
Abstract
Marek’s disease virus Chinese strain GX0101, isolated in 2001, is the first reported recombinant gallid herpesvirus type 2 (GaHV-2) field strain with one reticuloendotheliosis virus (REV) long terminal repeat (LTR) insert. We constructed an infectious bacterial artificial chromosome (BAC) clone of GX0101, which showed characteristics very similar to those of the parental virus in replication and pathogenicity. Using the GX0101 BAC clone, the complete genome of GX0101 was sequenced and analyzed. The length of the GX0101 genome is 178,101 bp, and it contains only one REV-LTR insert at a site 267 bp upstream of the sorf2 gene.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
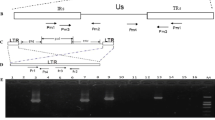
Marek’s disease (MD) is related to T-cell lymphomas in chickens. The causative agent is gallid herpesvirus type 2 (GaHV-2), a member of the genus Mardivirus within the subfamily Alphaherpesvirinae of the family Herpesviridae [1, 2]. GX0101 is the first natural recombinant GaHV-2 field strain isolated from birds showing tumors in China [3]. It contains a 538-bp reticuloendotheliosis virus (REV) long terminal repeat (LTR) inserted between nucleotide bases “C” and “A” numbered 153,175–153,176 (Md5 strain) or 154,507– 154,508 (RB1B strain) [4, 5]. GX0101 is a very virulent GaHV-2, with greater horizontal transmission ability than Md5, while other reported recombinant GaHV-2 strains with an REV-LTR [6], such as RM1 obtained from cell cultures, are attenuated and do not cause tumors [7–9].
Sequencing of 40 μg of GX0101-BAC [10] (200 ng/ul) was carried out commercially using a pyrosequencing platform, the Genome Sequencer 20 FLX System (454 Life Science Corporation). Problematic regions containing mononucleotide reiterations or repetitive regions were also scrutinized using PCR products derived from parental GX0101 DNA.
GX0101-BAC DNA sequences were assembled from 13,596 reads (average length 378 bp) using the Sequencher Program (Gene Codes, Ann Arbor, MI). On average, the final sequence represents a 28-fold coverage at each base pair. Ambiguities in the GX0101 BAC sequencing data were resolved by re-sequencing using an ABI-3730XL automated DNA sequencer (Applied Biosystems, Foster City, CA). Open reading frames (ORFs) and DNA regulatory sequences were maintained and analyzed using DNASTAR (Madison, WI), and other Web-based tools. The sequences of the GaHV-2 strains Md5, 814, pC12/130-10, pC12/130-15, CVI988/Rispens, GA, and RB1B used in comparisons were obtained from GenBank [1, 2, 11–14].
The complete sequence of GX0101 (GenBank no. JX844666) genome is 178,101 bp, not including the BAC vector sequence. The partial sequence in the US2 region that was lost during the construction of GX0101-BAC was added by sequencing PCR products of the parental GX0101 DNA [10]. The lengths of the terminal repeat long (TRL) region, the unique long (UL) region, the internal repeat long (IRL) region, the internal repeat short (IRS) region, the unique short (US) region, and the terminal repeat short (TRS) region are 12,758, 113,572, 12,741, 12,700, 11,695, and 13,134 bp, respectively. The genome contains about 200 ORFs and two copies of a 132-bp repeat sequence.
GX0101 genome contains only one REV-LTR insert of 538 bp (corresponding to nt 152,724-153,261) located within the sorf1 gene and 267 bp upstream of the sorf2 gene. Unlike Md5, GX0101 encodes only one SORF2 protein (MDV 087) [2], which is most similar to strains pC12/130-10, pC12/130-15, and GA. The sorf2 gene is nonessential for virus replication and tumor formation [15]. The LTR from REV may act as a strong promoter or enhancer, affecting the transcription of GaHV-2 sorf2 and probably enhancing the expression of the GaHV-2 SORF2 protein [16]. In the RM1 strain, the REV-LTR insertion site is farther upstream of both sorf1 and sorf2 [8]. More interestingly, the REV-LTR inserted in GX0101 is more stable than it is in other similar recombinant GaHV-2 strains. RM1 has obtained another REV-LTR repeat in its TRS during passage [8]. Furthermore, the RM1 REV-LTR insert is not stable during passage in chickens when inserted into Md5 at the same site and is lost after several passages in vivo [17]. In contrast, GX0101 is very stable in maintaining its REV-LTR insert even after 20 passages in chickens.
At least 160 ORFs, originally defined by annotating the GaHV-2 genomes GA, Md5, 814, pC12/130-10, pC12/130-15, CVI988/Rispens, and RB1B, were examined for mutations. Table 1 presents the percentage similarity and length difference relative to the homologs found in the genomes of attenuated (CVI988/Rispens, 814, pC12/130-15) and virulent (GA, RB1B, Md5, pC12/130-10) strains. Some ORFs are also noted with putative functions according to their homologs in other alphaherpesviruses or their protein-binding partners. ORFs containing frameshift mutations were also re-examined using PCR products derived from the parental GX0101 DNA. ORFs with high similarity scores (100 %) were omitted, while those with low similarity scores (<98 %) indicative of frameshift mutations, truncations, and non-synonymous amino acid substitutions were re-examined using multiple protein alignments with homologous ORFs from CVI988/Rispens, 814, GA, RB1B, Md5, pC12/130-10, and pC12/130-15 strains.
In order to identify amino acid changes that are important for horizontal transmission ability and pathogenicity, the nucleotide sequence of GX0101 was compared with sequences of the attenuated (CVI988/Rispens, 814, pC12/130-15) and virulent (RB1B, Md5, GA, pC12/130-10) strains by multiple sequence alignment. As listed in Table 2, in addition to the 14 GX0101-specific amino acid mutations identified in its ORFs, 61 single nucleotide polymorphisms in 23 genes were identified when compared with the virulent RB1B, Md5, and the vaccine strain CVI988/Rispens. pC12/130-10 and pC12/130-15 contained only two polymorphisms relative to GX0101 in the rlorf7 and icp4 genes.
When all of the ORFs of GX0101 were compared with those of other GaHV-2 strains (814, CVI988/Rispens, GA, RB1B, Md5, pC12/130-10, and pC12/130-15), only 11 of them differed significantly due to non-synonymous substitutions, as listed in Table 1 (in bold), including ORFs 5.0/76, 4.0/77, 49, and 86.6, encoding the MEQ protein, 23-kDa nuclear protein, large tegument protein, and SORF1 protein, respectively, as wells as ORFs 3.4/78.3, 3.6/78.2, 76.4, 80, 85/99, 86/98, and 91.5, encoding the putative proteins RLORF4, RLORF5, MLHG, MFAY, MNDR, MSWP, and MAHG, respectively.
When compared with the Md5, RB1B, GA, 814, and CVI988/Rispens strains, GX0101 had the highest sequence identity to two BAC clones, pC12/130-10 and pC12/130-15, from the UK strain C12/130 [18]. Among the mutated 76 ORFs of all GaHV-2 strains, GX0101 has 49 and 46 ORFs that are 100 % identical to pC12/130-10 and pC12/130-15, respectively, but only 7-27 ORFs that are 100 % identical to other strains (Table 2). pC12/130-10 is virulent, but pC12/130-15 is attenuated, although they are descended from the same original strain [11]. Since GX0101 and its BAC-rescued virus bac-GX0101 are very virulent [9], it is of consequence to compare these three strains for further understanding of genomic sequences related to their virulence and pathogenicity.
We also found five consecutive repeats of a 217-bp fragment in ORFs 97.3-97.6 in the TRS region and three repeats of the same 217 bp in ORFs 86.2- 86.4 in the IRS region of GX0101, while only 1-2 copies in both the TRS and IRS regions were found in other strains. GX0101 has a 486-bp deletion (corresponding to nt 164,033-164,518 of Md5) in its US region, which is also absent in GA, pC12/130-10, and pC12/130-15 [1, 12] but present in Md5, RB1B, and CVI988/Rispens [2, 13, 14]. It is not clear if these differences have an influence on the biological activity of GX0101.
Considering the above characteristics of pathogenicity, horizontal transmission ability, and genomic structure with a stable REV insert, this analysis of the complete sequence of GX0101 will be useful not only in studies of gene functions related to pathogenicity and transmission but also in understanding genomic mutations and evolutionary relationships of GaHV-2 from different geographical areas of the world.
References
Lee LF, Wu P, Sui D, Ren D, Kamil J, Kung HJ, Witter RL (2000) The complete unique long sequence and the overall genomic organization of the GA strain of Marek’s disease virus. Proc Natl Acad Sci USA 97:6091–6096
Tulman ER, Afonso CL, Lu Z, Zsak L, Rock DL, Kutish GF (2000) The genome of a very virulent Marek’s disease virus. J Virol 74:7980–7988
Zhang Z, Cui Z (2005) Isolation of recombinant field strains of Marek’s disease virus integration with reticuloendotheliosis virus genome fragments. Sci China C Life Sci 48:81–88
Su S, Cui N, Cui Z, Zhao P, Li Y, Ding J, Dong X (2012) Complete genome sequence of a recombinant Marek’s disease virus field strain with one reticuloendotheliosis virus long terminal repeat insert. J Virol 86:13818–13819
Cui Z, Zhuang G, Xu X, Sun A, Su S (2010) Molecular and biological characterization of a Marek’s disease virus field strain with reticuloendotheliosis virus LTR insert. Virus Genes 40:236–243
Kost R, Jones D, Isfort R, Witter R, Kung HJ (1993) Retrovirus insertion into herpesvirus: characterization of an attenuated Marek’s disease virus harboring a solo LTR. Virology 192:161–169
Isfort RJ, Jones D, Kost R, Witter R, Kung HJ (1992) Retrovirus insertion into herpesvirus in vitro and in vivo. Proc Natl Acad Sci USA 89:991–995
Jones D, Brunovskis P, Witter R, Kung HJ (1996) Retroviral insertional activation in a herpesvirus: transcriptional activation of US genes by an integrated long terminal repeat in a Marek’s disease virus clone. J Virol 70:2460–2467
Sun A, Xu X, Petherbridge L, Zhao Y, Nair V, Cui Z (2010) Functional evaluation of the role of reticuloendotheliosis virus long terminal repeat (LTR) integrated into the genome of a field strain of Marek’s disease virus. Virology 397:270–276
Sun A, Petherbridge L, Zhao Y, Li Y, Nair V, Cui Z (2009) A BAC clone of MDV strain GX0101 with REV-LTR integration retained its pathogenicity. Chinese Science Bulletin 54:2641–2647
Zhang F, Liu C, Zhang Y, Li Z, Liu A, Yan F, Cong F, Cheng Y (2012) Comparative full-length sequence analysis of Marek’s disease virus vaccine strain 814. Arch Virol 157:177–183
Spatz SJ, Smith LP, Baigent SJ, Petherbridge L, Nair V (2011) Genotypic characterization of two bacterial artificial chromosome clones derived from a single DNA source of the very virulent gallid herpesvirus-2 strain C12/130. J Gen Virol 92:1500–1507
Spatz SJ, Petherbridge L, Zhao Y, Nair V (2007) Comparative full-length sequence analysis of oncogenic and vaccine (Rispens) strains of Marek’s disease virus. J Gen Virol 88:1080–1096
Spatz SJ, Zhao Y, Petherbridge L, Smith LP, Baigent SJ, Nair V (2007) Comparative sequence analysis of a highly oncogenic but horizontal spread-defective clone of Marek’s disease virus. Virus Genes 35:753–766
Parcells MS, Anderson AS, Morgan TW (1995) Retention of oncogenicity by a Marek’s disease virus mutant lacking six unique short region genes. J Virol 69:7888–7898
Kim T, Mays J, Fadly A, Silva RF (2011) Artificially inserting a reticuloendotheliosis virus long terminal repeat into a bacterial artificial chromosome clone of Marek’s disease virus (MDV) alters expression of nearby MDV genes. Virus Genes 42:3693–3676
Mays JK, Silva RF, Kim T, Fadly A (2012) Insertion of reticuloendotheliosis virus long terminal repeat into a bacterial artificial chromosome clone of a very virulent Marek’s disease virus alters its pathogenicity. Avian Pathol 41:259–265
Barrow A, Venugopal K (1999) Molecular characteristics of very virulent European MDV isolates. Acta Virol 43:90–93
Acknowledgments
This work was supported by grants from Guangdong Joint Funds from the National Natural Science Foundation of China (U1131005).
Author information
Authors and Affiliations
Corresponding author
Additional information
S. Su and N. Cui contributed equally to this work.
Rights and permissions
About this article
Cite this article
Su, S., Cui, N., Sun, A. et al. Sequence analysis of the whole genome of a recombinant Marek’s disease virus strain, GX0101, with a reticuloendotheliosis virus LTR insert. Arch Virol 158, 2007–2014 (2013). https://doi.org/10.1007/s00705-013-1671-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-013-1671-1