
Abstract
Dendrolimus kikuchii Matsumura nucleopolyhedrovirus (DkNPV) is a novel nucleopolyhedrovirus strain that has exhibited high potential as biological control agent against D. kikuchii. In this work, a 1755-bp DkChi gene with sequence homology to a chitinase gene was cloned from the genomic DNA of DkNPV using a DNA fragment library. The DkChi gene, encoding 558 residues protein with a predicted mass of 61.6 kDa, was expressed at high levels in Escherichia coli and purified by affinity chromatography. We confirmed that the prepared protein was the DkChi protein by mass spectrometry analysis. Enzyme activity analysis showed that DkChi had both endo- and exo-chitinase activities. Interestingly, the DkChi protein displayed a strong insecticidal activity against Spodoptera exigua, Hyphantria cunea, Helicoverpa armigera and Lymantria dispar. The results suggest that DkChi is a good candidate protein for significantly contributing to pest control.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In insects, chitin is the major polysaccharide present in the insect cuticle, gut lining or peritrophic matrix, salivary gland, trachea, eggshells and muscle attachment points [1]. Chitinases catalyze the degradation of chitin, usually through hydrolysis of the β-1,4-linkage of the N-acetylglucosamine polymer of chitin to disrupt cuticle and gut physiology in many insect species [2]. Moreover, chitinases also play important roles in morphogenesis and cell division of organisms and inhibiting the growth of fungal mycelium in plants [3]. Chitinases, which have received close attention because of their potential application as bioinsecticides, are thought to be more environment-friendly than chemical pesticides in transgenic plants and biological control agents. Therefore, many chitinases have been isolated from natural sources such as animals, plants and bacteria. Based on their different amino acid sequences, molecular structures and hydrolytic mechanisms, chitinases are classified into two categories: family 18, using the mechanism of substrate-assisted catalysis, and family 19, demonstrating acid catalysis [3, 4]. Most of the prokaryotic and eukaryotic chitinases belong to family 18, whereas chitinases of higher plants and some Gram-positive bacteria are grouped in family 19 [2]. These two families use three primary mechanisms for degrading the chitin chain. Endochitinases cleave chitin randomly at internal sites [5], exochitinases cleave off chitobiose (GlcNAc)2 or chitotriose (GlcNAc)3 from the reducing or non-reducing end of the chitin microfibril [6] and N-acetylglucosaminidases, the third class of chitinolytic enzymes, release monomers of GlcNAc [7–9].
Many baculovirus chitinases belong to glycosyl hydrolase family 18 [10]. These enzymes retain high endo- and exo-chitinase activities between pH 3.0 and 10.0 and even higher alkaline conditions [11, 12]. Chitinases encoded by members of the family Baculoviridae are localized within the endoplasmic reticulum (ER) of infected cells because of the presence of a carboxy-terminal ER retention motif [13–15]. This motif is probably involved in hindering the secretion of chitinase and redistribution of chitinase within the cell during virus infection [11]. Deletion and mutation of the Autographa californica multicapsid nucleopolyhedrovirus (AcMNPV) chitinase KDEL motif resulted in the release of chitinase into the extracellular space and promoted the liquefaction of the host insect [14, 15]. A new recombinant AcMNPV with KDEL-deficient chitinase enhanced the insecticidal activity against Trichoplusia ni larvae and reduced the lethal dose and time until death associated with infection [15].
Baculovirus chitinases are believed to be responsible for the final liquefaction of infected host larvae [11, 12] and are used as a new tool for insect control. For example, treatment with the ChiA protein from AcMNPV resulted in 100 % mortality for Bombyx mori larvae and a significant increase in the number and size of perforations of the peritrophic membrane (PM) [16]. The deletion of chiA from B. mori nucleopolyhedrovirus (BmNPV) evidently delays cell lysis, with clear haemolymph and less degradation of the body in silkworm larvae [17]. Furthermore, chitinase and V-cathepsin, a cysteine protease involved in the degradation of the proteinaceous components of cadavers together promote the liquefaction of the host after death [18, 19]. Deletion of the chiA or cathepsin gene of AcMNPV has been shown to abolish the liquefaction process [12, 20].
Dendrolimus kikuchii Matsumura nucleopolyhedrovirus (DkNPV) is a new virus strain recently isolated from dead D. kikuchii larvae in Mile County, Yunnan Province, China [21]. The virus exhibited higher virulence against the larvae of D. kikuchii than D. Kikuchii Matsumura nuclear polyhedrosis virus (DKMNPV) previously isolated by Yang et al. [21, 22]. Although DkNPV showed insecticidal activity only against D. kikuchii, the insecticidal genes from the DkNPV genome have huge potential in biocontrol and transgenic engineering against other insects.
In this paper, we report the isolation and characterization of an insecticidal protein, DkChi, from DkNPV. We also show the expression of the DkChi gene in Escherichia coli and demonstrate its insecticidal activity against Spodoptera exigua, Hyphantria cunea, Helicoverpa armigera and Lymantria dispar. The results suggest that the DkChi has significant potential for use as a new tool for pest control.
Materials and methods
Viruses
The DkNPV was provided by the Research Centre of Forest Insect Virus, Chinese Academy of Forestry. Virus occlusion bodies (OBs) were purified by centrifugation in a 40-60 % (w/w) sucrose gradient of at 10,000g for 30 min at room temperature. The bands containing the virus were collected and washed with sterile water and then centrifuged three times at 12,000 rpm for 30 min at 4 °C.
Purification of viral DNA
Purified DkNPV was suspended in a buffer containing 0.1 M Na2CO3, 0.15 M NaCl and 0.05 M EDTA, pH 10.8, and incubated at 37 °C for 1 h to dissolve the polyhedron matrix. The pH of the suspension was adjusted to 7.0 with 0.1 M HCl, 0.5 % (w/v) SDS and proteinase K (50 mg/L) were then added successively, and digestion was carried out at 55 °C for 3 h. The solution was extracted with phenol-chloroform-isoamyl alcohol (25:24:1) and chloroform, respectively. DNA was precipitated with two volumes ethanol at −20 °C for 2 h, and pelleted by centrifugation at 12,000 rpm for 10 min. The precipitate was dissolved in TE buffer (pH 8.0) and stored at −20 °C.
Preparation of a DNA library
A DNA fragment library of DkNPV was constructed by shotgun cloning. Purified genomic DNA was sheared by ultrasonication into fragments with an average size of 1200 bp. The ends of each random fragment were repaired using T4 DNA polymerase (Klenow) according to the manufacturer’s protocols. Viral DNA fragments were then cloned into pUC19. Ligation products were introduced into E. coli XL1-Blue competent cells (Stratagene) by transformation. Recombinants were picked randomly. DNA templates for sequencing were further prepared using QIAprep Turbo Kits (QIAGEN) on a QIAGEN BioRobot 9600. Positive clones were sequenced at the Beijing Genomics Institute in China.
Sequence and phylogenetic analysis
The predicted amino acid sequence was determined using the DNAMAN tool. Sequence features, such as signal peptide, pI and molecular mass were evaluated using protein analysis tools (http://expasy.org/tools). The conserved domains and motifs were deduced using PredictProtein. Chitinase sequences were selected from NCBI. The mature protein sequences were aligned with Cluster X version 2.0, and gaps were removed from the alignments. The phylogenetic tree of those alignments was constructed by the neighbor-joining method using the MEGA 4 program, and bootstrap values from 1000 replicates are indicated at the branches.
Construction of bacterial expression plasmids
A truncated sequence of DkChi was amplified by PCR using primers (5′-AAAAGGATCCTTGCCGGGGACGCCACAAATCGA-3′ and 5′-AAAAGAGCTCAACGCGCAACACGACCTCAGA-3′) to generate BamHI and SacI restriction sites (underlined) at the 5′end and 3′end. The PCR mixture included 40 ng DNA, 0.1 μmol forward and reverse primers, 0.4 mM dNTP, 5 U of Expand High Fidelity Taq polymerase (Roche), and 1× polymerase buffer (containing MgCl2) and had a final volume of 50 μL. The reaction conditions for PCR were as follows: 94 °C for 5 min, followed by 33 cycles of 94 °C for 30 s, 60 °C for 45 s, and 72 °C for 1 min, and a final extension at 72 °C for 10 min. The resulting DNA fragment was subcloned into the pQE30 vector (Novagen) with 6×his-tag gene under the control of the T5 promoter.
Overexpression and purification of the recombinant protein
The DkChi protein was overexpressed as a fusion protein with the 6×his-tag in E. coli strain M15 (Novagen). 1 L cells were cultured in a rotary shaker at 37 °C until an OD600 of 0.6 was reached, and the expression of recombinant DkChi was induced by addition of 0.2 mM IPTG at 16 °C for 15 h. The cells were harvested by centrifugation, homogenized in 80 mL buffer A (20 mM Tris-HCl, 150 mM NaCl, 10 mM imidazole, pH 7.5), and sonicated on ice with a Sonifier (300 W, 3 s/2 s). After centrifugation, the soluble fraction was applied to an Ni Sepharose 6 Fast Flow (GE Healthcare) column. The column was equilibrated with buffer A and initially eluted with buffer B (20 mM Tris-HCl, 150 mM NaCl, 20 mM imidazole, pH 7.5). The adsorbed protein was eluted with buffer C (20 mM Tris-HCl, 150 mM NaCl, and 200 mM imidazole, pH 7.5) and buffer D (20 mM Tris-HCl, 150 mM NaCl, and 500 mM imidazole, pH 7.5), sequentially. The adsorbed protein fractions were pooled and dialysed against buffer E (20 mM Tris-HCl pH 7.5) using a 10-kDa Centricon concentrator (Millipore). The dialyzed solution was condensed by freeze drying and loaded onto AKTA FPLC Resource S column (GE Healthcare, NJ, USA). The chromatography was equilibrated with buffer E, and adsorbed protein was eluted in a linear gradient using NaCl from 0 to 0.2 M in buffer E. Finally, the protein was further purified on an AKTA FPLC Superdex 75 HR10/30 (GE Healthcare, NJ, USA) in buffer F (10 mM Tris-HCl, 150 mM NaCl, 2 mM DTT, pH 7.5). The identification and purity of the samples were confirmed by SDS-PAGE (12 % gel).
Mass spectrometry analysis
The protein band was removed from the SDS-PAGE gel with a scalpel, crushed, and destained by washing with 25 mM ammonium bicarbonate containing 50 % acetonitrile for 1 h. Then, the gel pieces were shrunk with 100 % acetonitrile and completely dried before tryptic digestion. The protein was digested with 25 mM NH4HCO3 containing 0.01 % sequencing-grade trypsin (Promega, Madison, WI, USA) at 30 °C overnight, and the mixture was then sonicated for 10 min and centrifuged. The supernatant was removed, and the peptide fragments were extracted twice with saturated matrix solution (α-cyano-4-hydroxycinnamic acid in 60 % acetonitrile and 0.1 % trifluoroacetic acid). The sample was spotted onto the MALDI target plate and air-dried before mass spectrometric analysis.
The peptide was identified by MALDI-TOF MS (ReFlex III, Bruker USA), and mass fingerprint spectra were acquired and analysed by the National Center of Biomedical Analysis, Academy of Military Medical Sciences, Beijing, China.
Protein identification was performed using the Mascot search engine (Matrix science) in the NCBI non-redundant database, and the monoisotopic, mass accuracy, 0.2 kDa and missed cleavages values were set to 1.
Enzymatic activity of DkChi produced in E. coli
Enzyme activity of the recombinant DkChi protein was quantified as described previously [5, 16] using 4-methylumbelliferyl β-D-N,N′ diacetilchitobioside [4MU-(GluNAC)2] and 4-methylumbelliferyl β-D-N, N′,N′′ triacetilchitotrioside [4MU-(GluNAC)3] substrates for estimating exo-chitinase and endo-chitinase activities, respectively. For each standard assay, 20 μL of McIlvaine’s buffer (0.1 M citric acid, 0.2 M dibasic sodium phosphate, pH 5.0) and 5 μL of the appropriate substrate were mixed, and different amounts of protein were added to each tube. After incubation at 30 °C for 30 min, the reaction was terminated by adding 120 μL of 1 M glycine/NaOH buffer, pH 10.6, for 5 min. Fluorescence was detected using a Fluoroskan fluorimeter (Thermo) with excitation at 360 nm and emission at 460 nm. All experiments were repeated three times.
Insect bioassays
S. exigua, H. cunea, H. armigera and L. dispar were provided by the Research Centre of Forest Insect Virus, Chinese Academy of Forestry. Larvae were fed on an artificial diet and reared at 26 ± 1 °C, and 60-70 % relative humidity, with a 14:10 h light:dark photoperiod. The insecticidal activity assay in vivo and determination of 50 % lethal concentration (LC50) were performed as described previously [23] and partly modified. Briefly, a 100-μL volume of each of five appropriate doses of DkChi protein solution (concentration in ng) dissolved in elution buffer were applied to the surface of artificial diet in each 2-cm2 well (Sterilin plates). Control diet was added with elution buffer F only. After the plates were dried, third-instar larvae were divided into groups of 20 larvae (3 per well) and used in the experiments. Mortality was monitored daily after 5 days, and LC50 values were estimated by Probit analysis [24].
Results
Characterization of DkChi
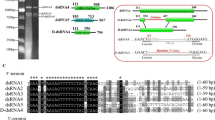
The target DNA was obtained from the DNA fragment library of DkNPV, named DkChi, GenBank accession no. JN680874 (Fig. 1). The CkChn134 gene was 1755 bp long and contained an open reading frame (ORF) of 1674 bp, encoding a 558-residue protein with a theoretical molecular mass of 61.68 kDa, pI 5.7. The deduced protein has the typical features of signal peptide at 1-18 aa. A consensus baculovirus late transcription initiation motif, ATAAG, was found 16 bp upstream from the putative translation start codon ATG. The typical poly(A) signal, AATAAA was not present downstream of the chitinase ORF. The enzyme contained a predicted polycystic kidney (PKD1) domain at aa 45-125 and a characteristic endoplasmic reticulum targeting sequence, REEL, at the C-terminus. Analysis of the DkChi functional motif revealed a conserved family 18 glycohydrolase motif (SIGGWT) and the consensus Prosite motif (FDGIDIDWE) containing the critical glutamate residue, which acts as a proton donor in the catalytic domain. Furthermore, the conserved aromatic residues of DkChi that are involved in the chitin-feeding mechanism and substrate binding were located on the surface of the protein at positions similar to those in SmChiA from S. marcescens and other chitinases [25, 26]. These aromatic residues included Trp-27 and Trp-60 along the immunoglobulin-like fold, Trp224, Trp237 and Tyr162, leading into the catalytic cleft, and Trp159, Typ531, Trp277, Tyr411 and Trp389, forming the catalytic binding site. Additionally, after analyzing the deduced amino acid sequence of DkChi by BLAST in NCBI, we found that DkChi shared a high degree of sequence identity with family 18 chitinases from H. cunea NPV (70 %), Orgyia pseudotsugata MNPV (70 %) and A. californica NPV (69 %). Phylogenetic analysis of baculovirus chitinases showed that DkChi was grouped with HcNPV, OpMNPV, BmNPV and AcMNPV (Fig. S1). This suggests that they may have similar features and functions.

Nucleotide sequence of the DkChi gene and the predicted amino acid sequence of its encoded protein. Capital letters and small letters indicate coding regions and non-coding regions, respectively. Nucleotides are numbered on the left, and amino acids on the right.*, termination codon. The signal peptide is underlined. The typical chitinase 18 glycohydrolase motif is indicated by double underlining. The consensus Prosite motif is shaded and in bold type. An endoplasmic reticulum targeting sequence is shown in bold. The putative motif for late transcription initiation is shaded
Expression and purification of the DkChi protein
In order to analyze the biological activity of DkChi, the protein was expressed in E. coli with a N-terminal 6×His-tag. The recombinant protein was obtained in the major soluble fraction after induction at 16 °C with 0.2 mM IPTG (Fig. 2, lane 2). The recombinant His-tagged DkChi was purified using Ni2+ affinity chromatography (Fig. 2, lane 5). Subsequently, the eluted fractions were collected and further fractionated on a cation exchange chromatography column (Fig. 3a). Finally, the partially purified concentrated protein was further purified by gel-filtration chromatography (Fig. 3b).

SDS-PAGE analysis for expression and Ni2+-affinity chromatography of the DkChi protein. Lane 1, the insoluble fraction of induced cells after sonication; lane 2, the soluble fraction of induced cells after sonication; lane M, molecular mass markers (from the top down, 94.0, 66.2, 43.0, 31.0, and 20.0 kDa); lane 4, culture pellet (uninduced); lane 5, culture pellet (induced with 0.2 mM IPTG at 16 °C). Lane 6, purified DkChi protein eluted with 20 mM Tris, 150 mM NaCl, 500 mM imidazole, pH 7.5

Purification of the DkChi protein from E. coli. a The DkChi protein was purified by cation exchange chromatography on an FPLC-Resource S column. b The DkChi protein was purified by gel-filtration chromatography on FPLC-Superdex 75 HR10/30 column. The purity was checked by SDS-PAGE analysis after each purification procedure
MALDI-TOF mass spectrometry analysis of the DkChi protein
The purified protein was identified by MALDI-TOF-MS on the basis of peptide mass fingerprinting, following in-gel digestion with trypsin. The peptide mass fingerprinting data were matched with the theoretical peptide masses of all proteins from viruses in the NCBI database, using Mascot. A representative spectrum of trypsin-digested protein is shown in Fig. 4, and only JN680874 protein from DkNPV was obtained as a result, with a score of 123. This demonstrated that the purified DkChi protein is the JN680874 protein from DkNPV.

Identification of the purified DkChi protein based on its MALDI-TOF peptide mass fingerprint (PMF). The PMF analysis was made from fragments of DkChi protein derived through trypsin digestion. The sequence coverage of these fragments is shown in bold red type
Enzyme activity analysis of DkChi
In order to test the DkChi exo- and endo-chitinase activities, we measured the enzyme activity of DkChi using 4-MU-(GlcNAC)2 and 4-MU-(GlcNAC)3 substrates, respectively. As shown in Fig. 5, both exo- and endo-chitinase activities increased with the DkChi protein concentration up to 4.7-fold and 3.7-fold, respectively, over their lowest levels,. These data indicate that the DkChi enzyme produced in E. coli was active and exhibited its native exo- and endo-chitinolytic activities.

Enzymatic assay of DkChi purified from E. coli cells. Increasing amounts of the recombinant ChiA were incubated either with 4-MU-(GlcNAc)2 to detect exo-chitinase activity (a) or with 4-MU-(GlcNAc)3 to detect endo-chitinase activity (b). Results are expressed as means ± SE (n = 3)
In vivo assays on larvae
The DkChi protein showed a strong insecticidal activity against S. exigua, H. cunea, H. armigera and L. dispar. The LC50 (50 % lethal concentration) values for the larvae were 192.4, 305.3, 378.9 and 431.7 ng/cm2, respectively (Table 1).
Discussion
In this paper, DkChi (JN680874) from Dendrolimus kikuchii nucleopolyhedrovirus was characterized. Based on the alignment with HcNPV and other NPVs, several functional consensus motifs identified in baculovirus chitinase genes were found in the deduced DkChi full-length sequence. It contained an N-terminal secretion signal that was cleaved upon translation and a C-terminal ER-retention sequence (Fig. 1) that is probably involved in retaining the enzyme inside the cell. ER-retention sequences have also been identified in the chitinases of B. mori NPV [27], AcNPV [14, 15] and HaSNPV [10]. Because of the ER retention motif (KDEL), chitinase of AcMNPV was localized in the ER during infection, and deletion of the KDEL motif resulted in earlier secretion into the medium from infected cells [14, 15]. Furthermore, the two major domains, a PKD1 domain and a catalytic domain indicative of the family 18 glycohydrolase, which were deduced from the DkChi sequence are similar to the known structures present in SmchiA [28]. The PKD1 domain, which forms an immunoglobulin-like fold, is involved in carbohydrate splitting and guiding the substrate into the catalytic groove [29, 30]. The catalytic domain forms a deep substrate-binding cleft [25, 26]. In addition, conserved tryptophan residues along the PKD1 fold and other aromatic residues in the catalytic domain have been found on the surface of SmChiA and other chitinases [25, 26]. The conserved residues of SmChiA, included Trp69 and Trp33 in the N-terminal domain and Trp245 in the catalytic domain, which play a vital role in chitin binding, and Phe-232 guides the chitin chain into the catalytic cleft [25]. Young et al. [26] reported that ORF110 from Epiphyas postvittana nucleopolyhedrovirus (EppoNPV) has conserved residues Trp223, Trp236 and Tyr161 leading into the catalytic cleft and Trp158, Trp529, Trp266, Tyr410 and Trp388 forming the catalytic binding site. Analogously, DkChi has Trp-27 and Trp-60 in the corresponding positions along the immunoglobulin-like fold, and Trp224, Trp237 and Tyr162 may also aid in feeding the insoluble chitin chain into the catalytic pocket and thus to the active site, and Trp159, Typ531, Trp277, Tyr411 and Trp389 probably form hydrophobic interactions with hydrophobic faces of the alternating glucosamine units of chitin, thus producing the binding sites.
To investigate its biological activity, the truncated DkChi gene lacking the N-terminal signal peptide sequence and C-terminal ER-retention sequence (REEL) was expressed in E. coli. A small amount of the recombinant DkChi protein was stored in inclusion bodies, and large amount was in soluble cytosolic components (Fig. 2). In order to avoid the multi-step renaturing processes required to recover the enzyme activity, the recombinant protein was efficiently purified in its native form and further identified by SDS-PAGE and MALDI-TOF MS (Figs. 3, 4). The DkChi protein showed both exo- and endo-chitinase activities using 4-MU-(GlcNAc)2 and 4-MU-(GlcNAc)3, respectively, as substrate, (Fig. 5). This is consistent with previous reports showing that baculovirus chitinases have both exo- and endo-chitinases activities [11, 12, 16].
The DkChi of DkNPV displayed an obvious insecticidal activity agianst S. exigua, H. cunea, H. armigera and L. dispar (Table 1). This indicates that DkChi has great potential for use in pest control. Previous studies have demonstrated the role of baculovirus chitinase. Rao et al. reported that ChiA of AcMNPV resulted in a decrease in larval body weight (LW) of B. mori at a dose of 0.56 μg/g LW and 100 % mortality at a dose of 1 μg/g LW [16]. ChiA from BmNPV could aid in degrading the chitinous PM lining the B. mori larval midgut [17]. Whether the insecticidal mechanism of DkChi is due to the damage of PM needs to be investigated further.
In summary, in this work, we have identified a new chitinase from DkNPV as a candidate protein capable of protecting crops and forests against insect pests.
References
Kramer KJ, Muthukrishnan S (2005) Chitin metabolism in insects. In: Gilbert LI, Iatrou K, Gill S (eds) Comprehensive molecular insect science. Elsevier, Oxford, pp 111–144
Cohen-Kupiec R, Chet I (1998) The molecular biology of chitin digestion. Curr Opin Biotechnol 9:270–277
Kasprzewska A (2003) Plant chitinases-regulation and function. Cell Mol Biol Lett 8:809–824
Sharma N, Sharma KP, Gaur RK, Gupta VK (2011) Role of chitinase in plant defense. Asian J Biochem 6:29–37
McCreath KJ, Gooday GW (1992) A rapid and sensitive microassay for determination of chitinolytic activity. J Microbiol Methods 14:229–237
Brurberg MB, Synstad B, Klemsdal SS, van Aalten DMF, Sundheim L, Eijsink VGH (2001) Chitinases from Serratia marcescens. Recent Res Dev Microbiol 5:187–204
Tews I, Vincentelli R, Vorgias CE (1996) N-Acetylglucosaminidase (chitobiase) from Serratia marcescens: gene sequence, and protein production and purification in Escherichia coli. Gene 170:63–67
Tews I, Perrakis A, Oppenheim A, Dauter Z, Wilsion KS, Vorgias CE (1996) Bacterial chitobiase structure provides insight into catalytic mechanism and the basis of Tay-Sachs disease. Nat Struct Biol 3:638–648
Williams SJ, Mark BL, Vocadlo DJ, James MN, Withers SG (2002) Aspartate 313 in the Streptomyces plicatus hexosaminidase plays a critical role in substrate-assisted catalysis by orienting the 2-acetamido group and stabilizing the transition state. J Biol Chem 277:40055–40065
Wang H, Wu D, Deng F, Peng H, Chen X, Lauzon H, Arif BM, Jehle JA, Hu Z (2004) Characterization and phylogenetic analysis of the chitinase gene from the Helicoverpa armigera single nucleocapsid nucleopolyhedrovirus. Virus Res 100:179–189
Hawtin RE, Arnold K, Ayres MD, Zanotto PM, Howard SC, Gooday GA, Chappell LH, Kitts PA, King LA, Possee RD (1995) Identification and preliminary characherization of a chitinase gene in the Autographa californica nuclear polyhedrosis virus genome. Virology 212:673–685
Hawtin RE, Zarkowska T, Arnold K, Thomas CJ, Gooday GW, King LA, Kuzio JA, Possee RD (1997) Liquefaction of Autographa californica nucleopolyhedrovirus infected insects is dependent on the integrity of virus-encoded chitinase and cathepsin genes. Virology 238:243–253
Thomas CJ, Brown HL, Hawes CR, Lee BY, Min M, King LA, Possee RD (1998) Localization of a baculovirus-induced chitinase in the insect cell endoplasmic reticulum. J Virol 72:10207–10212
Saville GP, Thomas CJ, Possee RD, King LA (2002) Partial redistribution of the Autographa californica nucleopolyhedrovirus chitinase in virus-infected cells accompanies mutation of the carboxy-terminal KDEL ER-retention motif. J Gen Virol 83:685–694
Saville GP, Patmanidi AL, Possee RD, King LA (2004) Deletion of the Autographa californica nucleopolyhedrovirus chitinase KDEL motif and in vitro and in vivo analysis of the modified virus. J Gen Virol 85:821–831
Rao R, Fiandra L, Giordana B, de Eguileor M, Congiu T, Burlini N, Arciello S, Corrado G, Pennacchio F (2004) AcMNPV ChiA protein disrupts the peritrophic membrane and alters midgut physiology of Bombyx mori larvae. Insect Biochem Mol Biol 34:1205–1213
Wang F, Zhang C, Shyam Kumar V, Wu X (2005) Influences of chitinase gene deletion from BmNPV on the cell lysis and host liquefaction. Arch Virol 150:981–990
Ohkawa T, Majima K, Maeda S (1994) A cysteine protease encoded by the baculovirus Bombyx mori nuclear polyhedrosis virus. J Virol 68:6619–6625
Hom LG, Volkman LE (2000) Autographa californica M nucleopolyhedrovirus chiA is required for processing of V-CATH. Virology 277:178–183
Slack JM, Kuzio J, Faulkner P (1995) Characterization of v-cath, a cathepsin L-like proteinase expressed by the baculovirus Autographa calfornica multiple nuclear polyhedrosis virus. J Gen Virol 76:1091–1098
Yang M, Li M, Wang Y, Qu L, Huai K, Nie X, Qiao L, Ding J, Zhang Y (2011) Virulence and characteristics of a new nucleopolyhedrovirus strain of Dendrolimus kikuchii (Lepidoptera: Lasiocampidae). Afr J Microbiol Res 5:2261–2265
Yang D, Wang Y, Li Y (1986) Studies on the nuclear polyhedrosis virus disease of simao pine caterpil-lcar infected by Dendrolimus kikuchii Matsumura (Lepidoptera: Lasiocampidae). J Yunnan Univ 1:105–110
Rajamohan F, Cotrill JA, Gould F, Dean DH (1996) Role of domain II, loop 2 residues of Bacillus thuringiensis CryIAb delta-endotoxin in reversible and irreversible binding to Manduca sexta and Heliothis virescens. J Biol Chem 271:2390–2396
Finney DJ (1971) Probit analysis, 3rd edn. Cambridge University Press, Cambridge
Uchiyama T, Katouno F, Nikaidou N, Nonaka T, Sugiyama J, Watanabe T (2001) Roles of the exposed aromatic residues in crystalline chitin hydrolysis by chitinase A from Serratia marcescens 2170. J Biol Chem 276:41343–41349
Young VL, Simpson RM, Ward VK (2005) Characterization of an exochitinse from Epiphyas postvittana nucleopolyhedrovirus (family Baculoviridae). J Gen Virol 86:3253–3261
Gomi S, Majima K, Maeda S (1999) Sequence analysis of the genome of Bombyx mori nucleopolyhedrovirus. J Gen Virol 80:1323–1337
Perrakis A, Tews I, Dauter Z, Oppenheim AB, Chet I, Wilson KS, Vorgias CE (1994) Crystal structure of a bacterial chitinase at 2.3 Å resolution. Structure 2:1169–1180
Bork P, Doolittle RF (1992) Proposed acquisition of an animal protein domain by bacteria. Proc Natl Acad Sci USA 89:8990–8994
Perrakis A, Ouzounis C, Wilson KS (1997) Evolution of immunoglobulin-like modules in chitinases: their structural flexibility and functional implications. Fold Des 2:291–294
Acknowledgments
This work was supported by the Program of National Nature Science Foundation of China (grant no. 31200491) and State 863 Project funded by Ministry of Science and Technology (grant no. 2012AA101503), P.R. China.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
705_2013_1775_MOESM1_ESM.tif
Fig. S1 Phylogenetic tree of chitinases from baculovirus. The tree was constructed by the neighbor-joining method, and bootstrap values are indicated at the branches. The amino acid sequences of chitinases were from the following viruses: Dendrolimus kikuchii NPV (JN680874), Hyphantria cunea NPV (YP473218), Orgyia pseudotsugata MNPV (NP046280), Bombyx mori NPV (NP047523), Autographa californica MNPV (NP054156), Spodoptera exigua MNPV (NP037779), Helicoverpa armigera SNPV (AAK28347) and Spodoptera litura NPV (NP258310) (TIFF 292 kb)
Rights and permissions
About this article
Cite this article
Wang, Q., Qu, L., Zhang, Z. et al. Characterization of a novel chitinase, DkChi, from Dendrolimus kikuchii nucleopolyhedrovirus. Arch Virol 158, 2523–2530 (2013). https://doi.org/10.1007/s00705-013-1775-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-013-1775-7