
Abstract
A real-time reverse-transcription PCR (rRT-PCR) that targets a region of the polymerase (L) gene was developed to detect all known lineages of avian paramyxovirus type 1 (APMV-1), also known as Newcastle disease virus (NDV). A panel of 23 viruses representing the current known phylogenetic diversity of the APMV-1 population with a bias towards the more recent European strains, which had been grown in embryonated fowls’ eggs, were tested. A range of positive and negative clinical samples (n = 350) provided by the National Reference Laboratory and International Reference Laboratory at VLA Weybridge were also tested. Positive clinical material included samples considered representative of lineages 3, 4 and 5 obtained from chickens, ducks, pigeons and partridges. The negative sample population was obtained from chickens, turkeys and ducks. The APMV-1 L gene rRT-PCR gave high relative sensitivity (96.05%) and specificity (98.18%) when compared with virus isolation in embryonated fowls’ eggs. It is proposed that this assay could provide a first-line screening tool for the detection of APMV-1 in clinical samples.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Avian paramyxovirus type 1 (APMV-1), also known as Newcastle disease virus (NDV), is an important avian pathogen that circulates throughout the world and affects many species of birds [6, 15]. Outbreaks caused by infections with APMV-1 in susceptible domestic poultry flocks may cause significant morbidity and mortality, the extent of which varies with virus strain, and levels range from asymptomatic for some avirulent strains to 100% mortality for virulent strains.
The major determinant of the virulence of these viruses is the amino acid motif in the precursor fusion protein (F0), which must be cleaved by host cell proteases to afford infectivity to virus particles [19]. Avirulent APMV-1 strains are activated by trypsin-like enzymes with a restricted distribution in the host and are therefore associated with localised or focal infections, usually in the respiratory and/or intestinal tract. In contrast, virulent viruses are activated by a ubiquitous host protease[s], and infections therefore result in systemic spread and disease. In addition to the variation in the disease presentation observed in domestic poultry infected with avirulent (asymptomatic or lentogenic infections) or virulent (mesogenic or velogenic infections) viruses, there is also significant antigenic and genetic variation across the APMV-1 population as a whole. This variation has been used for many years for the detection and differentiation of APMV-1, assigning viruses to specific monoclonal antibody groups [3, 4] or genetic lineages [1]. In different studies involving partial sequence analysis of the gene encoding the F protein (F gene), APMV-1 isolates have been placed in either eight main distinct groups (I–VIII) with sublineages within them [13] or six distinct genetic lineages (1–6), each with several sublineages [1]. More recently, Czegledi et al. [12] considered viruses placed in lineage 6 by Aldous et al. [1] to show sufficient variation to classify them as class I APMV-1 viruses, while the remainder became class II APMV-1 viruses.
The existence of the variation in disease presentation associated with individual virus strains means that diagnosis of notifiable/virulent APMV-1 generally requires multiple steps of detection and differentiation. The current ‘gold standard’ method of detection requires initial isolation of a haemagglutinating agent in embryonated fowls’ eggs, followed by confirmation of identification as an APMV-1 using serological techniques. The differentiation or pathotyping is then achieved either by in vivo techniques (such as the intracerebral pathogenicity index [ICPI] in day-old chicks) or molecular methods for the deduction of the F0 cleavage site amino acid motif [5].
In the United Kingdom (UK) and most European countries, ND outbreaks tend to be sporadic, and when they have occurred in recent years, they have been usually associated with virulent viruses from the genetic lineages 4 and 5. The two most recent outbreaks of ND in the UK were associated with a sublineage 5b virus in pheasants (Phasianus colchicus) in 2005 [2] and a sublineage 4b virus in grey partridges (Perdix perdix) in 2006 [14]. In both of these outbreaks, despite both remaining localised and not spreading from the initial infected premises (IP), the diagnostic response was very resource-intensive and required a number of days to confirm samples as negative, since passage in embryonated fowls’ eggs was required. As described by Crossley et al. [11], in response to the 2002–2003 outbreak of ND in southern California, in the event of widespread multiple IP outbreaks, the use of embryonated fowls’ eggs for screening was not feasible, and a much more rapid molecular technique needed to be employed.
In the present study, we report the development of a screening assay using real-time reverse-transcription PCR (rRT-PCR) incorporating hydrolysis probes that can detect ND isolates from both classes [12] and all lineages [1, 8] of APMV-1. The assay targets a region of the polymerase (L) gene, which was selected because of the high level of genetic conservation it exhibits in different strains of APMV-1. It is proposed that this assay could provide a first-line screening tool for the detection of APMV-1 in clinical samples.
Materials and methods
Viruses and sample preparation
All viruses used in the study were obtained from the International Reference Laboratory (IRL) for ND at VLA Weybridge; amplified samples had been grown in embryonated fowls’ eggs, and clinical samples were suspended in antibiotic solution prior to RNA extraction according to standard protocols [5]. The avian influenza virus, APMV-2 to -9, infectious bronchitis virus (IBV) and avian metapneumovirus (aMPV) isolates were grown in embryonated fowls’ eggs or chick embryo fibroblasts (CEFs) prepared by conventional methods using embryos from a specific-pathogen-free flock. Details of the isolates used for optimisation work are listed in Table 1. Positive and negative clinical material originated from East Africa (n = 40), the Middle East (n = 12) and Europe (n = 298). Full isolate details are not provided.
RNA extraction
Viral RNA was extracted from all samples using the QIamp viral RNA mini kit for manual extractions, and for robotic extractions the Qiagen BioRobot Universal, using the QIamp viral RNA Biorobot kit customised for the VLA, following the manufacturer’s instructions.
Primer and probe design
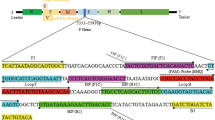
A total of 34 complete genomes were obtained from GenBank, aligned and used for the design and selection of primers and probes to be used in the assay. Three sets of primer pairs were selected for evaluation, and one pair, targeting the L gene, was selected for use. The primer and probe details are listed in Table 2. The probes were designed according to the manufacturer’s guidelines and were labelled with a 5′ fluorescent dye (FAM) and a 3′ black hole quencher (BHQ).
Assay details
The initial approach to designing this test involved identification and testing of existing APMV-1 screening assays that use used real-time technologies; two were identified [16, 20]. Both assays were evaluated and tested on four viruses considered to represent contemporary viruses from lineages 1, 4b, 5d and 6 of APMV-1 (Table 1). Neither assay detected all four samples; this confirmed that both tests were unsuitable for fulfilment of the aims of the present study, and the development of a new assay was required.
For the rRT-PCR assay, the Qiagen one-step RT-PCR kit was used. Each reaction mix, apart from the ‘no template control’ (NTC), in which the RNA was omitted and molecular grade water (2 μl) was added instead, was comprised of 5× buffer (5 μl), 10 mM of each dNTP (1 μl), 12.5 μM primer NDF1 (1 μl), 12.5 μM primer NDR2 (1 μl), 5 μM probe NDPro1 (1 μl), 5 μM probe NDPro2 (1 μl), 2 μM ROX (1 μl), 25 mM MgCl2 (3 μl), 5 U RNase inhibitor (1 μl), enzyme mix (1 μl), sterile dH2O (7 μl) and RNA (2 μl). In all experiments, NTCs were included to detect any viral RNA contamination between wells or non-specific probe breakdown. Cycling conditions for the rRT-PCR using the MX3000P (Stratagene) were one cycle at 50°C for 30 min, one cycle at 95°C for 15 min and 40 cycles at 95°C for 10 s, 50°C for 30 s and 72°C for 30 s.
For data analysis, the ‘MX PRO QPCR’ software version 3.20 (Stratagene) was used with three algorithm enhancements: the amplification-based threshold, the adaptive baseline and moving average to obtain the most accurate threshold cycle (Ct) value, the cycle at which a statistically significant increase in fluorescence is first detected. The level of fluorescence was ‘normalised’ to correct for fluctuations due to changes in concentration and volume and expressed as the dRn value, which reliably indicates the magnitude of the signal generated by a given set of PCR conditions.
Assay specificity and sensitivity
In order to estimate the sensitivity of the assay, eight serial ten-fold dilutions of six isolates, Ulster-2C, Hitchner-B1, Herts-33, pi/CY/819/07-3256/1-1, ck/ET/1028/07-762 and dk/UK/7800/06 at the determined median embryo infectious dose (EID50) per 0.1 ml of 108.1, 108.5, 106.9, 106.7, 107.1 and 108.3, respectively, were tested, and the mean Ct from four different tests was recorded. The limit of detection of the assay was determined for DNA copies of a full-genome APMV-1 plasmid (kindly provided by Ben Peeters [17]). The plasmid was quantified spectrophotometrically. To ensure that the assay was specific for APMV-1, ten strains representing each of the other APMV serotypes (APMV-2, three APMV-3, APMV-4, APMV-5, APMV-6, APMV-7, APMV-8, APMV-9), 18 strains of avian influenza virus representing all 16 of the known haemagglutinin subtypes (H1N1, H2N2, H3N1, H4N6, H5N1 highly pathogenic [HP], H5N2 low-pathogenic [LP], H6N5, H7N3 LP, H7N7 HP, H8N4, H9N2, H10N7, H11N3, H12N6, H13N6, H14N6, H15N6, H16N3), three strains of infectious bronchitis virus (IBV-H120, CC220 and 2119) and a reference strain for each of the three avian metapneumovirus (aMPV) groups A, B and C were also tested (data not shown).
Results
Based on the alignment of the 34 full APMV-1 genomes from GenBank, three possible primer pair combinations were selected and tested against four viruses considered to represent contemporary viruses from lineages 1, 4b, 5d and 6 of APMV-1 (Table 1). Only one primer pair, which amplified a 142-bp region of the large [L] (polymerase) gene, reliably detected all four viruses. Two probes were designed for inclusion in the assay to ensure detection of isolates from all lineages.
Optimisation of the assay parameters and components was completed on a panel of 23 viruses, which are detailed in Table 1. Through the optimisation of this assay, the threshold cycle (Ct), above which isolates are not considered definitely positive, was set at Ct 37. Any sample that yielded a Ct result >37 was repeat tested. If a Ct result of ≥37 was obtained for two consecutive runs with any isolate, it was considered an inconclusive result, which was recorded as negative for the purposes of this test analysis. The sensitivity of the assay was evaluated using both plasmid DNA and RNA extracted from virus samples that had been quantified by EID50. The assay could detect about 22 DNA copies of the APMV-1 genome plasmid. Detection limits were defined as the highest tenfold dilution that produced an average Ct of ≤37, and in terms of infectivity, these were 101.1 EID50/ml, 102.5 EID50/ml, 102.9 EID50/ml, 101.7 EID50/ml and 102.1 EID50/ml for viruses from lineages 1–5, respectively, and for the virus representing lineage 6, the detection limit was 105.3 EID50/ml.
The optimised assay was tested extensively using a range of positive and negative clinical samples (350 total) that were provided by the NRL and IRL at VLA Weybridge. Positive clinical material included samples considered representative of lineages 3, 4 and 5 obtained from chickens, ducks, pigeons and partridges. The negative sample population was obtained from chickens, turkeys and ducks. A summary of the isolate categories and results is listed in Table 3.
A two-by-two table is presented in Table 4 in which the results of our L gene rRT-PCR assay described in this report are compared to those obtained using the current ‘gold standard’ method (using embryonated fowls’ eggs). In comparison with the standard assay, the relative sensitivity is 96.05% (95% confidence interval 88.12, 98.97%), and the relative specificity is 98.18% (95% confidence interval 95.55, 99.33%), with an efficiency of 97.71%. The determined κ value of 0.93 (95% confidence interval 0.89, 0.98) indicates very good agreement between these two tests. For eight of the 350 isolates tested (~2%), the results of the two test results were not in agreement. Five samples were positive by rRT-PCR and negative by the ‘gold standard’. These samples originated from swab and tissue pools obtained from two groups of five partridges from the same infected premises. Three further samples recorded as negative by rRT-PCR (Ct values >37) were positive in the assay with embryonated eggs. One these three samples was obtained from the partridge swab samples described above, the remaining two were from brain homogenate from a chicken and a dove.
Discussion
The need for a specific and sensitive molecular test for the detection of APMV-1 has been identified especially when there are a large number of samples to be tested [11]. Currently, preliminary diagnosis and screening requires virus growth in embryonated fowls’ eggs. Whilst this procedure is reliable and robust, it is labour-intensive and can take several days for a diagnosis to be confirmed, a period that is largely unacceptable in the face of ever-increasing needs for improved efficiency, rapid diagnosis, surveillance capabilities and the option for dealing with large numbers of samples in the event of disease outbreaks. It is anticipated that through the application of real-time molecular technologies to the detection and characterisation of NDV in clinical samples, some of these requirements will be addressed. Development of an rRT-PCR assay for detection of APMV-1 also enables consistent application of this technology across the diagnostic test portfolio for avian notifiable disease with the current molecular assays employed for the rapid detection of AI in the EU diagnostic manual [9, 10].
The application of rRT-PCR assays for the detection of APMV-1 has been under investigation and development for a number of years. Many of the assays developed to date have been designed to detect specific groups of APMV-1s. Some examples of these include many diverse classical strains (matrix-gene-based), pre-1960s American genotypes (matrix-gene-based) and ‘exotic’ Newcastle disease (fusion-gene-based) [20]; class I (or lineage 6) APMV-1 isolates using a polymerase (L)-gene-based assay [16]; differentiation of lentogenic, mesogenic and velogenic strains targeting the fusion gene [18]; and seven of the eight lineages [13] targeting the fusion gene [7]. These studies have utilised hydrolysis probes, intercalating dyes with melting point analysis and light upon extension (LUX)-labelled primers. All of these studies present data confirming the success of the respective methods. However, with regard to a method that would provide the pan-APMV-1 assay that was the goal of the present study, it seemed unlikely that any of these existing tests could provide the level of sensitivity required.
Within the APMV-1 population, there exists considerable genetic variation that can be usefully applied to establish key epidemiological information but at the same time makes the development of a single molecular assay for detection of any APMV-1 a complex and challenging task. In an effort to establish the efficacy of the assay developed and presented in this study, the 23 viruses selected for use in the optimisation stages of development were specifically chosen to reflect the current known phylogenetic diversity of the APMV-1 population with a bias towards the more recent European strains and included seven from the UK, ten from other European Union (EU) member states, three from Asia, two from Africa and one from the Middle East. The L gene assay we have developed was successful in detecting this broad spectrum of viruses (from all recognised lineages and sub-lineages of APMV-1), indicating the potential of this assay as an initial screening test for the detection of any APMV-1 infection. The limit of detection for virus representing lineage 6 was only 105.3 EID50/ml, which suggests that the assay has reduced efficiency for viruses of this lineage.
All egg-amplified APMV-1 samples utilised in this work were diluted 1,000-fold to reduce the virus titre in the sample to a lower level. Positive and negative samples of clinical origin were obtained from more recent outbreaks of ND. These included both pooled tissue and swab samples; the positive samples corresponded to eight different outbreaks of ND from Western Europe, the Middle East, and Eastern Africa. There were multiple samples from each outbreak.
The APMV-1 L gene rRT-PCR gave high relative sensitivity (96.05%) and specificity (98.18%) when compared with virus isolation in embryonated fowls’ eggs (Table 4). Results for the clinical material indicate that the rRT-PCR may detect more positive samples than virus isolation in eggs, since five samples, originating from tissue pools from five partridges, were positive by rRT-PCR but negative in eggs. Using the 16 clinical samples from these partridges as an example, all samples were obtained from two groups of five birds from the same infected premises. Using virus isolation, 11 of 16 were positive, whereas in the L gene rRT-PCR assay, 15 of 16 were positive, and there was one inconclusive result, which was scored as negative. In this example, where samples are positive only by rRT-PCR, it is likely that the test is detecting non-infectious virus genomes present in the samples.
The three samples that were positive by virus isolation but recorded as negative by rRT-PCR all produced a Ct value of >37 that could not be repeated in subsequent tests. These samples were exposed to multiple freeze–thaw cycles after virus isolation had been completed and prior to rRT-PCR testing. It is probable that these anomalous results could be a consequence of poor sample handling and associated poor RNA integrity. All tests are dependent on the sample type and the way it has been handled to preserve the component to be detected.
It will be necessary to continually monitor the L gene rRT-PCR test against new, emerging, and evolving strains to ensure the assay remains fit for purpose. However, the results presented with the viruses tested to date demonstrate that with this assay we have produced a rapid and specific screening test capability for the detection of APMV-1 virus infection in clinical specimens from domestic poultry. The sensitivity of the assay varies depending on the virus strain being tested, with the test proving to be highly sensitive for isolates in genetic lineages 1–5 but of decreased sensitivity for isolates corresponding to lineage 6, with considerably higher levels of virus required in the sample to enable detection [1]. This reduced sensitivity is unsurprising, since the lineage 6 isolates are highly divergent and show considerable genetic heterogeneity when compared to the APMV-1 population as a whole. Further work may be required to increase the sensitivity of the assay for these specific isolates, which are more commonly associated with isolates obtained from wild birds.
The application of this assay is a ‘first step’ screening test for the detection of APMV-1 viruses only. Confirmation of notifiable disease would require virus identification by nucleotide sequencing and/or in vivo testing, in accordance with the OIE diagnostic manual [6]. Use of the L gene APMV-1 rRT-PCR assay has the potential to decrease the time taken to confirm the presence of suspected avian notifiable disease such as Newcastle disease, thus allowing the competent veterinary authority to more rapidly implement appropriate disease-control measures with a high degree of certainty. In turn, this can directly contribute to minimising the length of the ‘high-risk period’ between first incursion and detection of virus, a key determinant in affecting the scale and duration of trans-boundary animal disease outbreaks, and allow for timely implementation of appropriate disease-control strategies.
References
Aldous EW, Mynn JK, Banks J, Alexander DJ (2003) A molecular epidemiological study of avian paramyxovirus type 1 (newcastle disease virus) isolates by phylogenetic analysis of a partial nucleotide sequence of the fusion protein gene. Avian Pathol 32:239–257
Aldous EW, Manvell RJ, Cox WJ, Ceeraz V, Harwood DG, Shell W, Alexander DJ, Brown IH (2007) Outbreak of Newcastle disease in pheasants (Phasianus colchicus) in south-east England in July 2005. Vet Rec 160:482–484
Alexander DJ, Manvell RJ, Lowings JP, Frost KM, Collins MS, Russell PH, Smith JE (1997) Antigenic diversity and similarities detected in avian paramyxovirus type 1 (Newcastle disease virus) isolates using monoclonal antibodies. Avian Pathol 26:399–418
Alexander DJ, Banks J, Collins MS, Manvell RJ, Frost KM, Speidel EC, Aldous EW (1999) Antigenic and genetic characterisation of Newcastle disease viruses isolated from outbreaks in domestic fowl and turkeys in Great Britain during 1997. Vet Rec 145:417–421
Alexander DJ (2008) Newcastle disease. In: Manual of diagnostic tests and vaccines for terrestrial animals. OIE World Organisation for Animal Health, Paris, pp 576–589. http://www.oie.int/eng/normes/mmanual/A_summry.htm
Alexander DJ, Senne DA (2008) Newcastle disease, other Avian paramyxoviruses and pneumovirus infections. In: Saif YM (ed) Diseases of poultry. Iowa State University Press, Iowa, pp 75–100
Antal M, Farkas T, German P, Belak S, Kiss I (2007) Real-time reverse transcription-polymerase chain reaction detection of Newcastle disease virus using light upon extension fluorogenic primers. J Vet Diagn Invest 19:400–404
BallagiPordany A, Wehmann E, Herczeg J, Belak S, Lomniczi B (1996) Identification and grouping of Newcastle disease virus strains by restriction site analysis of a region from the F gene. Arch Virol 141:243–261
Communities CotE (2006) Council Directive 2005/94/EC of 20th December 2005 on Community measures for the control of avian influenza and repealing 92/40/EEC. Off J Eur Comm L10:16–65
Communities CotE (2006) Commission Decision 2006/437/EC approving a diagnostic manual for avian influenza as provided for in the Council Directive 2005/94/EC. Off J Eur Comm L237:1–27
Crossley BM, Hietala SK, Shih LM, Lee L, Skowronski EW, Ardans AA (2005) High-throughput real-time RT-PCR assay to detect the Exotic Newcastle Disease Virus during the California 2002–2003 outbreak. J Vet Diagn Invest 17:124–132
Czegledi A, Ujvari D, Somogyi E, Wehmann E, Werner O, Lomniczi B (2006) Third genome size category of avian paramyxovirus serotype 1 (Newcastle disease virus) and evolutionary implications. Virus Res 120:36–48
Herczeg J, Wehmann E, Bragg RR, Dias PMT, Hadjiev G, Werner O, Lomniczi B (1999) Two novel genetic groups (VIIb and VIII) responsible for recent Newcastle disease outbreaks in Southern Africa, one (VIIb) of which reached Southern Europe. Arch Virol 144:2087–2099
Irvine RM, Aldous EW, Manvell RJ, Cox WJ, Fuller CM, Wood AM, Milne JC, Wilson M, Hepple RG, Hurst A, Sharpe CE, Alexander DJ, Brown IH (2009) Outbreak of Newcastle disease due to pigeons paramyxovirus type 1 in grey partridges (Perdix perdix) in Scotland in October 2006. Vet Rec 165(18):531–535
Kaleta EF, Baldauf JP (1988) Newcastle disease in free living and pet birds. In: Alexander DJ (ed) Newcastle disease. Kluwer, Massachusetts, pp 197–246
Kim LM, King DJ, Suarez DL, Wong CW, Afonso CL (2007) Characterization of class I Newcastle disease virus isolates from Hong Kong live bird markets and detection using real-time reverse transcription-PCR. J Clin Microbiol 45:1310–1314
Peeters BPH, de Leeuw OS, Koch G, Gielkens ALJ (1999) Rescue of Newcastle disease virus from cloned cDNA: evidence that cleavability of the fusion protein is a major determinant for virulence. J Virol 73:5001–5009
Pham HM, Konnai S, Usui T, Chang KS, Murata S, Mase M, Ohashi K, Onuma M (2005) Rapid detection and differentiation of Newcastle disease virus by real-time PCR with melting-curve analysis. Arch Virol 150:2429–2438
Rott R, Klenk HD (1988) Molecular basis of infectivity and pathogenicity of Newcastle disease virus. In: Alexander DJ (ed) Newcastle disease. Kluwer, Massachusetts, pp 79–97
Wise MG, Suarez DL, Seal BS, Pedersen JC, Senne DA, King DJ, Kapczynski DR, Spackman E (2004) Development of a real-time reverse-transcription PCR for detection of Newcastle disease virus RNA in clinical samples. J Clin Microbiol 42:329–338
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fuller, C.M., Brodd, L., Irvine, R.M. et al. Development of an L gene real-time reverse-transcription PCR assay for the detection of avian paramyxovirus type 1 RNA in clinical samples. Arch Virol 155, 817–823 (2010). https://doi.org/10.1007/s00705-010-0632-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-010-0632-1