
Abstract
We determined the complete nucleotide sequence of EDRD-1, a Japanese strain of the North American type-Porcine reproductive and respiratory syndrome virus (PRRSV), and identified a novel 117-base deletion and 108-base insertion previously reported in the nsp2 gene of the SP strain, which contains the largest genome among PRRSV strains. Based on genetic analysis of the partial nsp2 gene in 30 additional Japanese isolates and 50 strains from various countries, we classified North American-type PRRSVs into three nsp2-types, represented by EDRD-1, which contains the 117-base deletion and 108-base insertion; prototypic VR-2332, which does not contain the deletion and insertion; and SP, which contains only the 108-base insertion. The three nsp2-types were phylogenetically separated, suggesting that these structural changes only occurred at earlier stages of viral evolution. In the nsp2 genes, we identified an additional 19 deletions ranging from 3 to 378 bases and 2 insertions of 3 and 21 bases which were not common within each nsp2-type, suggesting that these changes occurred at later stages of viral evolution. In addition, our results suggest that the three nsp2-types can be rapidly differentiated by RT-PCR using their polymorphisms as natural tags.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Porcine reproductive and respiratory syndrome virus (PRRSV) is the causative agent of porcine reproductive and respiratory syndrome (PRRS), which is typically characterized by acute reproductive and chronic respiratory diseases in sows and piglets, respectively [2]. PRRS emerged nearly simultaneously on two different continents, with its initial recognition in North America in 1987 [17], followed by Europe in 1990 [32, 42]. The two PRRSV isolates, whose evolutionary history remains unknown, show approximately 60% genome identity [1, 27] and are classified into two distinct genotypes, namely the North American-type (NA-type) and European-type (EU-type). PRRSV is characterized by a high mutation rate [8, 15], with a potential risk of new PRRSV strains emerging. Recently, a few reports indicate the emergence of potentially high pathogenic variants of PRRSV as the possible cause for large-scale outbreaks with high mortality in China in 2006 [22, 41].
PRRSV is a member of the order Nidovirales, family Arteriviridae, genus Arterivirus [3, 38]. PRRSV is an enveloped virus composed of a positive-sense, single-stranded RNA genome [25]. The 5′-capped and 3′-polyadenylated viral genome is approximately 15 kb in length and encodes nine open reading frames (ORFs), ORF1a, 1b, 2a, 2b, 3, 4, 5, 6 and 7, which are transcribed into a nested set of subgenomic mRNAs [5, 25, 43]. ORF1a and ORF1b, which cover the four-fifths of the 5′-terminal region of the genome, encode nonstructural proteins (nsps), whereas the remaining ORFs encode structural proteins. ORF1a and ORF1ab are translated into two polyproteins which are cleaved to produce nsp1–nsp8 and nsp9–nsp12, respectively. ORF1a- and ORF1ab-derived nsps possess protease [36, 46] and replicase-related activities, respectively.
Nsp2 was previously suggested to possess cysteine protease activity for autocleavage [36]. Other studies reported that nsp2 and nsp3 are involved in formation induction of double-membrane vesicles associated with the Equine arteritis virus replication complex, suggesting the creation of a suitable microenvironment for viral RNA synthesis [33, 37]. Further, nsp2 was also reported to be highly antigenic [4, 29]. The nsp2 gene shows the highest genetic diversity in the viral genome, as well as polymorphisms of considerable sizes, including a 108-base insertion [35] and various 3- to 333-base deletions [6, 7, 11, 13, 22, 28, 34, 41]. In particular, in potentially high pathogenic Chinese isolates, a discontinuous deletion of 90 bases was reported as an epidemiological genetic marker [22, 41].
PRRSV genotypes also show considerable genetic variation and contain several phylogenetic clusters [9, 12, 16, 23, 34, 39, 45]. Our previous study on ORF5 gene showed that the majority of Japanese isolates are classified into a distinct cluster, tentatively named cluster III, which also includes one Taiwanese and one Chinese isolate [45]. To date, no cluster III isolate has been genetically characterized in detail. Here, we genetically characterized the Japanese EDRD-1 strain [26], which was isolated earlier in Japan and belongs to ORF5-cluster III. Based on further genetic investigation of nsp2 gene polymorphisms, we provide a novel classification of NA-type PRRSV into three nsp2-types, which are suggested to be rapidly discriminated by RT-PCR using polymorphisms as natural tags.
Methods
Cells and viruses
Porcine alveolar macrophages (PAM) were obtained from approximately 4-week-old pigs, as previously described [24], and cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum and antibiotics. Monkey kidney cell line MARC-145 [19] was cultured in Eagle’s minimum essential medium supplemented with 5% fetal bovine serum.
The 31 Japanese PRRSV isolates used in this study are listed in Table 1. The 2000–2001 isolates were passaged twice in PAM, with the exception of Jtg1, which was passaged three times in MARC-145 and twice in PAM, whereas the 1992–1993 isolates had various passage-histories. All viruses were used in a previous analysis on genetic variation of the ORF5 gene [45], with ORF5-based phylogenetic clusters listed in Table 1. The EDRD-1 strain was cloned by three rounds of limiting dilution in PAM, and the virus of the 14th passage on PAM was used for sequencing analysis. The EDRD-1 strain, as well as the lung homogenate from which it had been isolated, were kindly provided by Dr. Y. Murakami, National Institute of Animal Health. Two Japanese isolates, Gu922M [20] and Jtg1, were previously provided by Dr. H. Kuwahara, Nippon Institute for Biological Science, and Dr. T. Saito, Tochigi Livestock Hygiene Service Center, respectively. An attenuated vaccine strain, Ingelvac® MLV (Boehringer Ingelheim Vetmedica), was propagated in MARC-145 cells, which were then used for nsp2-typing by RT-PCR.
RT-PCR and nucleotide sequencing
Primers used in this study are listed in Table 2. RNA was extracted from 200 μl of lung homogenate or cell culture supernatant of PRRSV-infected PAM or MARC-145 cells using a High Pure Viral RNA kit (Roche). The EDRD-1 genome was reverse transcribed using ReverTra Ace-alpha (Toyobo) and genome-specific primers, followed by amplification into five fragments using high-fidelity DNA polymerase KOD-plus (Toyobo) and primer pairs 153F/4137R, 4037F/7793R, 7117F/10062R, 9669F/12148R and 12040F/15325R. PCR products were purified using a High Pure PCR Product Purification Kit (Roche), and sequenced using the primer-walking method with a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) on an ABI 3100 genetic analyzer (Applied Biosystems). The 5′- and 3′-ends of the EDRD-1 genome were determined using a 5′ RACE kit version 2.0 (Invitrogen) and a 3′ RACE kit (Takara), respectively.
To determine the nucleotide sequence of the partial nsp2 genes from 30 additional Japanese isolates, amplification of the complete nsp2 gene (expected size: 3,277 bp in the SP strain, which possesses the largest genome among PRRSV strains by including an 118-base insertion) was first performed using the 1184F and 4343R primer pair, KOD-plus polymerase, and cDNA synthesized with random hexamers, followed by amplification of the partial nsp2 gene by nested PCR (expected size: 1,485 bp in the SP strain) using the 2543F and 3910R primer pair. PCR conditions consisted of preheating at 94°C for 2 min, followed by 35 cycles composed of 15 sec denaturation at 94°C, 30 s annealing at 53°C, 3.5 min extension at 68°C, and a final 10-min incubation at 68°C, whereas nested PCR conditions were similar to those of PCR, but consisted instead of 30 cycles and 1.5 min extension periods. Nucleotide sequencing of the partial nsp2 gene was then performed using the primers used for PCR (2543F and 3910R), as well as additional internal primers (3147F and 3306R). For nucleotide sequencing in isolate Jnt1, the full-size nsp2 PCR products were used as templates due to a deletion in the 2543F primer-binding site. The accession numbers for sequences of the complete EDRD-1 strain genome and partial nsp2 gene from the additional Japanese isolates are listed in Table 1.
Phylogenetic and statistical analysis
For comparative analysis with Japanese isolates, the partial nucleotide sequences of the nsp2 gene from 50 strains of the NA-type virus were used based on DDBJ/EMBL/GenBank DNA databases (Table 1). Amino acid alignment was performed using the Clustal W program in MEGA 4.0 [40] with default parameters for GAP opening and extension penalties of 10 and 0.2, respectively, or with parameters ranging from 1 to 30 and from 0.1 to 1, respectively. Phylogenetic trees were constructed in MEGA 4.0 based on the amino acid sequences using the neighbor-joining method with the Kimura 2-parameter model. dN/dS ratios were calculated in MEGA 4.0 using the Nei-Gojobori method with the Jukes–Cantor model for the entire region and B-cell epitope region of nsp2. To assess the presence of a statistically higher frequency of deletions and insertions in B-cell epitopes than would be expected by chance, a binominal test was performed.
Typing RT-PCR
Based on the partial nsp2 sequences from Japanese isolates and database strains, primers were designed at regions which include either the 117-base deletion of nsp2-type E or the 108-base insertion of nsp2-types E and S, named regions A and B, respectively. Region A (corresponding to nt 1,206–2,073 of the nsp2 gene in the SP strain) was amplified using the 2543F and 931R primer pair, and region B (nt 2,041–2,690) using the 899F and 3910R primer pair, as well as the KOD-plus polymerase from the full-size PCR products. PCR conditions for both regions consisted of preheating at 94°C for 2 min, followed by 30 cycles composed of 15 s denaturation at 94°C, 30 s annealing at 53°C, 1 min extension at 68°C, and a final 10-min incubation at 68°C. The expected sizes for region A were 751 bp for EDRD-1 and 868 bp for SP and VR-2332, and for region B 650 bp for EDRD-1 and SP and 542 bp for VR-2332. The nsp2-type was determined based on the pattern of band sizes in regions A and B: short and long (SL) for nsp2-type E, long and short (LS) for nsp2-type V, and long and long (LL) for nsp2-type S.
Results
Genetic characterization of EDRD-1 strain
The complete genome sequence of the Japanese EDRD-1 strain was compared with that of the prototypic VR-2332 strain, as well as the SP strain, which possesses the largest genome among NA-type PRRSV strains. Excluding the poly A tail, the EDRD-1 genome was found to be 15,401 bases in length, which is 10 and 119 bases shorter than the length of the VR-2332 and SP sequences, respectively. Comparison between EDRD-1 and both VR-2332 and SP showed 87.0 and 87.4% nucleotide identity, respectively, for the whole genome and 78.7 and 81.2%, respectively, for the nsp2 gene. Remarkably, compared to the VR-2332 strain, the nsp2 gene of EDRD-1 contained a novel deletion of 117 bases (39 aa) in addition to an insertion of 108 bases (36 aa), which was also previously reported in the SP strain [35] (Supplemental Fig. 1a, Fig. 1; Table 1). Because of the sensitivity to varying parameters, namely opening and extension penalties, the position of the 117-base deletion could not be precisely determined, which is suggested to be due to the presence of direct repeat sequences in surrounding regions of the deletions, as described below. Further, these deletions are possibly located in the region between these direct repeats. To exclude the possibility that the deletion and insertion occurred during cell-passage, the EDRD-1 sequence was determined directly from the lung homogenate from which it was isolated. Results showed that the original viral genome also contained the 117-base deletion and 108-base insertion, indicating that the structural changes in nsp2 were intrinsic. In the partial nsp2 region of 1,216 bases in length, however, only one non-synonymous substitution (aa 676 in the SP strain) was found between original (proline) and PAM-passaged viral genome (serine).

Schematic representation of amino acid positions of 20 deletions and 3 insertions in nsp2. Amino acid positions are shown for the nsp2 of the SP strain. Lines between closed triangles represent the partial amino acid sequences of Japanese isolates determined in this study, except for EDRD-1, whose complete genome sequence was determined. Gaps in lines, numbers above lines and negative numbers in parenthesis represent deletions, possible sequence position ranges and sizes, respectively. Open triangles numbers above lines and positive numbers in parenthesis represent insertions, sequence positions and sizes, respectively. Black boxes represent regions corresponding to the 39-aa deletion in nsp2-type E and 36-aa insertion in nsp2-type S
Polymorphism of nsp2 gene in NA-type PRRSV
To confirm whether the deletion and insertion found in the nsp2 gene of the EDRD-1 strain were unique to this strain or not, partial nsp2 sequences (approximately 1.3 kb) from 30 additional Japanese isolates (Table 1) were determined and compared with 50 NA-type PRRSV strain sequences available from DDBJ/EMBL/GenBank DNA databases. The partial nsp2 region corresponds to nt 1,267–2,599 in the SP strain and covers the 117-base deletion and 108-base insertion found in the EDRD-1 strain. The 31 Japanese isolates and 50 strains from various countries were classified into three types and tentatively termed nsp2-types E, V and S. Type E, represented by the EDRD-1 strain, contains both the 117-base deletion and 108-base insertion and includes 21 Japanese isolates; type V, represented by the VR-2332 strain, does not contain the deletion or insertion and is composed of 58 strains, including 9 Japanese isolates; and type S, represented by the SP strain, contains only the 108-base insertion and is composed of 2 strains, including 1 Japanese isolate (Fig. 1a; Table 1). Approximately two-thirds (21/31) of Japanese isolates were classified into nsp2-type E, strains from various countries into nsp2-type V, and the SP strain was classified into nsp2-type S. All 20 Japanese isolates of ORF5-cluster III belonged to nsp2-type E, which also unexpectedly included one ORF5-cluster I isolate, Kagoshima-N14 (Table 1). This remarkable inconsistency in the Kagoshima-N14 isolate is possibly due to recombination occurring between nsp2 and ORF5 genes. In contrast, nsp2-types V and S contained several ORF5-clusters.
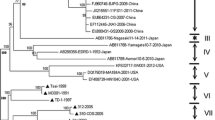
To clarify the phylogenetic relationships among the three nsp2-types, a phylogenetic tree was constructed using the neighbor-joining method based on the partial nsp2 aa sequences of the 31 Japanese isolates and the 50 NA-type strains. Analysis showed phylogenetic separation among the three nsp2-types, which are characterized by both or either the 119-base deletion and the 108-base insertion (Fig. 2). These results suggest that nsp2-type E diverged from an ancestral gene containing the 117-base deletion and the 108-base insertion. Two nsp2-type V strains, MN184A and MN184B, which were characterized by the absence of this deletion and insertion, were distantly and fairly closely related to other nsp2-types V and to nsp2-type S, respectively, which suggests a distinct evolutionary history.

Phylogenetic relationships among 31 Japanese isolates and 50 strains from various countries, based on partial amino acid sequences corresponding to aa 423–866 of the nsp2 gene in the SP strain. Bootstrap values on major nodes are shown as percentages
To exclude the possibility that nsp2-type S was a recombinant between nsp2-types E and V, two phylogenetic trees were constructed and compared. One tree was based on approximately half of the partial nsp2 sequence from the 5′-terminal end covering the 117-base deletion (nt 1,206–2,073 in the SP strain), whereas the other on approximately half from the 3′-terminal end covering the 108-base insertion (nt 2,041–2,690). Analysis showed that nsp2-type S formed a distinct branch in both trees, with 80 and 60% bootstrap values in 5′ and 3′ regions, respectively (data not shown). These results suggest that nsp2-type S is not a recent recombinant but instead an independent lineage.
In addition to the deletion and insertion mentioned above, the partial nsp2 sequences contained an additional 10 deletions ranging from 3 to 378 bases and 2 insertions of 3 and 21 bases at various positions in Japanese isolates, as well as 9 deletions previously reported in HB-2 [11], P129, BJ-4, 19407B [28], MN184A, MN184B [13], and potentially high pathogenic Chinese strains [22, 41] (Fig. 1; Table 1). However, the position of eight deletions could not be precisely determined. Undefined positions for these deletions were therefore represented as a possible position range. In addition, analysis showed that Jnt1 possesses the largest deletion, consisting of 378 bases (126 aa) (Supplemental Fig. 1b). In contrast with the determined 117-base deletion and 108-base insertion, the additional deletions and insertions were not common among sequences within each nsp2-type.
Development of RT-PCR-based genetic typing
To further distinguish the three nsp2-types, we performed RT-PCR based on the sizes of two regions, respectively composed of the 117-base deletion and 108-base insertion. Based on the size pattern of the two products amplified from the two regions, where nsp2-types E, V and S typically show the SL, LS and LL pattern, respectively, typing was successfully performed for 27 out of 31 Japanese isolates, as well as for Ingelvac® MLV vaccine strain (Fig. 3; Table 1). For the other four Japanese isolates Jtg1, Jyc1, Jnt1 and Jyt3, no or differentially sized PCR products were obtained due to an additional 93-, 57- and 378-base deletion and 21-base insertion, respectively.

Identification of the three nsp2-types by RT-PCR based on sizes of two regions, which cover a 117-base deletion and a 108-base insertion named region A and region B, respectively. Open triangles indicate sizes of amplified products. “L”, “S”, “M” and “–” indicate long, short and middle amplification sizes, and no amplification, respectively. Asterisks indicate no amplification or the different sizes in the four isolates. Asterisk Jnt1 has a 378-base deletion, including the 2543F primer-binding site. Double asterisk Jyc1 has 3- and 57-base deletions in region A. Triple asterisk Jyt3 has a 21-base insertion in region B. Four asterisk Jtg1 has a 93-base deletion in region B
Repeated sequences in deletions and insertions
Areas containing the 117-base deletion in nsp2-type E and 19 additional deletions were investigated for repeated sequences, which were previously shown to induce nucleotide deletion by jumping over a target sequence flanked by direct repeats or by skipping a stem structure formed through inverted repeats [31]. Results in the SP strain showed direct repeats of 18 bases flanking the 117-base deletion of nsp2-type E (Fig. 4). Further, direct repeats were also found in an additional 10 deletions (Fig. 4).

Direct repeats found in 11 deletions in the nsp2 gene. Direct repeat sequences are enclosed in shaded boxes. Dots and hyphens represent identical nucleotides and gapped positions, respectively. Asterisks indicate matched nucleotides between direct repeats. Open boxes represent the possible range of gapped positions
Deletions and insertions at B-cell epitopes in nsp2
Nsp2 is highly antigenic and contains a cluster of B-cell epitopes [4, 29, 44]. B-cell epitope regions, which were identified in the NVSL97-7895 [4] and BJ-4 strains [44] of nsp2-type V, were examined by analyzing the 20 deletions and 2 insertions found in nsp2. Results revealed 17 deletions and 2 insertions positioned at B-cell epitopes (Supplemental Fig. 1a, b; Table 1). A total of 19 indels were found in a 205 aa-long B-cell epitope region, whereas 2 indels were found in a 365 aa-long non-epitopic region. Statistical analysis showed that the frequency of indels is significantly higher in B-cell epitopic than non-epitopic regions (binominal test, P < 0.00001).
To determine the presence of positive or negative selection in nsp2, dN/dS ratios were calculated for the entire and B-cell epitope regions of nsp2. Results showed that ratios were 0.264 and 0.437, respectively, indicating that both regions were subjected to negative selection.
Discussion
In the present study, we investigated polymorphisms in the nsp2 gene of NA-type PRRSV. Based on polymorphisms characterized by a 117-base deletion and 108-base insertion, we have genetically classified NA-type PRRSV into three nsp2-types, which we have tentatively termed nsp2-types E, S and V. The 108-base sequence in nsp2-types E and S was originally reported in the SP strain as a unique insertion [35]. The present study, however, shows that this sequence is also predominantly found in Japanese isolates. The absence of the 117-base sequence in nsp2-type E, which is commonly present in subtypes V and S, was also observed. Consistent results from size polymorphism and phylogenetic clustering suggest that structural changes in each ancestral nsp2-type have only occurred at earlier stages of viral evolution. However, inconsistent results between size polymorphism and phylogenetic clustering were found for two isolates, namely MN184A and MN184B, which suggest that the 108-base insertion was not a single event in the evolutionary history of these viruses. Although we classified MN184 strains into nsp2-type V based on our nsp2-typing definition, the phylogenetic relationship with other nsp2-type V strains was distant. Further genetic investigation may reveal a fourth nsp2-type, characterized by a 111-base deletion, as found in MN184 strains. In contrast to the common 117-base deletion and 108-base insertion, 19 and 2 additional deletions and insertions, respectively, were occasionally found within each nsp2-type, suggesting the emergence of polymorphisms at later stages of viral evolution.
Forsberg [10] reported that MRCA, the most recent common ancestor of all PRRSV isolates, which includes both NA and EU genotypes, existed near 1,880, approximately 100 years before the emergence of PRRSV in pigs. During this period, PRRSV has possibly developed affinity and pathogenicity in domestic pigs, leading to the evolution of three nsp2-types of NA-type PRRSV. Our previous [45] and present studies show that in 1992–1993, when PRRSV was initially recognized in Japan, nsp2-type E was distributed in the eastern part of Japan, whereas nsp2-type V was limited to the western part. These findings suggest that the clear geographic separation of nsp2-types E and V in Japan was possibly due to multiple introduction of the virus into the country, rather than initial emergence.
We demonstrated that polymorphisms of the nsp2 gene are natural genetic markers. The three nsp2-types were typically defined by the pattern of two product sizes which were amplified from two regions containing either the 117-base deletion or 108-base insertion. The molecular typing method developed in this study may facilitate the differentiation of nsp2-type E field isolates, which are predominant in Japan, from the nsp2-type V vaccine strain of Ingelvac® MLV, which is widely used in Japan. In addition, potentially high pathogenic nsp2-type V Chinese isolates, in which primer-binding sites for nsp2-typing were conserved, contain a 90-base deletion in region A and no deletion or insertion in region B, and are expected to show an SS pattern in RT-PCR typing. Confirmation of this pattern would enable discrimination of these Chinese isolates from other nsp2-type V isolates, as well as nsp2-types E and S isolates. Further results, however, showed that the molecular typing method cannot type all isolates. For example, in four exceptional cases, no or differentially sized PCR products were obtained due to additional deletions or an insertion. In contrast, using these additional deletions or the insertion as additional genetic markers may facilitate discrimination of these isolates from other typical isolates of the same nsp2-type.
Analysis showed that the majority of deletions and insertions in nsp2 correspond to B-cell epitope regions, which suggests that these antigenic regions are potential serological markers. In particular, the 39-aa deletion in nsp2-type E is a potential marker to distinguish pigs infected with nsp2-type E from those vaccinated for nsp2-type V. Better understanding of the antigenicity of these regions requires further investigation.
The considerable flexibility of nsp2 was demonstrated using reverse genetics. A previous deletion mutagenesis analysis revealed that 403 and 86 aa in the central region of nsp2, which corresponds to aa positions 324–726 and 727–813 in the VR-2332 strain, are not critical for viral replication [14]. Further, all deletions and insertions observed in nsp2 were found to be contained in these regions. These findings suggest that additional deletions can possibly occur in the nsp2 region. Another study showed that nsp2 tolerates artificial insertions of several polyprotein tags, including FLAG, enhanced green fluorescent protein and luciferase [18], in large dispensable regions, providing the opportunity to develop a marker vaccine. In the present study, we showed that about a half of deletions are associated with direct repeats, suggesting that the region flanked by these repeats should be avoided for more stable retention of a foreign gene. A previous report showed that nucleotide deletions are induced by repeated sequences which jump over a sequence flanked by direct repeats [31], using a mechanism proposed to involve detachment of an extending sense (or antisense) RNA strand from the RNA template after completely or partially copying the 5′ (or 3′) direct repeat sequence, followed by jumping to the 3′ (or 5′) direct repeat sequence on the same or different template molecule and continuing extension. We speculate that deletions in the nsp2 gene partially involves direct repeats through this mechanism.
Although genetic markers have been identified, vaccine efficacy against the three nsp2-types remains unknown. One study reported low efficiency of an EU-type vaccine of the Lelystad cluster against the challenge of an evolutionarily distinct Italian isolate [21]. Another study reported that genomic homology between a vaccine strain and challenge NA-type PRRSV isolates was not a good predictor of vaccine efficacy [30]. To date, for the NA-type PRRSV, live nsp2-type V (Ingelvac® MLV and Ingelvac® ATP) and nsp2-type S (PrimePac®) vaccines are available, but no nsp2-type E vaccine has yet been developed. Assessment of the efficacy of vaccines against the evolutionarily distinct nsp2-type E will be necessary.
In conclusion, our study suggests a novel classification of NA-type PRRSV into three nsp2-types based on polymorphisms of the nsp2 gene. By using deletions and insertions as natural tags, rapid discrimination among these nsp2-types can be potentially facilitated. In addition, our findings provide new insight into the significant evolutionary history of NA-type PRRSV.
References
Allende R, Lewis TL, Lu Z, Rock DL, Kutish GF, Ali A, Doster AR, Osorio FA (1999) North American and European porcine reproductive and respiratory syndrome viruses differ in non-structural protein coding regions. J Gen Virol 80:307–315
Benfield DA, Collins JE, Dee SE, Halbur PG, Joo HS, Lager KM, Mengeling WL, Murtaugh MP, Rossow K, Stevenson GW, Zimmerman JJ (1999) Porcine reproductive and respiratory syndrome, 8th edn. Iowa State Press, Iowa
Cavanagh D (1997) Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch Virol 142:629–633
de Lima M, Pattnaik AK, Flores EF, Osorio FA (2006) Serologic marker candidates identified among B-cell linear epitopes of Nsp2 and structural proteins of a North American strain of porcine reproductive and respiratory syndrome virus. Virology 353:410–421
Dea S, Gagnon CA, Mardassi H, Pirzadeh B, Rogan D (2000) Current knowledge on the structural proteins of porcine reproductive and respiratory syndrome (PRRS) virus: comparison of the North American and European isolates. Arch Virol 145:659–688
Fang Y, Kim DY, Ropp S, Steen P, Christopher-Hennings J, Nelson EA, Rowland RR (2004) Heterogeneity in Nsp2 of European-like porcine reproductive and respiratory syndrome viruses isolated in the United States. Virus Res 100:229–235
Fang Y, Schneider P, Zhang WP, Faaberg KS, Nelson EA, Rowland RR (2007) Diversity and evolution of a newly emerged North American Type 1 porcine arterivirus: analysis of isolates collected between 1999 and 2004. Arch Virol 152:1009–1017
Forsberg R, Oleksiewicz MB, Petersen AM, Hein J, Botner A, Storgaard T (2001) A molecular clock dates the common ancestor of European-type porcine reproductive and respiratory syndrome virus at more than 10 years before the emergence of disease. Virology 289:174–179
Forsberg R, Storgaard T, Nielsen HS, Oleksiewicz MB, Cordioli P, Sala G, Hein J, Botner A (2002) The genetic diversity of European type PRRSV is similar to that of the North American type but is geographically skewed within Europe. Virology 299:38–47
Forsberg R (2005) Divergence time of porcine reproductive and respiratory syndrome virus subtypes. Mol Biol Evol 22:2131–2134
Gao ZQ, Guo X, Yang HC (2004) Genomic characterization of two Chinese isolates of porcine respiratory and reproductive syndrome virus. Arch Virol 149:1341–1351
Goldberg TL, Hahn EC, Weigel RM, Scherba G (2000) Genetic, geographical and temporal variation of porcine reproductive and respiratory syndrome virus in Illinois. J Gen Virol 81:171–179
Han J, Wang Y, Faaberg KS (2006) Complete genome analysis of RFLP 184 isolates of porcine reproductive and respiratory syndrome virus. Virus Res 122:175–182
Han J, Liu G, Wang Y, Faaberg KS (2007) Identification of nonessential regions of the nsp2 replicase protein of porcine reproductive and respiratory syndrome virus strain VR-2332 for replication in cell culture. J Virol 81:9878–9890
Hanada K, Suzuki Y, Nakane T, Hirose O, Gojobori T (2005) The origin and evolution of porcine reproductive and respiratory syndrome viruses. Mol Biol Evol 22:1024–1031
Kapur V, Elam MR, Pawlovich TM, Murtaugh MP (1996) Genetic variation in porcine reproductive and respiratory syndrome virus isolates in the midwestern United States. J Gen Virol 77:1271–1276
Keffaber KK (1989) Reproductive failure of unknown etiology. Am Assoc Swine Pract Newsl 1:1–9
Kim DY, Calvert JG, Chang KO, Horlen K, Kerrigan M, Rowland RR (2007) Expression and stability of foreign tags inserted into nsp2 of porcine reproductive and respiratory syndrome virus (PRRSV). Virus Res 128:106–114
Kim HS, Kwang J, Yoon IJ, Joo HS, Frey ML (1993) Enhanced replication of porcine reproductive and respiratory syndrome (PRRS) virus in a homogeneous subpopulation of MA-104 cell line. Arch Virol 133:477–483
Kuwahara H, Nunoya T, Tajima M, Kato A, Samejima T (1994) An outbreak of porcine reproductive and respiratory syndrome in Japan. J Vet Med Sci 56:901–909
Labarque G, Van Reeth K, Nauwynck H, Drexler C, Van Gucht S, Pensaert M (2004) Impact of genetic diversity of European-type porcine reproductive and respiratory syndrome virus strains on vaccine efficacy. Vaccine 22:4183–4190
Li Y, Wang X, Bo K, Tang B, Yang B, Jiang W, Jiang P (2007) Emergence of a highly pathogenic porcine reproductive and respiratory syndrome virus in the Mid-Eastern region of China. Vet J 174:577–584
Mateu E, Martin M, Vidal D (2003) Genetic diversity and phylogenetic analysis of glycoprotein 5 of European-type porcine reproductive and respiratory virus strains in Spain. J Gen Virol 84:529–534
Mengeling WL, Lager KM, Vorwald AC (1995) Diagnosis of porcine reproductive and respiratory syndrome. J Vet Diagn Invest 7:3–16
Meulenberg JJ, Hulst MM, de Meijer EJ, Moonen PL, den Besten A, de Kluyver EP, Wensvoort G, Moormann RJ (1993) Lelystad virus, the causative agent of porcine endemic abortion and respiratory syndrome (PEARS), is related to LDV and EAV. Virology 192:62–72
Murakami Y, Kato A, Tsuda T, Morozumi T, Miura Y, Sugimura T (1994) Isolation and serological characterization of porcine reproductive and respiratory syndrome (PRRS) viruses from pigs with reproductive and respiratory disorders in Japan. J Vet Med Sci 56:891–894
Nelsen CJ, Murtaugh MP, Faaberg KS (1999) Porcine reproductive and respiratory syndrome virus comparison: divergent evolution on two continents. J Virol 73:270–280
Nielsen HS, Oleksiewicz MB, Forsberg R, Stadejek T, Botner A, Storgaard T (2001) Reversion of a live porcine reproductive and respiratory syndrome virus vaccine investigated by parallel mutations. J Gen Virol 82:1263–1272
Oleksiewicz MB, Botner A, Toft P, Normann P, Storgaard T (2001) Epitope mapping porcine reproductive and respiratory syndrome virus by phage display: the nsp2 fragment of the replicase polyprotein contains a cluster of B-cell epitopes. J Virol 75:3277–3290
Opriessnig T, Pallares FJ, Nilubol D, Vincent AL, Thacker EL, Vaughn EM, Roof M, Halbur PG (2005) Genomic homology of ORF 5 gene sequence between modified live vaccine virus and porcine reproductive and respiratory syndrome virus challenge isolates is not predictive of vaccine efficacy. J Swine Health Prod 13(5):246–253
Parvin JD, Wang LH (1984) Mechanisms for the generation of src-deletion mutants and recovered sarcoma viruses: identification of viral sequences involved in src deletions and in recombination with c-src sequences. Virology 138:236–245
Paton DJ, Brown IH, Edwards S, Wensvoort G (1991) ‘Blue ear’ disease of pigs. Vet Rec 128:617
Pedersen KW, van der Meer Y, Roos N, Snijder EJ (1999) Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J Virol 73:2016–2026
Ropp SL, Wees CE, Fang Y, Nelson EA, Rossow KD, Bien M, Arndt B, Preszler S, Steen P, Christopher-Hennings J, Collins JE, Benfield DA, Faaberg KS (2004) Characterization of emerging European-like porcine reproductive and respiratory syndrome virus isolates in the United States. J Virol 78:3684–3703
Shen S, Kwang J, Liu W, Liu DX (2000) Determination of the complete nucleotide sequence of a vaccine strain of porcine reproductive and respiratory syndrome virus and identification of the Nsp2 gene with a unique insertion. Arch Virol 145:871–883
Snijder EJ, Meulenberg JJ (1998) The molecular biology of arteriviruses. J Gen Virol 79:961–979
Snijder EJ, van Tol H, Roos N, Pedersen KW (2001) Non-structural proteins 2 and 3 interact to modify host cell membranes during the formation of the arterivirus replication complex. J Gen Virol 82:985–994
Snijder EJ, Brinton MA, Faaberg KS, Godeny EK, Gorbalenya AE, MacLachlan NJ, Mengeling WL, Plagemann PG (2005) Family Arteriviridae. Elsevier Academic Press, California
Stadejek T, Oleksiewicz MB, Potapchuk D, Podgorska K (2006) Porcine reproductive and respiratory syndrome virus strains of exceptional diversity in eastern Europe support the definition of new genetic subtypes. J Gen Virol 87:1835–1841
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Tian K, Yu X, Zhao T, Feng Y, Cao Z, Wang C, Hu Y, Chen X, Hu D, Tian X, Liu D, Zhang S, Deng X, Ding Y, Yang L, Zhang Y, Xiao H, Qiao M, Wang B, Hou L, Wang X, Yang X, Kang L, Sun M, Jin P, Wang S, Kitamura Y, Yan J, Gao GF (2007) Emergence of fatal PRRSV variants: unparalleled outbreaks of atypical PRRS in China and molecular dissection of the unique hallmark. PLoS ONE 2:e526
Wensvoort G, Terpstra C, Pol JM, ter Laak EA, Bloemraad M, de Kluyver EP, Kragten C, van Buiten L, den Besten A, Fea Wagenaar (1991) Mystery swine disease in The Netherlands: the isolation of Lelystad virus. Vet Q 13:121–130
Wu WH, Fang Y, Farwell R, Steffen-Bien M, Rowland RR, Christopher-Hennings J, Nelson EA (2001) A 10-kDa structural protein of porcine reproductive and respiratory syndrome virus encoded by ORF2b. Virology 287:183–191
Yan Y, Guo X, Ge X, Chen Y, Cha Z, Yang H (2007) Monoclonal antibody and porcine antisera recognized B-cell epitopes of Nsp2 protein of a Chinese strain of porcine reproductive and respiratory syndrome virus. Virus Res 126:207–215
Yoshii M, Kaku Y, Murakami Y, Shimizu M, Kato K, Ikeda H (2005) Genetic variation and geographic distribution of porcine reproductive and respiratory syndrome virus in Japan. Arch Virol 150:2313–2324
Ziebuhr J, Snijder EJ, Gorbalenya AE (2000) Virus-encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol 81:853–879
Acknowledgments
We thank Dr. H. Kuwahara, Nippon Institute for Biological Science, and Dr. T. Saito, Tochigi Livestock Hygiene Service Center, for providing Japanese isolates Gu922 M and Jtg1, respectively. We also thank Dr. Y. Murakami, National Institute of Animal Health, for providing the EDRD-1 strain and the lung homogenate from which it had been isolated. We also thank Dr. Y. Kaku, National Institute of Health, and Dr. T. Sekizaki, Dr. M. Osaki, and Dr. I. Yamane, National Institute of Animal Health, and Dr. K. Tamura, Tokyo Metropolitan University, and Dr. N. Saito, National Institute of Genetics, for their helpful suggestion.
Author information
Authors and Affiliations
Corresponding author
Additional information
The GenBank/EMBL/DDBJ accession numbers of the sequences reported in this paper are AB288110–AB288139 and AB288356.
Electronic supplementary material
Below is the link to the electronic supplementary material.
705_2008_98_MOESM1_ESM.ppt
Amino acid alignment of the three nsp2-types in nsp2. Dots and hyphens represent identical amino acids and gapped positions, respectively. B-cell epitopes identified in NVSL97-7895 [1] and BJ-4 strains [2] are underlined. Shaded areas represent positions of possible deletion ranges. a Alignment of nsp2 aa positions 423-866 (based on the SP strain) in 31 Japanese isolates and 50 strains from other countries. b Alignment of nsp2 aa 297-444 (based on the SP strain) in Jnt1 and representative strains. (PPT 62 kb).
Rights and permissions
About this article
Cite this article
Yoshii, M., Okinaga, T., Miyazaki, A. et al. Genetic polymorphism of the nsp2 gene in North American type-Porcine reproductive and respiratory syndrome virus . Arch Virol 153, 1323–1334 (2008). https://doi.org/10.1007/s00705-008-0098-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-008-0098-6