
Abstract
Serotonin, in addition to its fundamental role as a neurotransmitter, plays a critical role in the cardiovascular system, where it is thought to be involved in the development of cardiac hypertrophy and failure. Indeed, we recently found that mice with deletion of monoamine oxidase A had enhanced levels of blood and cardiac 5-HT, which contributed to exacerbation of hypertrophy in a model of experimental pressure overload. 5-HT2A receptors are expressed in the heart and mediate a hypertrophic response to 5-HT in cardiac cells. However, their role in cardiac remodeling in vivo and the signaling pathways associated are not well understood. In the present study, we evaluated the effect of a selective 5-HT2A receptor antagonist, M100907, on the development of cardiac hypertrophy induced by transverse aortic constriction (TAC). Cardiac 5-HT2A receptor expression was transiently increased after TAC, and was recapitulated in cardiomyocytes, as observed with 5-HT2A in situ labeling by immunohistochemistry. Selective blockade of 5-HT2A receptors prevented the development of cardiac hypertrophy, as measured by echocardiography, cardiomyocyte area and heart weight-to-body weight ratio. Interestingly, activation of calmodulin kinase (CamKII), which is a core mechanism in cardiac hypertrophy, was reduced in cardiac samples from M100907-treated TAC mice compared to vehicle-treated mice. In addition, phosphorylation of histone deacetylase 4 (HDAC4), a downstream partner of CamKII was significantly diminished in M100907-treated TAC mice. Thus, our results show that selective blockade of 5-HT2A receptors has beneficial effect in the development of cardiac hypertrophy through inhibition of the CamKII/HDAC4 pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Serotonin (5-HT), in addition to its fundamental role as a neurotransmitter, is a vasoactive substance that elicits several effects on the cardiovascular system. The myriad of biological actions of 5-HT are mediated by no fewer than 15 receptors divided into seven subtypes (5-HT1 to 5-HT7). Among those, 5-HT2 receptors, which are coupled to the Gq/11/PLC signaling pathway, are known to be critical both in the central nervous and cardiovascular systems. 5-HT2 receptors have generated strong therapeutic interest in a diverse array of pathophysiological implications such as sleep, hallucinogenesis, schizophrenia, appetite control, neuroendocrine secretions, hypertension, migraine and depression.
In the heart, accumulating evidences point toward 5-HT2 receptors as potential candidates in the development of cardiac hypertrophy. Cardiac hypertrophy is an adaptive response to pressure overload, initiated by hypertension or valvular diseases. When prolonged, cardiac hypertrophy can lead to heart failure, a condition where the diseased heart is no longer able to meet the circulatory requirements of the body. 5-HT levels have been demonstrated to be increased in blood from heart failure patients and to be associated with disease severity, which support a role for this neurohormone in cardiac failure (Nigmatullina et al. 2009).
Recently, the consequences of monoamine oxidase A (MAO-A) deletion in blood and cardiac serotonin as well as in cardiac remodeling were evaluated (Lairez et al. 2009). MAO-A constitutes the major metabolic pathway for 5-HT and during substrate degradation, hydrogen peroxide (H2O2) is generated, acting as a signaling intermediate in cell hypertrophy and/or death (Mialet-Perez et al. 2007). We hypothesized that MAO-A would play an important role during cardiac hypertrophy, either by regulating 5-HT levels or by its generation of H2O2 in the heart. In our study, we found that MAO-A knock-out mice were constantly exposed to elevated levels of blood and cardiac 5-HT, and demonstrated exacerbation of cardiac hypertrophy in response to pressure overload. We identified the 5-HT2A receptor as a major component involved in the exacerbation of hypertrophy in response to elevated levels of 5-HT (Lairez et al. 2009). In vitro, additional evidences demonstrated that 5-HT2A receptors were expressed in cardiac cells and triggered a hypertrophic response to 5-HT, through the involvement of transient receptor potential canonical 1 (TRPC1) channels and calcineurin/NFAT activation (Bush et al. 2004; Villeneuve et al. 2009; Vindis et al. 2010). In addition, further studies demonstrated that during cardiac remodeling, increased expression of 5-HT2A receptors generated a functional inotropic response to 5-HT (Qvigstad et al. 2005).
At present, the consequences of selective inhibition of 5-HT2A receptors in vivo on the development of cardiomyocyte hypertrophy and the signaling pathways associated are not well defined. To address this question, we used M100907, an antagonist with high potency and selectivity toward 5-HT2A receptors, and measured its effect on the development of cardiac hypertrophy in a model of pressure overload induced by transverse aortic constriction (TAC) in mice. We found that selective inhibition of 5-HT2A receptors with M100907 prevented the development of cardiac hypertrophy following pressure overload and inhibited the activation of the calmodulin-dependent kinase II (CaMKII)–histone deacetylase 4 (HDAC4) hypertrophic pathway.
Methods
Aortic banding
C57Bl6J mice (Janvier, France) were housed in a pathogen-free facility and handled in accordance with the procedures outlined in Council Directive 86/609/EEC. 8-week-old male mice were anesthetized with isoflurane (4 %), ventilated, and the left thorax was opened through a small thoracic window. Aortic constriction (TAC) was performed by ligating the transverse aorta under a dissecting microscope. For this purpose, two knots were made on the thread which was carefully introduced around the transverse aorta. Both knots were tied together to perform aortic constriction (27-gauge needle circumference). Age-matched animals underwent identical surgical procedure except for ligation of the aorta (sham-operated mice).
Experimental protocol
Animals were randomly categorized into four groups: (a) sham vehicle (n = 5), (b) TAC vehicle (n = 10), (c) sham M100907 (n = 5), and (d) TAC M100907 (n = 10). Mice were treated for 3 months with the 5-HT2A receptor antagonist M100907 ((R)-(+)-a-(2,3-dimethoxyphenyl)-1-[2-(4-fluorophenyl)ethyl]-4-pipidinemethanol) (0.1 mg/kg/day, dissolved in a minimum volume of 0.01 N hydrochloric acid, further diluted to its final concentration using 0.9 % saline, and adjusted to pH 7.5) or vehicle, administered with 0.2-ml Alzet mini-osmotic pumps (Charles River labs, L’Arbresle, France) placed intraperitoneally. Mini-osmotic pumps were replaced every month. Animals were sacrificed 1 or 3 months after TAC.
Echocardiography
Animals were anesthetized with 2 % isoflurane and examined with non-invasive echocardiography (echocardiograph Vivid 7 ultrasound, GE). Cardiac ventricular dimensions were measured on M-mode images at least five times for the number of animals indicated.
Invasive hemodynamic study
Animal anesthesia was induced by intraperitoneal injection of ketamine (125 mg/kg) and xylazine (5 mg/kg) and maintained with 1 % isoflurane in oxygen flow (0.1 l/min). Mice were orally intubated with a 20-G catheter and ventilated using a Minivent 845 respirator (Harvard Apparatus, Les Ulis, France); the tidal volume and respiration rate were, respectively, 200 μl and 150 bpm. Left ventricular apex was reached through the left 5th intercostal space and punctured with a 26-G needle, following the septal axis. A high-fidelity Millar Mikro-Tip pressure catheter (SPR-671, ADInstruments Ltd., Oxford, UK) was then inserted into this pre-hole and left ventricular pressure was recorded for 10 min using a Bridge Amp (ML221) connected to a PowerLab 8/30 acquisition system (ML870, ADInstruments). Data were analyzed with LabChart Pro v7.3.3 software (ADInstruments), using five consecutive 30 s periods.
Western blot
Ventricular homogenates were lysed in RIPA buffer (10 mM Tris pH 7.4, 150 mM NaCl, 1 % Triton X-100, 1 % sodium deoxycholate, 0.1 % sodium dodecyl sulfate, 1 mM sodium orthovanadate, 1 mM sodium pyrophosphate, 5 mM sodium fluoride, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml aprotinin). Protein extracts were resolved by SDS-polyacrylamide gel electrophoresis, transferred onto PVDF membranes (Millipore, Molsheim, France). Then, membranes were probed with the indicated primary antibodies and revealed with the secondary antibodies coupled to horseradish peroxidase using the ECL chemoluminescence kit. Membranes were then stripped and reprobed with anti-GAPDH antibody to control equal loading of proteins. Primary antibodies for phospho-Ser632–HDAC4, HDAC4 and CamKII were from Santa-Cruz Biotechnology (Heidelberg, Germany). Primary antibody for 5-HT2A receptor was from GenScript (Paris, France). Primary antibody for phospho-Thr286-CamKII was from Cell Signaling (Saint-Quentin, France).
Histological analysis
Ventricles were incubated in Carnoy’s fixative solution (ethanol 60 %, chloroform 30 %, acetic acid 10 %), embedded in paraffin and transversally sectioned. 5 μm tissue sections were stained with H&E. Cardiomyocyte diameter was evaluated after coloration with H&E (250–300 cells counted per heart) on left ventricle.
Immunohistochemistry
Paraffin-embedded tissue sections were first de-waxed in toluene and rehydrated through a series of graded ethanol washes before endogenous peroxidase blockage. Then, they were incubated in blocking buffer (1 % BSA in TBS-T) for 10 min. Specific primary antibodies were incubated (1 h at room temperature) on mouse sections for the detection of 5-HT2A receptor (Genscript, 1:50). For visualization, we used (DAB) chromogen detection system with secondary HRP anti-rabbit antibody. Negative controls for the immunohistochemical procedures included substitution of the primary antibody with non-immune sera.
Real-time RT-PCR
Extraction of RNA was performed using column affinity purification (Macherey-Nagel, Hoerdt, France). First-strand cDNA was synthesized using the superscript II RT-PCR system (Invitrogen, Cergy Pontoise, France) with random hexamers. Negative controls without reverse transcriptase were made to verify the absence of genomic DNA contamination. Real-time PCR was performed on a StepOnePlus system (Applied Biosystem, Courtaboeuf, France) in 96-well plates. A 1/10 dilution of cDNA (5 μl) from RT reaction was mixed with specific primers and SYBR green mix (Eurogentec, Angers, France). ANP, NppaF: 5′-AGAGTGGGCAGAGACAGCAAA-3′, NppaR: 5′-AAGGCCAAGACGAGGAAGAAG-3′; β-MHC, Myh7F: 5′-AGGTGGCTCCGAGAAAGGAA-3′, Myh7R: 5′-TGAGCCTTGGATTCTCAAACGT-3′; αSK-actin, Acta1F: 5′-TACCACCGGCATCGTGTTG-3′, Acta1R: 5′-CCAGGTCCAGACGCATGAT-3′; Collagen III, Coll 3F: 5′-ACGTAGATGAATTGGGATGCAG-3′, Coll 3R: 5′-GGGTTGGGCAGTCTAGTG-3′.
Statistical analysis
Results are expressed as mean ± SEM. Experimental groups were compared using Student’s t test, 1-way ANOVA or 2-way ANOVA followed by Bonferroni’s post hoc test, as appropriate. A value of p < 0.05 was considered significant.
Results
Transient upregulation of 5-HT2A receptor during the development of cardiac hypertrophy
To better understand the relevance of 5-HT2A receptor in cardiac hypertrophy, we followed the time course of regulation of this receptor by Western blot in the model of pressure overload induced by TAC in mice. Pressure overload causes progressive left ventricle hypertrophy, which can lead to subsequent cardiac dilatation and failure in the later stage. As shown in Fig. 1a, 5-HT2A receptor expression was significantly increased in ventricles of TAC-operated mice at 1 month, compared to sham-operated hearts. However, in the later stage of hypertrophy, 3 months after TAC, 5-HT2A receptor expression was slightly decreased (non-significant) compared to sham mice. Next, we investigated the localization of cardiac 5-HT2A receptors by immunohistochemistry in ventricles from sham or TAC mice at 1 month. A good labeling of 5-HT2A receptors was observed in ventricular myocytes of sham-operated mice, which was compatible with binding values measured in previous studies (Fig. 1b) (Jaffre et al. 2009). During cardiac hypertrophy, 5-HT2A receptor staining became more pronounced in cardiomyocytes (Fig. 1b). In conclusion, overexpression of 5-HT2A receptors in cardiomyocytes is transient and occurs during the early phase of hypertrophy.

Transient regulation of 5-HT2A receptor expression in a mouse model of cardiac hypertrophy induced by aortic constriction. 8-week-old mice were subjected to transverse aortic constriction (TAC) or sham operation to trigger pressure-overload hypertrophy of the heart. a 5-HT2A receptor levels were analyzed by immunoblotting on cardiac homogenates at 1 and 3 months after TAC. Immunoblots were performed on cardiac homogenates with anti-5-HT2A, and GAPDH expression serves as a loading control. Graph represents densitometric analysis of 5-HT2A receptor expression in sham or TAC hearts (n = 3). b 5-HT2A receptor expression by immunohistochemistry in ventricles from sham or TAC hearts at 1 month (n = 5) (×200 magnitude). (*p < 0.05 vs. indicated value)
M100907 treatment significantly reduced the development of pressure-overload hypertrophy
As we observed significant upregulation of 5-HT2A receptors in cardiomyocytes during the development of hypertrophy, we evaluated the role of these receptors using a selective 5-HT2A receptor antagonist, M100907. We treated mice with 0.1 mg/kg/day of M100907, a dose selective for 5-HT2A receptors in vivo and maximally efficient in behavioral tests (Marek et al. 2005).
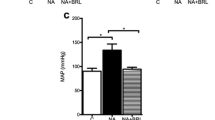
Cardiac morphology and function were analyzed by non-invasive echocardiography and invasive hemodynamics (Table 1). Following 2 months of TAC, significant increases in interventricular septum thickness (IVST) and posterior wall thickness (PWT) were observed in TAC-vehicle mice compared to sham-vehicle mice, which is a consequence of pressure-overload hypertrophy (Table 1). Interestingly, TAC-M100907 mice presented significantly lower IVST and PWT values compared to TAC-vehicle mice, indicating that hypertrophy was reduced. Administration of M100907 in sham mice did not alter any of the echocardiographic parameters. At 3 months, TAC-vehicle mice had increased end-diastolic (EDD) and end-systolic diameters (ESD) compared to sham-vehicle mice, with preserved cardiac function since fractional shortening (FS%) was not significantly modified (Table 1). However, in TAC-M100907 mice at 3 months, cardiac dilatation was not observed (EDD and ESD) and FS was higher than in TAC-vehicle mice. Thus, M100907 treatment prevents the development of hypertrophy and limits the evolution to cardiac dilatation at a later stage.
Left ventricular function was assessed using a high-fidelity Millar catheter inserted through the cardiac apex. A significant decrease in the maximal left ventricular pressure was found following M100907 treatment, in both sham (−17.2 mmHg) and TAC (−21.1 mmHg) mice (Table 1). This drop in maximal pressure induced by 5-HT2A blockade should cause a similar reduction in systolic arterial blood pressure. End-diastolic left ventricular pressure, dP/dt max and dP/dt min were not significantly modified, indicating that M100907 treatment did not affect left ventricular contractility or relaxation.
M100907 treatment significantly reduced other parameters associated with ventricle hypertrophy. Morphometric analysis in post-mortem animals demonstrated that heart/body weight (HW/BW) ratios were significantly lower in TAC-M100907 animals compared to TAC-vehicle animals (Fig. 2a). Concerning ventricle/body weight ratios, the same difference was observed, albeit non-significant. Next, we performed specific measurements of cardiomyocyte cross-sectional areas on transverse sections of the hearts (Fig. 2b). Again, we found a significant reduction in cardiomyocyte size in TAC-M100907 treated mice compared to TAC-vehicle mice. Modification of genes involved in cardiac remodeling, such as foetal gene expression and fibrosis, was evaluated by real-time RT-PCR (Fig. 2c). In TAC animals, M100907 treatment decreased the expression of αSK-actin and β-MHC mRNA, compared to TAC-vehicle animals. Expression of ANP was not statistically different. In addition, expression of collagen I mRNA, but not collagen III mRNA, was decreased by M100907 treatment in TAC mice.

M100907 prevents the development of cardiac hypertrophic parameters induced by TAC in mice. Mice were subjected to TAC or sham operation in the presence of 0.1 mg/kg/day of M100907 or vehicle, by osmotic mini-pumps. Hearts were collected 2 months after TAC for analysis. a Ventricle-to-body weight and heart-to-body weight ratios in sham (n = 5) and TAC (n = 10) vehicle-treated or M100907-treated mice. b Cardiomyocytes cross-sectional areas determined on hematoxylin–eosin stained sections using SigmaScan Pro (~200 cells/heart) with representative image on the left panel (×400 magnitude) and histogram on the right panel (n = 5 for sham and n = 10 for TAC mice). c Real-time PCR in ventricles from sham (n = 5) and TAC (n = 10) vehicle-treated or M100907-treated mice. Expression of the gene of interest is normalized to HPRT by the method of 2ΔCT, and results are expressed as fold over sham-vehicle mice (*p < 0.05; **p < 0.01; ***p < 0.001 vs. sham mice, #p < 0.05; ##p < 0.01; ###p < 0.001 vs. TAC-vehicle-treated mice, 1-way analysis with post hoc Turkey test)
Taken together, our data show that 5-HT2A selective antagonist M100907 prevents the development of pressure-overload ventricle remodeling in mice.
M100907 inhibits the activation of the CamKII–HDAC4 hypertrophic pathway
The CamKII pathway is a core mechanism that promotes myocardial hypertrophy and heart failure. CamKII is known to be activated by Gq-coupled receptors through autophosphorylation at Thr286 in response to elevation of Ca2+. Thus, we evaluated the activation of CamKII in hearts from TAC mice with a phospho-Thr286 antibody by Western blotting (Fig. 3). We found that hypertrophic hearts had enhanced levels of phosho-Thr286 CaMKII compared to sham hearts. Interestingly, the 5-HT2A antagonist M100907 prevented the activation of CamKII during cardiac hypertrophy.

M100907 attenuates TAC-induced activation of calcium-related proteins. Mice were subjected to TAC or sham operation in the presence of 0.1 mg/kg/day of M100907 or vehicle, by osmotic mini-pumps. Hearts were collected 3 months after TAC and heart homogenates were prepared for immunoblot analysis. Activation of CamKII and HDAC4 were evaluated with anti-phospho-Thr286–CamKII and anti-phospho-Ser632–HDAC4 antibodies, respectively. Expression of CamKII was measured with anti-CamKII antibody. GAPDH serves as a loading control. Densitometric analysis were performed on n = 5 mice and were normalized to GAPDH as represented in the histogram. (*p < 0.05 vs. indicated value)
In the heart, CamKII is able to phosphorylate HDAC4 to promote pathological cardiac remodeling. Phosphorylation of HDAC4 on its serine residue by CamKII regulates its nucleocytoplasmic shuttling, relieving and derepressing hypertrophic gene expression. Thus, we evaluated HDAC4 phosphorylation with a phospho-Ser632 specific antibody in hearts from TAC and sham-operated animals (Fig. 3). We found that pressure overload increased the level of phospho-HDAC4. However, in M100907-treated animals, the phosphorylation of HDAC4 was inhibited. In conclusion, the 5-HT2A selective antagonist M100907 prevents the activation of the CamKII–HDAC hypertrophic pathway in vivo.
Discussion
In the present study, we demonstrate that M100907, a selective 5-HT2A receptor antagonist, prevents the development of myocardial hypertrophy in a model of pressure overload. Mice with experimental hypertrophy induced by TAC demonstrated reduced wall thickness, decreased cardiomyocyte area and reduced expression of genes involved in cardiac remodeling, when treated with M100907. In addition, 5-HT2A receptor blockade inhibited the activation of the CamKII–HDAC4 pathway in the heart.
To our knowledge, this is the first study that addresses specifically the role of the 5-HT2A receptor subtype in cardiac hypertrophy in vivo. Previous studies mainly focused on the 5-HT2B receptor subtype, taking advantage of the availability of gene-targeted mice for this receptor. Indeed, authors nicely demonstrated that fibroblasts 5-HT2B receptors played an important role in the secretion of pro-hypertrophic cytokines during cardiac remodeling (Jaffre et al. 2009; Monassier et al. 2008). In addition, pharmacological blockade of 5-HT2B receptors with SB206553 or SB215505 confirmed their function in cardiac hypertrophy and failure in mice and rats (Jaffre et al. 2004; Liang et al. 2006; Monassier et al. 2008). Concerning the role of 5-HT2A receptors in cardiac remodeling, some interesting findings have been published but they rely on indirect evidences or on the use of antagonists that lack specificity toward the 5-HT2A receptor. Indeed, in a mouse model of enhanced 5-HT levels, hypertrophic response to pressure overload was exacerbated due to over-stimulation of 5-HT2A receptors (Lairez et al. 2009). In addition, a number of studies demonstrated that the 5-HT2A receptor antagonist ketanserin decreased cardiac hypertrophy in an animal model of hypertension (Xie et al. 2005) and in hypertensive patients (Cobo et al. 1990; Vyssoulis et al. 1990). However, ketanserin demonstrates combined blockade of 5-HT2A/5-HT2B receptors, along with high affinity for α1-adrenergic receptors, which make it difficult to conclude on the specific role mediated by 5-HT2A receptors. Another 5-HT2A antagonist, sarpogrelate, showed beneficial effect in the model of ischemia–reperfusion and myocardial infarction in the rat, which supports a role for this receptor in cardioprotection (Rajesh et al. 2006; Sanganalmath et al. 2008). Here, we used M100907, a potent and selective 5-HT2A receptor antagonist showing a >100-fold separation from activities at 5-HT2C and >1000-fold separation with 5-HT2B receptors, and we provided evidence that 5-HT2A receptors participate in the development of myocardial hypertrophy (Kehne et al. 1996; Knight et al. 2004).
Using immunohistochemistry in mouse heart, we observed that cardiomyocytes expressed 5-HT2A receptors, as previously defined using radioligand binding assays (Jaffre et al. 2009). Interestingly, the increase in 5-HT2A receptor expression observed in the early stage after pressure overload could indicate a transient role for 5-HT2A in the initial development of hypertrophy, since 3 months after TAC, there was no upregulation of 5-HT2A receptor in TAC heart. Our observations are supported by previous findings, showing that, in a rat model of hypertrophy induced either by pressure overload or by myocardial infarction, a 5-HT2A-mediated inotropic response appeared in the heart, and that the response was strongly correlated with the degree of hypertrophy but not heart failure (Brattelid et al. 2007). At present, the mechanisms of regulation of 5-HT2A receptors in cardiac hypertrophy are unknown.
The presence of 5-HT2A receptors in cardiomyocytes and their role in mediating 5-HT-induced hypertrophic response in vitro (Mialet-Perez et al. 2012; Villeneuve et al. 2009) support a direct effect of M100907 inhibition on cardiac cells during pressure-overload hypertrophy. However, it is well known that 5-HT2A receptor expression is not restricted to cardiomyocytes. 5-HT2A receptors are expressed in cardiac fibroblasts, where they participate in TGFβ secretion and myofibroblasts transformation (Yabanoglu et al. 2009). Moreover, they are widely distributed among the body, in smooth muscle cells (where they participate in vasoconstriction) but also in platelets (where they mediate platelet aggregation) and in the central nervous system (where they induce sympatho-excitation) (Cote et al. 2004). Intraventricular hemodynamic parameters indicated that M10907 treatment did not affect cardiac contractility or diastolic function but induced, in both sham and TAC animals, a reduction in the maximal left ventricular pressure, which should result in a parallel drop in systolic arterial blood pressure. This phenomenon would thus probably participate in the reduction of myocardial hypertrophy evoked by M100907, by an indirect mechanism. Therefore, to determine the specific contribution of cardiomyocyte 5-HT2A receptors among other cell types, cardiomyocyte-targeted KO mice will need to be developed.
The CamKII signaling pathway appears to be central to the pathological hypertrophic response in the heart (Anderson et al. 2011). CamKII is known to be activated by Gq-coupled receptors through autophosphorylation at Thr286 in response to sustained rise in intracellular Ca2+. CamKII activity is constantly increased in hypertrophied and failing myocardium from animal models and patients (Ling et al. 2009). In addition, inhibition of this Ca2+-sensitive kinase by drugs or gene deletion improves myocardial hypertrophy, which shows that CamKII constitute a central event during cardiac remodeling. Interestingly, in the present study, M100907 treatment significantly reduced CamKII Thr286 phosphorylation in cardiac homogenates from TAC mice. Thus, this decrease in CamKII activation likely contributes to the prevention of myocardial hypertrophy in M100907-treated TAC mice. In support of this, we found that one of the downstream signaling partners of CamKII, HDAC4, was also inhibited by M100907 treatment in TAC mice. HDAC4 is regulated by phosphorylation on Ser632 which induces the exclusion of this enzyme from the nucleus and allows the transcription of hypertrophic factors in the heart. In our model, HDAC4 phosphorylation was significantly reduced by M100907 treatment. This effect of 5-HT2A receptor on activation of myocardial CamKII/HDAC4 signaling pathway has not been reported previously. Only in cultured cardiomyocytes, a link between 5-HT2A/B receptors activation and HDAC5 export from the nucleus was demonstrated previously (Bush et al. 2004). Thus, our results bring new knowledge in the signaling mechanisms associated with 5-HT2A-mediated hypertrophy.
In conclusion, this study demonstrates that M100907, an anti-psychotic drug with potential use in insomnia and depression, has beneficial effects on cardiac remodeling through inhibition of 5-HT2A-mediated cardiac hypertrophy.
References
Anderson ME, Brown JH, Bers DM (2011) CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol 51(4):468–473
Brattelid T, Qvigstad E, Birkeland JA, Swift F, Bekkevold SV, Krobert KA, Sejersted OM, Skomedal T, Osnes JB, Levy FO, Sjaastad I (2007) Serotonin responsiveness through 5-HT2A and 5-HT4 receptors is differentially regulated in hypertrophic and failing rat cardiac ventricle. J Mol Cell Cardiol 43(6):767–779
Bush E, Fielitz J, Melvin L, Martinez-Arnold M, McKinsey TA, Plichta R, Olson EN (2004) A small molecular activator of cardiac hypertrophy uncovered in a chemical screen for modifiers of the calcineurin signaling pathway. Proc Natl Acad Sci USA 101(9):2870–2875
Cobo C, Alcocer L, Chavez A (1990) Effects of ketanserin on left ventricular hypertrophy in hypertensive patients. Cardiovasc Drugs Ther 4(Suppl 1):73–76
Cote F, Fligny C, Fromes Y, Mallet J, Vodjdani G (2004) Recent advances in understanding serotonin regulation of cardiovascular function. Trends Mol Med 10(5):232–238
Jaffre F, Callebert J, Sarre A, Etienne N, Nebigil CG, Launay JM, Maroteaux L, Monassier L (2004) Involvement of the serotonin 5-HT2B receptor in cardiac hypertrophy linked to sympathetic stimulation: control of interleukin-6, interleukin-1beta, and tumor necrosis factor-alpha cytokine production by ventricular fibroblasts. Circulation 110(8):969–974
Jaffre F, Bonnin P, Callebert J, Debbabi H, Setola V, Doly S, Monassier L, Mettauer B, Blaxall BC, Launay JM, Maroteaux L (2009) Serotonin and angiotensin receptors in cardiac fibroblasts coregulate adrenergic-dependent cardiac hypertrophy. Circ Res 104(1):113–123
Kehne JH, Baron BM, Carr AA, Chaney SF, Elands J, Feldman DJ, Frank RA, van Giersbergen PL, McCloskey TC, Johnson MP, McCarty DR, Poirot M, Senyah Y, Siegel BW, Widmaier C (1996) Preclinical characterization of the potential of the putative atypical antipsychotic MDL 100,907 as a potent 5-HT2A antagonist with a favorable CNS safety profile. J Pharmacol Exp Ther 277(2):968–981
Knight AR, Misra A, Quirk K, Benwell K, Revell D, Kennett G, Bickerdike M (2004) Pharmacological characterisation of the agonist radioligand binding site of 5-HT(2A), 5-HT(2B) and 5-HT(2C) receptors. Naunyn Schmiedebergs Arch Pharmacol 370(2):114–123
Lairez O, Calise D, Bianchi P, Ordener C, Spreux-Varoquaux O, Guilbeau-Frugier C, Escourrou G, Seif I, Roncalli J, Pizzinat N, Galinier M, Parini A, Mialet-Perez J (2009) Genetic deletion of MAO-A promotes serotonin-dependent ventricular hypertrophy by pressure overload. J Mol Cell Cardiol 46(4):587–595
Liang YJ, Lai LP, Wang BW, Juang SJ, Chang CM, Leu JG, Shyu KG (2006) Mechanical stress enhances serotonin 2B receptor modulating brain natriuretic peptide through nuclear factor-kappaB in cardiomyocytes. Cardiovasc Res 72(2):303–312
Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D, Brown JH (2009) Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest 119(5):1230–1240
Marek GJ, Martin-Ruiz R, Abo A, Artigas F (2005) The selective 5-HT2A receptor antagonist M100907 enhances antidepressant-like behavioral effects of the SSRI fluoxetine. Neuropsychopharmacology 30(12):2205–2215
Mialet-Perez J, Bianchi P, Kunduzova O, Parini A (2007) New insights on receptor-dependent and monoamine oxidase-dependent effects of serotonin in the heart. J Neural Transm 114(6):823–827
Mialet-Perez J, D’Angelo R, Villeneuve C, Ordener C, Negre-Salvayre A, Parini A, Vindis C (2012) Serotonin 5-HT2A receptor-mediated hypertrophy is negatively regulated by caveolin-3 in cardiomyoblasts and neonatal cardiomyocytes. J Mol Cell Cardiol 52(2):502–510
Monassier L, Laplante MA, Jaffre F, Bousquet P, Maroteaux L, de Champlain J (2008) Serotonin 5-HT(2B) receptor blockade prevents reactive oxygen species-induced cardiac hypertrophy in mice. Hypertension 52(2):301–307
Nigmatullina RR, Kirillova VV, Jourjikiya RK, Mukhamedyarov MA, Kudrin VS, Klodt PM, Palotas A (2009) Disrupted serotonergic and sympathoadrenal systems in patients with chronic heart failure may serve as new therapeutic targets and novel biomarkers to assess severity, progression and response to treatment. Cardiology 113(4):277–286
Qvigstad E, Sjaastad I, Brattelid T, Nunn C, Swift F, Birkeland JA, Krobert KA, Andersen GO, Sejersted OM, Osnes JB, Levy FO, Skomedal T (2005) Dual serotonergic regulation of ventricular contractile force through 5-HT2A and 5-HT4 receptors induced in the acute failing heart. Circ Res 97(3):268–276
Rajesh KG, Suzuki R, Maeda H, Murio Y, Sasaguri S (2006) 5-HT2 receptor blocker sarpogrelate prevents downregulation of antiapoptotic protein Bcl-2 and protects the heart against ischemia-reperfusion injury. Life Sci 79(18):1749–1755
Sanganalmath SK, Barta J, Takeda N, Kumamoto H, Dhalla NS (2008) Antiplatelet therapy mitigates cardiac remodeling and dysfunction in congestive heart failure due to myocardial infarction. Can J Physiol Pharmacol 86(4):180–189
Villeneuve C, Caudrillier A, Ordener C, Pizzinat N, Parini A, Mialet-Perez J (2009) Dose-dependent activation of distinct hypertrophic pathways by serotonin in cardiac cells. Am J Physiol Heart Circ Physiol 297(2):H821–H828
Vindis C, D’Angelo R, Mucher E, Negre-Salvayre A, Parini A, Mialet-Perez J (2010) Essential role of TRPC1 channels in cardiomyoblasts hypertrophy mediated by 5-HT2A serotonin receptors. Biochem Biophys Res Commun 391(1):979–983. doi:10.1016/j.bbrc.2009.12.001
Vyssoulis GP, Karpanou EA, Pitsavos CE, Paleologos AA, Kourtis TK, Toutouzas PK (1990) Left ventricular hypertrophy regression and function changes with ketanserin in elderly hypertensives. Cardiovasc Drugs Ther 4(Suppl 1):81–84
Xie HH, Shen FM, Cao YB, Li HL, Su DF (2005) Effects of low-dose ketanserin on blood pressure variability, baroreflex sensitivity and end-organ damage in spontaneously hypertensive rats. Clin Sci (Lond) 108(6):547–552
Yabanoglu S, Akkiki M, Séguélas MH, Mialet-Perez J, Parini A, Pizzinat N (2009) Platelet derived serotonin drives the activation of rat cardiac fibroblasts by 5-HT2A receptors. J Mol Cell Cardiol 46(4):518–525
Acknowledgments
This work was supported by grants from Institut National de la Santé et de la Recherche Médicale (INSERM), Agence Nationale pour la Recherche (ANR CARDIOMAO), Région Midi-Pyrénées and Université Paul Sabatier (Bonus Qualité Recherche). We thank J.J. Maoret (IFR150, Molecular Biology Platform), C. Delage (IFR 150, Microsurgery Platform) for their excellent technological assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lairez, O., Cognet, T., Schaak, S. et al. Role of serotonin 5-HT2A receptors in the development of cardiac hypertrophy in response to aortic constriction in mice. J Neural Transm 120, 927–935 (2013). https://doi.org/10.1007/s00702-013-1011-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-013-1011-3