
Abstract
Infecting ducks with duck hepatitis B virus (DHBV) is widely accepted as a relevant model for studying aspects of human HBV infection. However, efficient and sensitive diagnostic methods for the various infection models are limited. In order to provide a more simple and convenient method for serologic diagnosis, we improved the production of recombinant DHBV viral capsid protein (core protein) and then used it to develop an indirect enzyme-linked immunosorbent assay (ELISA) for detecting anti-DHBc antibodies (DHBcAg ELISA) in DHBV-infected ducks. Given the positive/negative cut-off value, the maximum dilution of duck sera in which anti-DHBc antibodies could be detected was 1:12,800. In addition, the DHBcAg ELISA displayed no cross reactivity with duck antisera against duck circovirus (DuCV), duck plague virus (DPV), duck hepatitis virus (DHV), duck swollen head septicemia virus (DSHSV), avian influenza virus (AIV), Riemerella anatipestifer, Salmonella anatum, or Escherichia coli. Furthermore, the coefficients of variation (CVs) of inter-assay and intra-assay experiments were both below than 10 %. When compared to PCR for accuracy on clinical samples from cases of suspected DHBV infection, the DHBcAg showed 95.45 % coincidence with PCR. In conclusion, recombinant DHBc was readily produced and used to establish a simple DHBcAg ELISA that provided a highly specific and sensitive method for analysis of clinical samples.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Duck hepatitis B virus (DHBV), discovered in 1980 [14], is a member of the genus Avihepadnavirus, family Hepadnaviridae, and is the causal agent of duck hepatitis B. The viral envelope of DHBV contains the viral surface antigen (DHBsAg). The icosahedral nucleocapsid within this envelope is composed of the viral core antigen (DHBcAg), which surrounds the DNA genome and viral polymerase. DHBV infection does not cause severe clinical disease in infected ducks or a drop in productivity, but as an animal infection model, it has been widely used for comparative studies of human hepatitis B virus (HBV) infection.
In recent years, a variety of methods have been developed for diagnosing DHBV infection. Most of these methods have focused on detecting viral DNA through routine PCR [24], real-time PCR [21], or Southern blot assays [4, 6], or detecting viral proteins via western blot and immunohistochemistry [23]. In addition, some diagnostic methods that quantify the immune response activated by DHBV infection have also been developed. These approaches include neutralizing antibody tests (NTs) [9] and various ELISAs. NTs have been regarded as the gold standard in virus diagnosis due to their specificity, but this method has a lower sensitivity than other analytical tools, and the results often take several days to acquire [12]. Alternatively, ELISA has been considered the predominant serologic assay in diagnostic research, due to its flexibility and the large number of samples accommodated by this method.
ELISAs for detecting the presence of anti-DHBs and anti-DHBc antibodies have already been developed [11, 17]. Even though anti-DHBs antibodies are present at high levels in the sera of ducks with resolved DHBV infection, the levels of these antibodies in the sera of congenitally and experimentally infected ducks with persistent DHBV infection are low [16, 17]. This makes the use of DHBsAg ELISA for DHBV infection detection less sensitive in these circumstances. Although anti-DHBc antibodies can be detected readily in congenitally or experimentally DHBV-infected ducks using the available DHBcAg ELISA methods [10, 11], a major factor contributing to the limitation of the current protocols is the lack of simple and efficient purification methods for the recombinant DHBc (rDHBc). Therefore, the goal of conducting this study was to simplify the current DHBcAg ELISA protocols.
Materials and methods
Strains, viruses and serum samples
E. coli DH5α and Rosetta (DE3) pLys, expression vector pET-32a(+), and DHBV (CHv strains) were kept in our laboratory. A confirmed DHBV-positive serum sample, used in the development and evaluation of the DHBcAg ELISA, was obtained from artificially infected ducks. Clinical samples from cases of suspected DHBV infection were collected from different farms (Sichuan, China).
Primer design, amplification and cloning of the DHBV PreC/C gene
Based on the DHBV genome sequence (GenBank no. EU429325), primers P1 and P2 were designed to amplify the entire PreC/C gene, which encodes the core protein of DHBV. The sequence of P1 was 5′-CCATGGCTATGGATATCAATGCTTCTAGA-3′, which introduced a restriction enzyme NcoI site (underlined). The sequence of P2 was 5′-GAGCTCTTTCCTAGGCGAGGGAGA-3′, which introduced a site for SacI (underlined) restriction digestion. The viral DNA extracted from DHBV-positive serum was used as the DNA template. The PCR amplification was performed in a 20-μl mixture containing 2 U LA Taq, 2 μl 10× LA PCR Buffer, 2 μl MgCl2 (25 mM), 3 μl dNTP (2.5 mM each), 2 μl DNA template, and 1 μl each of primers P1 and P2 (10 μM). The amplification procedure consisted of denaturation at 95 °C for 5 min followed by 35 cycles of denaturation at 95 °C for 40 s, annealing for 30 s at 60 °C, and extension at 72 °C for 50 s, and then a final extension at 72 °C for 5 min. The PCR product was resolved by electrophoresis in a 1 % (W/V) agarose gel and then purified and ligated into pMD18-T to generate the recombinant plasmid pMD18-T/PreC/C [3]. Insertion was confirmed by digestion with restriction enzymes NcoI and SacI.
Construction of the expression plasmid
pMD18-T/PreC/C was digested with NcoI and SacI and then subjected to electrophoresis in a 1.2 % agarose gel. The DNA fragments of PreC/C gene were purified and ligated into the previously NcoI and SacI-digested expression vector pET-32a(+) to generate the recombinant expression vector pET-32a(+)/PreC/C. pET-32a(+)/PreC/C were then introduced by transformation into component DH5α cells for propagation as described above. After pET-32a(+)/PreC/C was identified by sequencing, it was then introduced into competent E. coli Rosetta (DE3) pLys cells for expression of rDHBc.
Expression and purification of rDHBc
A positive colony of Rosetta (DE3) pLys cells containing pET-32a(+)/PreC/C was collected and inoculated into 5 ml of LB culture medium with 100 μg/ml ampicillin (Amp-LB) for propagation overnight at 37 °C. The cultures were used to inoculate 500 ml of Amp-LB, and expression was induced by addition of IPTG to a final concentration of 1 mM and incubation at 37 °C for 4 h until the optical density value of the cultures at 600 nm (OD600) reached approximately 0.8. Following the incubation, cells were collected and lysed. The cell lysate was then subjected to 12 % SDS-PAGE and analyzed by Coomassie brilliant blue R-250 staining [8]. To increase the yield of rDHBc, the induction conditions for protein expression in E .coli were optimized by testing various temperatures (25, 34 and 37 °C), concentrations of IPTG (0.2, 0.4, 0.6, 0.8, 1.0, 1.2 and 1.4 mM), and durations of induction (0, 1, 2, 3, 4, 5, 6 and 7 h). The expression level achieved under different conditions was assessed by 12 % SDS-PAGE.
Meanwhile, the solubility of the rDHBc was assessed in parallel with the temperature optimization experiments. Briefly, the lysate of the induced cells was centrifuged at 12,000×g, 4 °C, for 20 min. The clear supernatant (soluble fraction) was collected, and the precipitate (insoluble fraction) containing inclusion bodies was suspended in PBS. Both the soluble and insoluble fractions were then analyzed by SDS-PAGE. Based on the result of the solubility analysis, the fractions containing rDHBc were purified using Ni2+-agarose. In brief, the fractions were dissolved in binding buffer (20 mM Tris-HCl pH 7.9, 5 mM imidazole, 0.5 M NaCl, 8 M urea) and loaded into a Ni2+-agarose column. Then, the rDHBc that bound to the Ni2+-agarose was eluted using elution buffer (20 mM Tris-HCl, pH 7.9, 300 mM imidazole, 0.5 M NaCl, 8 M urea). The concentration of the purified protein was measured by the Bradford method [2], and its purity was analyzed by SDS-PAGE.
Development and optimization of the DHBcAg ELISA
A checkerboard titration was performed to determine the optimal working concentrations of coating antigen and sera. Ninety-six-well ELISA plates (Corning, USA) were coated for 12 h at 4 °C with 100 μl of purified rDHBc, that had been diluted 1:10, 1:20, 1:40, 1:80, 1:160, 1:320 and 1:640 with coating buffer (15 μM Na2CO3, 35 μM NaHCO3, pH 9.6). The wells were then washed three times with 200 μl of PBS containing 0.05 % Tween-20 (PBST) and blocked with 1 % BSA (BSA in PBS) at 37 °C for 2 h. DHBV-positive or negative serum at a dilution of 1:10, 1:20, 1:40, 1:80, 1:160, 1:320 or 1:640 was then added to the respective wells, and the plate was incubated for 2 h at 37 °C. Following this incubation, the wells were washed three times with 200 μl of PBST, incubated with 100 μl of 1:2000-diluted peroxidase-labeled goat anti-duck IgG antibody (KPL, USA) for 1.5 h at 37 °C, washed again, and then colorized by addition of 100 μl of TMB (KPL, USA) at 37 °C for 10 min. The reaction was stopped by adding 100 μl of 2 M H2SO4, and the absorbance of each well was measured at 450 nm (OD450). The dilutions that gave the maximum difference in absorbance at 450 nm between positive and negative serum (P/N) were selected for testing the experimental serum samples [26].
Determining the cutoff value of DHBcAg ELISA
Using the optimal conditions, 27 DHBV-negative serum samples from non-immune ducks were used to assess the cutoff value of the DHBcAg ELISA. Each sample was tested in triplicate, and the OD450 value plus three times the standard deviation (SD) was used as the cutoff [7]. All experimental samples were considered positive if the OD450 value was higher than this cutoff value.
Diagnostic sensitivity of the DHBcAg ELISA
Flat-bottom 96-well plates were coated with optimal concentrations of purified rDHBc as described above. DHBV-positive serum samples with twofold serial dilutions from 1:200 to 1:102,400 were subsequently added to each well and incubated with the antigen. PBS and confirmed DHBV-negative and DHBV-positive serum samples were used as blank, negative and positive controls, respectively. The sensitivity of the DHBcAg ELISA was defined as the highest dilution of the positive serum that produced an OD450 value higher than the cutoff value.
Specificity of the DHBcAg ELISA
Confirmed antisera against duck circovirus (DuCV), duck plague virus (DPV), duck hepatitis virus (DHV), duck swollen head septicemia virus (DSHSV), avian influenza virus (AIV), Riemerella anatipestifer, Salmonella anatum and Escherichia coli (stored in our laboratory) were used to evaluate the antigenic cross-reactivity of rDHBc in the DHBcAg ELISA. The OD450 value obtained from each sample was measured and compared to the cutoff value [25]. All samples were tested in triplicate, and antisera of DHBV and PBS served as positive and blank controls, respectively.
Repeatability and reproducibility of the DHBcAg ELISA
Four positive serum samples were used to evaluate the repeatability and reproducibility of the DHBcAg ELISA. Each sample was tested in a single experiment to analyze intra-assay variability (repeatability) or in three independent experiments at different times to analyze inter-assay variability (reproducibility). Each sample was tested in triplicate, and the mean OD450, standard deviation (SD), and coefficient of variation (CV) were calculated for each sample.
Comparison of DHBcAg ELISA and PCR for detection of DHBV
To validate the DHBcAg ELISA as a clinical diagnostic tool, 75 clinical samples from cases of suspected of DHBV infection were tested using our DHBcAg ELISA and routine PCR. PCR detection was performed using a pair of specific primers (P3, 5′-TAGTCACGCTGTCTGCTCTTTT-3′; P4, 5′-CATTTCCAGTCATACCATTCTC-3′). The amplification was initiated by denaturation at 95 °C for 5 min, followed by 34 consecutive cycles of denaturation at 94 °C for 35 s, annealing for 30 s at 46 °C, and extension at 72 °C for 40 s, and then a final extension at 72 °C for 5 min. Confirmed DHBV-positive and negative sera served as controls, and the results obtained from DHBcAg ELISA and routine PCR were compared to evaluate their detection coincidence.
Results
Amplification and cloning of the DHBV PreC/C gene
Given that current ELISA protocols are limited by the lack of methods for obtaining adequate amounts of rDHBc, we first sought to develop a novel means of expressing and purifying this protein. We began by amplifying the PreC/C gene and cloning it into the pMD18-T vector. Agarose gel electrophoresis revealed that the PCR product of PreC/C gene amplification corresponded to the predicted length of 789 bp. Furthermore, the lengths of DNA fragments obtained by digestion of pMD18-T/PreC/C with NcoI and SacI all coincided with the expected lengths (data not shown). These results indicated that the PreC/C gene was successfully amplified and cloned.
Construction of the expression plasmids
The recombinant expression plasmid pET-32a(+)/PreC/C was next generated by ligating the PreC/C gene (789 bp) into the pET-32a(+) vector (5,900 bp). Proper ligation was confirmed by digestion with the restriction enzymes NcoI and SacI. The size of the released fragment corresponded with the expected size of the cloned insert. Sequencing analysis also confirmed that the PreC/C gene was successfully inserted into pET-32a(+), and the PreC/C gene was in frame, without any sequence errors (data not shown).
Expression and purification of the DHBV core protein
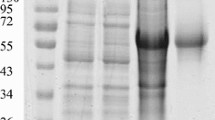
We first induced rDHBc expression by treatment of transformed E. coli cells with 1 mM IPTG at 37 °C for 4 h. A distinct band of approximately 50 kDa (core protein ≈ 30 kDa, His-tags = 20 kDa, His-tagged core protein ≈ 50 kDa), corresponding to the expected size of the rDHBc was observed (Fig. 1a). In addition, rDHBc was not present in the negative control culture to which no IPTG was added. These results confirmed that rDHBc was expressed in E. coli Rosetta (DE3) pLys. The protein expression conditions were also optimized to obtain the maximum amount of rDHBc. The expression level of rDHBc was highest at 37 °C (Fig. 1a), with IPTG concentrations between 0.2 mM and 1.4 mM (Fig. 1b), and incubation for 5 h (Fig. 1c). Thus, for the rest of our study we generated rDHBc at 37 °C in the presence of 0.8 mM IPTG for 5 h.

SDS-PAGE analysis of rDHBc expression and purification (a) E. coli Rosetta/pET-32a(+)/PreC/C was incubated with (+) or without (−) IPTG at different temperatures (25, 34, and 37 °C) and the supernatant (Sup), precipitate (Pre), and total protein of the cell lysate were analyzed. The arrow indicates the His-tagged core protein. (b) E. coli Rosetta/pET-32a(+)/PreC/C was incubated with different concentrations of IPTG (0.2-1.4 mM) at 37 °C for 6 h, and the total protein of each group of induced cells was analyzed. (c) E. coli Rosetta/pET-32a(+)/PreC/C was incubated with 0.8 mM IPTG for 0, 1, 2, 3, 4, 5, 6 and 7 h at 37 °C, and the total protein of each group of induced cells was analyzed. (d) Lane 1, total protein from induced cells; lane 2, purified rDHBc
Next, the distribution of rDHBc was examined in the soluble and insoluble fractions. Most of the recombinant DHBc predominantly was present in the insoluble fraction (precipitate) in the form of inclusion bodies (Fig. 1a). The insoluble fractions were then dissolved and loaded into a Ni2+-agarose column, which should bind the 6×His-tag of the rDHBc construct. Following incubation, the bound protein was eluted and analyzed by SDS-PAGE. A single band corresponding to the molecular weight of rDHBc (50 kDa) was observed (Fig. 1d). The concentration of rDHBc was 2.15 mg/ml, and the final yield of the purified rDHBcAg was approximately 16 mg/liter.
Development of the DHBcAg ELISA
After optimizing the conditions and obtaining sufficient rDHBc, we next sought to develop a more efficient ELISA. To this end, the optimal working dilutions of antigen and serum samples were tested using checkerboard titrations with known positive and negative sera at twofold serial dilutions. The OD450 values of the positive and negative sera gave a maximum difference (P/N = 4.65) when the dilutions of antigen and serum were 1:320 (6.7 μg each well) and 1:160, respectively (Table 1). The optimal dilution for the goat anti-duck HRP antibodies was 1:2000.
Determination of the cutoff value
We next determined the cutoff value for a positive result for the DHBcAg ELISA by analyzing 27 DHBV-negative serum samples. The mean OD450 value of these negative samples was 0.272, with a standard deviation (SD) of 0.04. The cutoff value of the DHBcAg ELISA was then calculated by taking the mean OD450 value plus three times the SD as follows: cutoff value = 0.272 + 3 × 0.04 = 0.392.
Diagnostic sensitivity of the DHBcAg ELISA
After establishing the cutoff value, we next determined the sensitivity of our DHBcAg ELISA using serial dilutions of DHBV-positive serum. The maximum dilution of serum that still had an OD450 value above the cutoff value of 0.392 was 1:12,800 (Fig. 2a). In a parallel experiment, positive and negative DHBV samples were used as controls (data not shown).

Sensitivity and specificity analysis of the DHBcAg ELISA. (a) Twofold serial dilutions from 1:200 to 1:102,400 of DHBV-positive serum were tested by DHBcAg ELISA, and the highest dilution that could be detected using the cutoff value (0.392) was found to be 1:12,800. (b) DHBV-positive serum, PBS and antisera against other duck pathogens were measured using the DHBV DHBcAg ELISA, and the OD450nm values with all antisera except the one against DHBV were lower than the cutoff value (0.392)
Specificity of the DHBcAg ELISA
Given the high sensitivity of our ELISA using rDHBc, we next wanted to determine the specificity of this assay. To this end, the cross-reactivity of the DBHcAg ELISA was evaluated by testing serum samples known to have antibodies against DuCV, DPV, DHV, DSHSV, AIV, R. anatipestifer, S. anatum and E. coli. The OD450 values of all antisera tested were below the cutoff value (Fig. 2b). These results indicate that the DHBcAg ELISA specifically detected DHBV and that rDHBc did not cross-react with antisera against other common duck pathogens.
Repeatability and reproducibility of the DHBcAg ELISA
Having confirmed the sensitivity and specificity of the DHBcAg ELISA, we next performed multiple parallel and sequential assays to test the intra-assay variability (repeatability) and inter-assay variability (reproducibility). First, three replicates with four separate specimens were conducted in the same assay to test repeatability. The OD450 values had CVs between 3.07 % and 6.34 % with a mean value of 4.99 % (Table 2, left). Next, each replicate was evaluated at different times to evaluate the reproducibility of the DHBcAg ELISA. This inter-assay analysis gave a mean CV of 6.15 %, and individual CVs varied from 4.49 % to 8.27 % (Table 2, right). The coefficients of variation for both the intra- and inter-assay comparisons were lower than 10 %, indicating that the DHBcAg ELISA is highly reproducible and stable.
Comparison of DHBcAg ELISA and PCR for DHBV detection
Finally, we wanted to evaluate the reliability of our DHBcAg ELISA protocol in a clinical diagnostic setting. To accomplish this, anti-DHBc antibody levels in 75 clinical serum samples were measured using our DHBcAg ELISA and compared to PCR analysis (Table 3). Serum from 42 samples tested positive for DHBV by DHBcAg ELISA (42/75), and 44 samples tested positive for DHBV (44/75) by PCR. The detection rate was 58.67 % for PCR, and 56 % for ELISA. Furthermore, all 42 samples that tested positive by DHBcAg ELISA were also positive when evaluated by PCR. Thus, the DHBcAg ELISA showed a 95.45 % (42/44) congruence with the PCR assay. These results, suggest that our DHBcAg ELISA is an efficient and reliable diagnostic tool for detecting DHBV.
Discussion
Infection of ducks with DHBV is widely used to model human HBV infection. As a result, many methods have been developed to detect virus levels or measure the immune response following DHBV infection. Currently, DHBsAg and DHBcAg ELISAs are the most common methods for evaluating the host humoral response to DHBV infection. These assays were developed for detecting anti-DHBsAg and anti-DHBcAg antibodies, respectively. Considering that the DHBsAg ELISA has many limitations with regard to clinical implementation, DHBcAg ELISA may be more feasible for clinical application. However, the method used for acquiring antigen in earlier DHBcAg ELISA procedures was a cumbersome process, and the protocols of the DHBcAg ELISA could be simplified by using another HRP-labeled antibody. Therefore, we developed a high-efficiency method for purifying the rDHBc, based on His-tag-targeted affinity chromatography. This method takes less time and has a higher yield than the method used previously to purify rDHBc. Moreover, goat anti-duck HRP antibodies were used in our study to replace the rabbit anti-duck and goat anti-rabbit antibodies that were used in the previous DHBcAg ELISA [16]. These changes make our newly developed DHBcAg ELISA have fewer operational steps and require less time for the entire assay.
The core protein, which is the principal protein component of the DHBV capsid, was successfully expressed in E. coli using a prokaryotic expression vector pET-32a(+) containing a T7lac promoter [5, 22]. For maximum recovery of rDHBc, expression was optimized by testing different induction conditions. Generally, high-level expression of recombinant protein in E. coli often results in the formation of insoluble aggregates called inclusion bodies [18, 20]. As expected, the rDHBc was also contained within inclusion bodies. Hence, a strong denaturant, 8 M urea, was required to solubilize the rDHBc [15]. In addition, a 6×His tag was introduced to both the N-terminal and C-terminal ends of rDHBc. This strategy enhanced the affinity of our rDHBc for Ni2+-agarose columns and increased the efficiency of protein purification. Together, these steps produced an amount of rDHBc that was 50 % of the total E. coli proteins. Moreover, compared with sucrose gradient centrifugation [9], our method of rDHBc purification was simpler and provided a higher yield of purified rDHBc.
To determine if the rDHBc produced under these conditions could be used for serologic diagnosis, an indirect ELISA was developed and evaluated for anti-DHBc antibodiy detection. We first determined the optimal working concentrations of both the coating antigen and sample sera, because these parameters greatly influence background readings and can prevent accurate analysis [13]. Using a checkerboard titration approach, we determined that the least nonspecific binding was observed when rDHBc was coated at 6.7 μg per well and the serum sample was diluted to 1:160. Because anti-DHBc antibody levels can vary between infected ducks, false-negative or false-positive results are possible. To overcome this problem, 27 DHBV-negative serum samples were used to establish a positive cutoff value using the classical method “mean + 3 × SD’’ [19]. In addition, this ELISA exhibited high sensitivity with antisera against DHBV detected at dilutions as low as 1:12,800.
Alonso et al. [1] reported that proteins from E. coli might be incorporated into the coating antigen when a fusion protein expressed by E. coli is used to coat the wells of an ELISA plate. As a result, we specially chose duck antisera positive for E. coli in the specificity analysis. Even with these samples, however, no cross-reactivity was observed. Moreover, the specificity analysis revealed that the DHBcAg ELISA did not cross-react with anti-sera of DuCV, DPV, DHV, DSHSV, AIV, R. anatipestifer or S. anatum [7]. In addition, The CVs for inter-assay and intra-assay variability were both lower than 10 %, suggesting that our DHBcAg ELISA exhibits both high repeatability and reproducibility. Together, these data indicate that the DHBcAg ELISA used for DHBV detection is specific and stable.
Before application of a new assay, it is critical that the performance is evaluated against the current standard using various different clinical samples. In the present study, 75 clinical serum samples of unknown DHBV status were evaluated using our DHBcAg ELISA, and the results were compared with those obtained using a PCR assay. Using our DHBcAg ELISA, 42 samples (56 %) were identified as positive for DHBV, while 44 samples (58.7 %) were identified as positive by PCR. The small variation was most likely due to the ability of PCR to detect DHBV in the early phase of DHBV infection. During this phase, viral DNA is present in sera, but anti-DHBc antibodies have not been produced. Despite this difference, the number of positive samples detected by DHBcAg ELISA was 95.45 % congruent with routine PCR.
In conclusion, this study describes a novel strategy for easily and efficiently purifying rDHBc and provides a highly sensitive and specific DHBcAg ELISA protocol. Moreover, the operational steps of the DHBcAg ELISA have been simplified, making it a useful supplement for clinical implementation for diagnosis of DHBV infection and monitoring host immune responses.
References
Alonso A, Gomes M, Martins M, Sondahl M (1990) Detection of foot-and-mouth disease virus infection-associated antigen antibodies: comparison of the enzyme-linked immunosorbent assay and agar gel immunodiffusion tests. Prev Vet Med 9:223–240
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Cai MS, Cheng AC, Wang MS, Zhao LC, Zhu DK, Luo QH, Liu F, Chen XY (2009) His6-tagged UL35 protein of duck plague virus: expression, purification, and production of polyclonal antibody. Intervirology 52:141–151
Chen YX, Huang AL, Qi ZY, Guo SH (2004) Establishment and assessment of two methods for quantitative detection of serum duck hepatitis B virus DNA. World J Gastroenterol 10:2666–2669
Dubendorff JW, Studier FW (1991) Creation of a T7 autogene. Cloning and expression of the gene for bacteriophage T7 RNA polymerase under control of its cognate promoter. J Mol Biol 219:61–68
Guo H, Mao R, Block TM, Guo JT (2010) Production and function of the cytoplasmic deproteinized relaxed circular DNA of hepadnaviruses. J Virol 84:387–396
Jia R, Cheng A, Wang M, Qi X, Zhu D, Ge H, Luo Q, Liu F, Guo Y, Chen X (2009) Development and evaluation of an antigen-capture ELISA for detection of the UL24 antigen of the duck enteritis virus, based on a polyclonal antibody against the UL24 expression protein. J Virol Methods 161:38–43
Jia R, Cheng A, Wang M, Zhu D, Ge H, Xin H, Liu F, Luo Q, Guo Y, Qi X, Yin Z, Chen X (2009) Cloning, expression, purification and characterization of UL24 partial protein of duck enteritis virus. Intervirology 52:326–334
Jilbert AR, Wu TT, England JM, Hall PM, Carp NZ, O’Connell AP, Mason WS (1992) Rapid resolution of duck hepatitis B virus infections occurs after massive hepatocellular involvement. J Virol 66:1377–1388
Jilbert AR, Botten JA, Miller DS, Bertram EM, Hall PM, Kotlarski J, Burrell CJ (1998) Characterization of age- and dose-related outcomes of duck hepatitis B virus infection. Virology 244:273–282
Jilbert AR, Kotlarski I (2000) Immune responses to duck hepatitis B virus infection. Dev Comp Immunol 24:285–302
Lambert V, Fernholz D, Sprengel R, Fourel I, Deleage G, Wildner G, Peyret C, Trepo C, Cova L, Will H (1990) Virus-neutralizing monoclonal antibody to a conserved epitope on the duck hepatitis B virus pre-S protein. J Virol 64:1290–1297
Li C, Cheng A, Wang M, Zhang N, Shen C, Yang J, Zhu D, Jia R, Luo Q, Chen X (2010) Development and validation of an indirect enzyme-linked immunosorbent assay for the detection of antibodies against duck swollen head hemorrhagic disease virus. Avian Dis 54:1270–1274
Mason WS, Seal G, Summers J (1980) Virus of Pekin ducks with structural and biological relatedness to human hepatitis B virus. J Virol 36:829–836
Middelberg AP (2002) Preparative protein refolding. Trends Biotechnol 20:437–443
Miller DS, Bertram EM, Scougall CA, Kotlarski I, Jilbert AR (2004) Studying host immune responses against duck hepatitis B virus infection. Methods Mol Med 96:3–26
Miller DS, Boyle D, Feng F, Reaiche GY, Kotlarski I, Colonno R, Jilbert AR (2008) Antiviral therapy with entecavir combined with post-exposure “prime-boost” vaccination eliminates duck hepatitis B virus-infected hepatocytes and prevents the development of persistent infection. Virology 373:329–341
Oneda H, Inouye K (1999) Refolding and recovery of recombinant human matrix metalloproteinase 7 (matrilysin) from inclusion bodies expressed by Escherichia coli. J Biochem 126:905–911
Pinto PS, Vaz AJ, Germano PM, Nakamura PM (2000) ELISA test for the diagnosis of cysticercosis in pigs using antigens of Taenia solium and Taenia crassiceps cysticerci. Rev Inst Med Trop Sao Paulo 42:71–79
Schein CH (1990) Solubility as a function of protein structure and solvent components. Biotechnology (NY) 8:308–317
Schmid B, Rösler C, Nassal M (2011) A high level of mutation tolerance in the multifunctional sequence encoding the RNA encapsidation signal of an avian hepatitis B virus and slow evolution rate revealed by in vivo infection. J Virol 85:9300–9313
Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol 185:60–89
Tohidi-Esfahani R, Vickery K, Cossart Y (2010) The early host innate immune response to duck hepatitis B virus infection. J Gen Virol 91:509–520
Walters KA, Joyce MA, Addison WR, Fischer KP, Tyrrell DL (2004) Superinfection exclusion in duck hepatitis B virus infection is mediated by the large surface antigen. J Virol 78:7925–7937
Wen Y, Cheng A, Wang M, Ge H, Shen C, Liu S, Xiang J, Jia R, Zhu D, Chen X, Lian B, Chang H, Zhou Y (2010) A Thymidine Kinase recombinant protein-based ELISA for detecting antibodies to Duck Plague Virus. Virol J 7:77–85
Wu Y, Cheng A, Wang M, Zhang S, Zhu D, Jia R, Luo Q, Chen Z, Chen X (2011) Serologic detection of duck enteritis virus using an indirect ELISA based on recombinant UL55 protein. Avian Dis 55:626–632
Acknowledgments
This research was supported by China Agricultural Research System (CARS-43-8), the Ministry of Education Program (20125103110013), Sichuan Province Research Programs (2013HH0042/ 2013TD0015/ 11ZA084/ 12TD005/ 2011ZO0034/ 2011JO0040), and China 973 program (2011CB111606).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Q. Liu, R. Jia, M. Wang, and J. Huang contributed equal to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Liu, Q., Jia, R., Wang, M. et al. Cloning, expression and purification of duck hepatitis B virus (DHBV) core protein and its use in the development of an indirect ELISA for serologic detection of DHBV infection. Arch Virol 159, 897–904 (2014). https://doi.org/10.1007/s00705-013-1897-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-013-1897-y