
Abstract
The complete genome sequence for an isolate of the Ugandan and Tanzanian strain types of Cassava brown streak virus have been determined using the novel approach of non-directed next generation sequencing. Comparison of the genome sequences revealed that CBSV is highly heterogeneous at the isolate level as well as the strain level. The isolate of the Ugandan strain was found to have a genome 9,070 nucleotides long coding for a polypeptide with 2,902 amino acid residues. The isolate of the Tanzanian strain was 9,008 nucleotides long and coded for a polypeptide with 2,916 amino acid residues. Nucleotide identity between the isolates across the genome was 76%, with protein encoding regions 57–77% and individual proteins had 65–91% amino acid similarity. In addition between the two strains four protein products (PIPO, CI, NIa-Vpg and coat protein) varied in size and an unusual HAM1-like protein, whilst of identical nucleotide length, was found to have the lowest homology. The implication of diversity of CBSV is discussed in the context of speciation, evolution, development of diagnostics, and breeding for resistance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Cassava brown streak virus (CBSV) is found to infect cassava (Manihot esculenta) in most cassava-growing areas of sub-Saharan East Africa [3]. In recent years, CBSV has become an increasing problem in the region, with the wider distribution of a purportedly more aggressive strain [3]. To date, more than 70 sequences are available for this virus, including one full genome [6]; the sequences appear to cluster as two distinct types, or strains [7], named Tanzanian and Ugandan based on the countries from which partial sequences of isolates were first found [3, 8, 9]. An alternative informal naming for the strains is also in use that describes a broader geographic distribution of the coastal/lowland strain (=Tanzanian) and the highland strain (=Ugandan); both of these commonly used strain descriptors, however, are misleading due to the current broad distribution of both strain types. Throughout this manuscript, we will use the more commonly used Ugandan and Tanzanian strain definition.
Here, we present the first complete genome sequence of an isolate of CBSV from the Tanzanian strain, acquired using a novel non-directed next-generation sequencing approach, and present results comparing the sequence of this isolate with sequences from the Ugandan strain type.
Isolates CBSV-Ug (Nkokojeru in the Mukono district, Uganda in 2006) and isolate CBSV-Tan (Kiabakri, Musoma district of Tanzania in 2008) were collected from symptomatic cassava and air-dried between sheets of paper for storage. Additional isolates, TanAB (Mogabiri, Tarime District, Tanzania, 2008), TanC (Kyamunyorwa, Muleba District, Tanzania 2008), TanT (Mwanjombo, Misungwi District, Tanzania 2008), and KenE (Namudura, Samia District, Kenya 2008) were randomly collected as part of the survey work of the GLCI project.
The isolates were sequenced using the next-generation sequencing method described previously [1]. Sequencing was performed using a GS-FLX Genome Sequencer (Advanced Genomics Facility, Liverpool University, UK). Specific primers were designed to these sequences (Table 1) to enable the amplification of overlapping PCR products that enabled the construction of the remainder of the genome. The 5′ end of the genome was amplified using rapid amplification of cDNA ends with the SMART RACE kit following the manufacturer’s protocols (Clontech, USA). Specific primers for the HAM1-like protein (Ug-NIb/HamA and Ug-CP/HamB; Tan-NIb/HamB and Tan-CP/HamA) were designed within the sequence of the coat protein and polymerase region and used to generate sequence for a further four isolates.
Following sequencing of the samples using the GS-FL, large numbers of individual fragments of sequence data were generated. For the isolate CBSV-Tan, 41127 sequence reads, totalling 12,323,878 nt were produced after de novo assembly with the CLCBio Genomics Workbench (CLCBio, Denmark); 136 contigs were generated leaving 6,406 unassembled reads. For the isolate CBSV-Ug, 103,337 sequence reads, totalling 18,573,034 nt were produced, resulting in 107 contigs and 12365 unassembled reads. Analysis using BLAST-N and BLAST-X assigned sequences to probabilistic phylogenies for the CBSV-Tan/CBSV-Ug data as follows: viral sequences accounted for 0.9/0.7%, with the remaining sequence accounted for by host plant (80.1/49.3%), bacterial (0.01/5.2%), fungal (0.2/25.9%), metazoan (3.8/3.3%) or unidentified (14.9/15.7%) sources. For both isolates, the individual sequence reads and contigs were spread across the genome and not clustered in any particular part of the genome sequence. The dispersed distribution enabled PCR to be used effectively to close the gaps in the sequence (Fig. 1). The host plant cassava (rather than mechanically inoculated indicator host) was used for the pyrosequencing to enable the identification of any other viruses that may be present. There has been some debate over the years about whether CBSD is caused by one or two viruses, due to observations of short virus particles in tissue, suggesting a carlavirus, and pinwheel inclusion bodies characteristic of members of the family Potyviridae [10]. In this study, no virus-like sequences were unaccounted for by the CBSV sequence, which strongly suggest that an ipomovirus alone is likely to be the cause of CBSD. A total of 3,200 nucleotides of the genome of CBSV-Tan were over-sequenced using both pyrosequencing and PCR followed by Sanger sequencing. Surprisingly, the two sequencing methods showed discordance at only two nucleotide positions. At position 7692 the coverage within the pyrosequencing data was low (five reads), two pyrosequencing reads had a T and three had a deletion causing a frameshift in the polyprotein. On visual inspection of the contig constructing this region, the reads with the deletion were of lower quality, and Sanger sequencing indicated the presence of a T at nt 7692. At nt 7260, the Sanger sequencing indicated a T, whereas the pyrosequencing data indicated a C, since the depth of coverage at this position was greater in the pyrosequencing data, the consensus was C. Where the depth of the pyrosequencing data was low, PCR and Sanger sequencing were used to confirm the sequence.

Schematic representation of the genome sequences of (a) CBSV-Tan and (b) CBSV-Ug illustrating sequence generated using different methods. Sequence generated using next-generation sequencing is shown in blue, sequence generated following amplification using 5′ RACE is shown in red, and sequence generated following specific PCR is shown in green (color figure online)
The genome of the isolate CBSV-Tan of the Tanzanian strain had the same number and location of protein coding regions as the Ugandan strain described previously [6]. Overall, the genome was shorter by 61 nucleotides, accountable in the 5′ UTR (8 nt) and 3′UTR (96 nt), though three protein coding regions were longer CI (6 nt), Nia-Vpg (3 nt) and the CP region (33 nt). The genome sequences of the two Ugandan strain isolates differed in size by a single nucleotide in the 3′UTR.
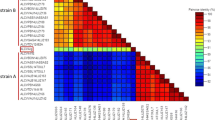
Pairwise sequence alignments between the Ugandan and Tanzanian strain sequences (CBSV-Tan [GQ329864] and CBSV-Ug [FJ185044]) revealed a genome-wide nucleotide identity of 71%, and the polyprotein has an amino acid sequence identity of 74%. Significant variation in sequence identity is evident between individual protein coding regions (Table 1), varying between 57% (HAM1) and 77% (6K2) sequence identity. Comparisons between the two Ugandan genome sequences (CBSV-Ug [FJ185044] and MLB3 [NC_012698]) also reveals a surprisingly low level of nucleotide sequence identity (86%) across the whole genome, with identity for individual genes ranging from 81 to 92% (Table 1).
The recently described PIPO [4] open reading frame and putative protein sequence could be identified within the sequence of both isolates sequenced (CBSV-Ug and CBSV-Tan). For this region, translation of the ORF would result in a polypeptide 78 and 79 amino acids in length (CBSV-Tan and CBSV-Ug, respectively), fused to the N-terminal region of the P3 protein. The sequence for CBSV-Tan, however, does not contain the highly conserved G1-2A6-7 motif [4] identified in all other sequences from members of the family Potyviridae; instead the sequence is replaced by TAAAAAAA. The conservation of this sequence amongst isolates of the Tanzanian strain was supported by obtaining the same sequence from another isolate of the Tanzanian strain (data not shown).
The only other virus in which a HAM1-like protein is found is one infecting another Euphorbiacea plant, Euphorbia ringspot virus (EuRSV) (accession number AY697300). Although only the 3′ end of the genome of EuRSV has been sequenced, the physical characteristics of this virus (particle size, genome length, vector species) appear to be typical for a member of the genus Potyvirus (Potyviridae) [4] distinct from the ipomoviruses. It is perhaps surprising that CBSV-Ug and CBSV-Tan, share lower sequence identity at the amino acid level with EuRSV (38 and 39%) than with eukaryotic HAM 1 homologues (e.g. 48 and 52%, respectively, with Zea mays (accession number ACG38801). Taken together, this information might suggest that the integration of HAM1 into these viruses has occurred as two independent events, though it is perhaps more likely that the integration of the HAM1 homologue is an ancient event in a common ancestor and that the two viruses then evolved separately.
The function of the HAM1 protein in both viruses remains unknown, although it has been suggested that because the HAM1 protein plays a role in protection against mutation, the homologue in viruses may reduce mutation of the viral RNA [6]. Considering that there are almost 6,500 sequences available for members of the Potyviridae, including complete genomes for 80 species, it is perhaps surprising that, if HAM1 has an advantageous role in mutation suppression, why it has not been acquired from hosts by other viruses or recombined amongst other potyviruses. It is perhaps interesting to speculate that this could be a reflection of how relatively few viruses are able to infect Euphorbiaceae hosts; the advantages of the protein may only be evident within certain environments, and perhaps Euphorbiaceae present a challenging host environment for viruses to colonise.
The present ICTV species demarcation criteria for potyviruses and ipomoviruses state that a nucleotide sequence identity of less than 85% over the whole genome or different polyprotein cleavage sites are suitable characteristics for the demarcation of a new species [2, 11]. Based on the comparisons of the full genome sequences now available, it could be argued that the whole genomes for the Ugandan and Tanzanian strain types display species-level differences. The same criteria, if applied to the two isolates of the Ugandan strain, would similarly identify isolates at the boundaries of speciation and certainly representing different strains. However, when taking into account an absence of data for symptom and epidemiological dissimilarity amongst the CBSV isolates and strains, it is apparent that CBSV is better described as a single virus species that is unusually variable at the nucleotide level, and currently there is no coherent argument to suggest sub-speciation. The strain descriptors currently used are equally misleading, since isolates currently referred to as belonging to the Ugandan/highland strain or Tanzanian/coastal/lowland strains are not limited to these regions. However, it has also been suggested that the Ugandan strain of the virus is becoming more widespread due to heightened virulence, resulting in greater crop losses [3]. Our data substantiate ideas on strain types and isolate diversity and provide a further basis upon which epidemiological studies need to be structured to allow for strain and isolate effects. For example, the scope for host/strain interactions is clearly broad, and this should be allowed for in breeding programmes.
There are no current improved varieties of cassava that have been found to be resistant to CBSV, and although landraces may possess useful traits for breeding, it is unclear if these materials possess resistance or tolerance traits [5]. Schemes based on distribution of clean planting materials must rely heavily on the availability of reliable diagnostics for virus detection. It is a particular challenge to detect CBSV, as the disease is not characterised by pronounced symptoms, and symptoms which may be only weakly correlated to the presence of virus. Diagnostic tools such as PCR or ELISA are therefore required for detection rather than visual inspection. The unusually low titre of the virus in some infected samples presents a scarce target for ELISA, and the sequence diversity detracts from nucleic acid approaches such as PCR. The data presented in this paper indicate that, in the development of reliable diagnostics, it will be necessary to allow for the variability observed, not just between different strains, but also between isolates from the same strain. Thus, it is likely that future diagnostics for CBSV will require highly degenerate reagents (probes, primers or antibodies) for the detection of each strain separately in order to achieve good specificity. The variability of the sequence shows that this is unlikely to be achieved using a single reagent for both strains. It is also clear that much more sequence is needed from a very broad range of isolates from different geographical locations, and ideally from different hosts, so that effective tools can be developed and validated to enable screening of clean planting material.
Cassava is a plant that originates from South America and was introduced to West Africa by the Portuguese in the late sixteenth century. Given the extensive production of cassava worldwide and the fact that this virus has only been found in Africa, it appears likely that the virus did not originate as a pathogen of cassava. It is therefore speculated that the virus is native to East Africa, where it has an as yet unknown plant host or hosts, and has jumped to cassava as a first encounter pathogen. The two strain variants of CBSV may then identify two independent jumps from a native host or an evolution of CBSV in cassava. Worryingly, this scenario does present the strong possibility that other variants of CBSV may exist in the natural host/s and could at some point in the future also be transmitted to cassava. In order to begin to evaluate this potential risk, identifying the natural host of CBSV in Africa must remain a high priority.
References
Adams IP, Glover RH, Monger WA, Mumford R, Jackeviciene E, Navalinskiene M, Samuitien M, Boonham N (2009) Next generation sequencing and metagenomic analysis: a universal diagnostic tool in plant virology. Mol Plant Pathol 10:537–545
Adams MJ, Antoniw JF, Fauquet CM (2005) Molecular criteria for genus and species discrimination within the family Potyviridae. Arch Virol 150:459–479
Alicai T, Omongo CA, Maruthi MN, Hillocks RJ, Baguma Y, Kawuki R, Bua A, Otim-Nape GW, Colvin J (2007) Re-emergence of cassava brown streak disease in Uganda. Plant Dis 91(1):24–29
Chung BYW, Miller WA, Atkins JF, Firth AE (2008) An overlapping essential gene in the Potyviridae. Proc Nat Acad Sci 105(15):5897–5902
Hillocks RJ, Jennings DK (2003) Cassava brown streak disease: a review of present knowledge and research needs. Int J Pest Manag 49:225–234
Mbanzibwa DR, Tian Y, Mukasa SB, Valkonen JPT (2009) Cassava brown streak virus (Potyviridae encodes a putative Maf/HAM1 pyrophosphatase implicated in reduction of mutations and a P1 proteinase that suppresses RNA silencing but contains no HC-Pro. J Virol 83(13):6934–6940
Mbanzibwa DR, Tian Y, Tugume AK, Mukasa SB, Tairo F, Kyamanywa S, Kullaya A, Valkonen JPT (2009) Genetically distinct strains of Cassava brown streak virus in the Lake Victoria basin and the Indian Ocean coastal area of East Africa. Arch Virol 154:353–359
Monger WA, Seal S, Issac AM, Foster GD (2001) Molecular characterization of Cassava brown streak virus coat protein. Plant Pathol 50:527–534
Monger WA, Seal S, Cotton S, Foster GD (2001) Identification of different isolates of Cassava brown streak virus and development of a diagnostic test. Plant Pathol 50:768–775
Shukla DD, Frenkel MJ, Ward CW (1991) Structure and function of the potyvirus genome with special reference to the coat protein coding region. Can J Plant Pathol 13:178–191
Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA (eds) (2005) Virus taxonomy, eighth report of the International Committee on Taxonomy of Viruses. Elsevier, Amsterdam, p 822, p 830
Acknowledgments
The authors wish to acknowledge financial support from the Bill and Melinda Gates Foundation and thank the numerous individuals involved in the Great Lakes Cassava Initiative, from which some of the infected cassava material reported in this paper was obtained. We also wish to thank Dr M. N. Maruthi (NRI, Greenwich University, UK) for providing additional positive material against which findings could be checked.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Monger, W.A., Alicai, T., Ndunguru, J. et al. The complete genome sequence of the Tanzanian strain of Cassava brown streak virus and comparison with the Ugandan strain sequence. Arch Virol 155, 429–433 (2010). https://doi.org/10.1007/s00705-009-0581-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-009-0581-8