genomic organization of Beauveria bassiana virus 1, related amalgamycoviruses, and members of the family Amalgaviridae. The untranslated regions are indicated as solid black lines. Putative ribosomal frameshifting sites are indicated by curved arrows
The genome consists of a 5′ leader region 283 nt long, two ORFs separated by a 59-nt spacer and a 3′ terminal region 110 nt long. The GC content is 53.6 % without extraordinary stretches of either AU or GC, except that GC values exceed 60 % in the few dozen nucleotides nearest the terminus of the genome. RT-PCR amplification of cDNA with combinations of specific primers and an oligo-dT primer produced no products, thereby confirming the absence of a polyadenylated tail. Both ORFs start with a double methionine residue. BLAST analysis of both in silico-translated ORFs revealed that ORF2 contains several motifs characteristic of RNA-dependent RNA polymerases (Cd01699).
A BLAST search using the entire nt sequence of BbV1-A24 revealed sequence similarity to isolate HN28 of Alternaria longipes RNA virus 1 (AlRV1-HN28; GenBank accession number KJ817371, [8]), with 56.7 % aa sequence identity in the putative RdRp. A BLAST search returned no results for the putative protein encoded by ORF1. In silico comparison of the ORF1-encoded proteins of other amalgaviruses with coat proteins of toti- and partitiviruses revealed no similarities in their predicted secondary structure. Extensive beta-strand-rich structures were predicted in the CPs of toti- and partitiviruses, while the ORF1s of amalgaviruses were calculated to consist almost entirely of alpha helices and coils [9–11]. Viral particles were not seen in virion preparations carried out as described by Klootwijk et al. [12] or in ultrathin sections viewed by electron microscopy.
A phylogenetic tree (Fig. 2, Online Resource 1) was constructed based on RdRp aa sequences of related viruses, including partiti-, toti- and chrysoviruses, by the PhyML method using the Phylogeny.fr service (http://phylogeny.lirmm.fr) with default parameters [13]. A multiple alignment was built using Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/) and then curated by removing gaps and statistically testing the alignment using aLTR [14].

Condensed maximum-likelihood phylogenetic tree constructed based on RdRp aa sequences with collapsed lineages of partiti-, chryso- and totiviruses (see uncollapsed phylogenetic tree in Online Resource 1). The analyzed dataset included 216 positions. The support of minimum SH-like and Chi2-based aLTR tests is indicated at the nodes; values lower than 0.5 are not shown
The genomic organization of the amalgaviruses has been described as two overlapping ORFs, with potential “slippery” sequences [9–11] (Fig. 1) where a +1 ribosomal frameshift is likely to occur. The fusion protein translated through this strategy would be a long “gag/pol-like” product [9–11]. The consensus sequence for the slippery sequences of southern tomato virus, rhododendron virus A, and blueberry latent virus is “GGGRRRXR,” where R is a purine base and X is any base. In the case of BbV1-A24, that motif does not lead to in silico translation of any long products via any frameshift mechanism, as there are multiple stop codons downstream. In AlRV-HN28, the sequence GGGAAGAG located at the 3′ terminus of ORF1 could potentially lead to the translation of a fusion protein via a −1 or +2 frameshift (Fig. 1, Online Resource 2). A frameshift mechanism has also been suggested for Ustilaginoidea virens nonsegmented virus 1 [15], while it is obviously not applicable for Ustilaginoidea virens RNA virus M, which is phylogenetically related to BbV1 (Figs. 1 and 2). These data challenge the hypothesis that amalgaviruses infecting fungi use a ribosomal frameshift strategy during their replication. These BbV1-related viruses together with a group of unclassified dsRNA viruses (Cryphonectria parasitica bipartite mycovirus 1, Fusarium graminearum dsRNA mycovirus 4, Heterobasidion RNA virus 6, Rhizoctonia solani dsRNA virus 1) and other amalgaviruses are phylogenetically grouped and closely related to members of the genera Gammapartitivirus and Deltapartitivirus (Fig. 2), which infect plants and fungi, respectively. Maximum-likelihood phylogenetic analysis strongly supports three distinct clades within this group, with RdRp aa identity of 33-53 %, and with Ustilaginoidea virens nonsegmented virus 1 and Zygosaccharomyces bailii virus Z being orphans (Fig. 2, Online Resource 3). Pairwise amino acid analysis of the RdRp genes of partitiviruses shows similar intragenus identity, generally exceeding 40 % [1].
After submission of a revised version of this article, another isolate apparently belonging to the same virus species was reported in B. bassiana and named B. bassiana non-segmented virus (BbNV)-1 [16]. BbV1-A24 and BbNV-1 share 91 % overall nt sequence identity and 98 % aa identity in both predicted proteins.
BbV1 and the group that consists of amalgaviruses and a number of unclassified dsRNA viruses that infect fungal hosts (Fig. 2) together are provisionally called “amalgamycoviruses”. In spite of being phylogenetically related, the viruses within this clade differ in a number of genomic segments and in their organization (Figs. 1 and 2). We therefore propose that BbV1 be placed in the family Amalgaviridae while anticipating future creation of a new taxon that will encompass the amalgamycoviruses. Missing information about genomic organization, replication, the ability to form virus particles, and availability of new sequence data will contribute to accurate classification of amalgamycoviruses and their evolutionary relationships.
References
Nibert ML, Ghabrial SA, Maiss E et al (2014) Taxonomic reorganization of family Partitiviridae and other recent progress in partitivirus research. Virus Res 188C:128–141. doi:10.1016/j.virusres.2014.04.007
Xiao G, Ying S-H, Zheng P et al (2012) Genomic perspectives on the evolution of fungal entomopathogenicity in Beauveria bassiana. Sci Rep 2:483. doi:10.1038/srep00483
Herrero N, Dueñas E, Quesada-Moraga E, Zabalgogeazcoa I (2012) Prevalence and diversity of viruses in the entomopathogenic fungus Beauveria bassiana. Appl Environ Microbiol 78:8523–8530. doi:10.1128/AEM.01954-12
Yie SW, Khalifa ME, Hahn T, Pearson MN (2013) Molecular characterization of a novel victorivirus from the entomopathogenic fungus Beauveria bassiana. Arch Virol 159:1321–1327. doi:10.1007/s00705-013-1938-6
Castillo A, Cottet L, Castro M, Sepúlveda F (2011) Rapid isolation of mycoviral double-stranded RNA from Botrytis cinerea and Saccharomyces cerevisiae. Virol J 8:38. doi:10.1186/1743-422X-8-38
Darissa O, Willingmann P, Adam G (2010) Optimized approaches for the sequence determination of double-stranded RNA templates. J Virol Methods 169:397–403. doi:10.1016/j.jviromet.2010.08.013
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols. A guide to methods and applications. Academic Press, New York, pp 315–322
Lin Y, Zhang H, Zhao C et al (2014) The complete genome sequence of a novel mycovirus from Alternaria longipes strain HN28. Arch Virol. doi:10.1007/s00705-014-2218-9
Sabanadzovic S, Abou Ghanem-Sabanadzovic N, Valverde RA (2010) A novel monopartite dsRNA virus from rhododendron. Arch Virol 155:1859–1863. doi:10.1007/s00705-010-0770-5
Sabanadzovic S, Valverde RA, Brown JK et al (2009) Southern tomato virus: the link between the families Totiviridae and Partitiviridae. Virus Res 140:130–137. doi:10.1016/j.virusres.2008.11.018
Liu W, Chen J (2009) A double-stranded RNA as the genome of a potential virus infecting Vicia faba. Virus Genes 39:126–131. doi:10.1007/s11262-009-0362-1
Klootwijk J, Klein I, Zabel P, van Kammen A (1977) Cowpea mosaic virus RNAs have neither m7G pppN … nor mono-, di- or triphosphates at their 5’ ends. Cell 11:73–82
Dereeper A, Guignon V, Blanc G et al (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:W465–W469. doi:10.1093/nar/gkn180
Guindon S, Dufayard J-F, Lefort V et al (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi:10.1093/sysbio/syq010
Zhang T, Jiang Y, Dong W (2014) A novel monopartite dsRNA virus isolated from the phytopathogenic fungus Ustilaginoidea virens and ancestrally related to a mitochondria-associated dsRNA in the green alga Bryopsis. Virology 462–463:227–235. doi:10.1016/j.virol.2014.06.003
Kotta-Loizou I, Sipkova J, Coutts RHA (2015) Identification and sequence determination of a novel double-stranded RNA mycovirus from the entomopathogenic fungus Beauveria bassiana. Arch Virol 160:873–875. doi:10.1007/s00705-014-2332-8
Acknowledgments
The authors would like to thank the anonymous reviewers for their valuable comments and suggestions. This work was supported by Postdok_BIOGLOBE (CZ.1.07/2.3.00/30.0032), co-financed by the European Social Fund and the State Budget of the Czech Republic (to I.K and K.P) and project LH-13136 Kontakt II - “Mycovirus-host interactions in diseased isolates of phyto- and entomopathogenic micromycetes” from the Ministry of Education, Youth and Sports of the Czech Republic (to K.P).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Koloniuk, I., Hrabáková, L. & Petrzik, K. Molecular characterization of a novel amalgavirus from the entomopathogenic fungus Beauveria bassiana . Arch Virol 160, 1585–1588 (2015). https://doi.org/10.1007/s00705-015-2416-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-015-2416-0