
Abstract
Folate is necessary for DNA and mtDNA integrity and via folate/B12-dependent methionine cycle for methylation of multiple substrates (epigenetic DNA and enzymes) and methylation of homocysteine. During embryogenesis, folate deficiency is a risk factor for neural tube defects and late in life for cognitive decline and Alzheimer’s dementia (AD). It induces several Alzheimer pathomechanisms like oxidative stress, Ca++ influx, accumulation of hyperphosphorylated tau and β-amyloid. But impact of folic acid supplementation on prevention or delay of dementia is a matter of debate. Six out of seven randomized controlled trials (RCT) with B vitamin intervention periods between 2 and 5.4 years reported about cognitive benefits in the supplemented groups mainly for those subjects with high homocysteine or low folate levels at baseline. This review tries to demonstrate the connection between folate deficiency and AD, analyses selected epidemiologic studies and RCT on folate/B12/homocysteine with long-observation periods (≥2 years RCT; ≥4 years observational) and attempts to find explanations for the controversy in literature like short follow-up, heterogeneity of subjects concerning age, recruitment, baseline cognition, inclusion criteria and probably “misleading”(not representative for the past) folate/B12/homocysteine levels due to not reported short-term use of multivitamins or food-fortification. Population-based studies—epidemiologic and interventional—starting in the fourth decade would provide the best information about the impact of folate on later development of AD. Mandatory folate fortification areas will be important future field studies for—like neural tube defects—hopefully declining AD incidence and disproving safety concerns.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
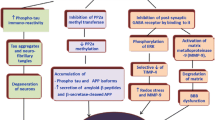
Folate deficiency induces several pathophysiologic changes supposed to be pathogenetic in Alzheimer′s dementia (AD) (Fig. 1) like mitochondrial dysfunction leading to oxidative stress, loss of calcium regulation, neuronal and synaptic impairment and accumulation of hyperphosphorylated tau and β-amyloid (Querfurth and LaFerla 2010). These specific features make low folate a probable candidate for long-term contribution to AD development.

Folate deficiency and Alzheimer’s dementia. For depicting this figure, the following references were used: (Fenech 2010; Fleming et al. 2011; Fuso and Scarpa 2011; Ho et al. 2003; Kruman et al. 2002; Kruman et al. 2005; Mattson and Shea 2003; Pierrot et al. 2006; Sontag et al. 2007, 2008). Only direct folate and/or homocysteine-dependent actions are considered. dUMP deoxy-uridine monophosphate, dTMP deoxy-tymidine monophosphat, mtDNA mitochondriale DNA, APP amyloid precursor protein, BACE1 beta site amyloid precursor protein cleavage enzyme 1, PSEN1 presenilin1, PP2A protein phosphatase 2A
Folate, the most important methyl group donator, is essential for maintaining integrity of DNA and mtDNA (Fig. 2). Multiple acceptors, like enzymes or DNA are methylated through the folate- and vitamin B12-dependent methionine–homocysteine cycle (Fig. 2). Folate is known to foster proper neural tube development during human embryogenesis (Mattson and Shea 2003) and lifelong neuronal cell growth and repair (Iskandar et al. 2004; Kruman et al. 2002, 2005). As a consequence, folic acid fortification of flour and grain has been introduced in several countries (USA and Canada 1998, Australia 2009, but not in Europe), reducing inadequate serum folate status from 16–26 % to 0.5–1.7 %, inadequate erythrocyte folate—a marker of folate intake over the past 90–120 days—from 39 to 3.7 %, high (>13 μmol/L) homocysteine from 18.7 to 9.8 % of the US population and reducing neural tube defects by 26 % (Dietrich et al. 2005; Jacques et al. 1999; Pfeiffer et al. 2005; Yetley and Johnson 2011). But fortification also has raised the problem of confusing ongoing trials (Bostom et al. 2001), appearance of unmetabolized folic acid of unknown significance in the blood and probably jeopardizing vitamin B12-deficient people (Morris et al. 2010).

Folate-mediated one-carbon metabolism. For depicting this figure, the following references were used: (Appling 1991; Herbig et al. 2002; MacFarlane et al. 2009; Mattson and Shea 2003; Morris and Tangney 2007; Quinlivan et al. 2005; Ulrey et al. 2005). The chemical reactions are simplified and incomplete. Dietary “folate” intake, highest with “Cretan diet”, provides the body with pteroyl-polyglutamate.Cleavage by several intestinal y-Glutamyl-Carbopeptidases (1) from gut, liver and pancreas into pteroyl-monoglutamate is necessary before resorption (only about 50 %). Supplemented “folic acid” (pteroyl-monoglutamate) is readily absorbed (100 %), converted to dihydrofolate (DHF Glu 1 dihydro-pteroyl-monoglutamat) (both able to enter mitochondria) and tetrahydrofolate (THF tetrahydro-pteroyl-monoglutamat)—the bioactive starting point of methylation—by FR and DHFR. Glu 1 (monoglutamat) is only explicitly written for DHF (dihydrofolate), and omitted for all other active THF-metabolites. THF-polyglutamate and 5-methyl-THF-polyglutamate are inactive intracellular storage forms not shown in this figure. For activation, they have to be transformed into monoglutamates by folyl-polyglutamyl-hydrolase first. Cytoplasm: In cytoplasm the trifunctional enzyme MTHFD1 bidirectionally catalyzes transformation into 10-formyl THF (cFTHFS/MTHFD1), the starting point for purine synthesis supplying C2 and C8 of adenine and guanine and for protein synthesis producing fMet-tRNA. cMTHFC/MTHFD1 and MTHFD1 bidirectionally transform 10-formylTHF into 5,10-methyleneTHF, starting point for thymidilate synthesis. Mitochondria: The analog enzyme found in mitochondria is only bifunctional: MTHFD2 with mMTHFC activity. mFTHFS bidirectionally tranforms 10-formylTHF and THF and generates formate and fMet-tRNA. THF is bidirectionally transformed into 5,10-methyleneTHF by vitamin B6 highly dependent cSHMT or B6 less dependent mSHMT using serine as a methyl group donor. Formate, serine, folic acid and DHF can pass mitochondrian membrane. Thymidilate synthesis: 5,10methylenTHF is methylating dUMP by TS forming dTMP and DHF Glu1, which is again reduced to THF by DHFR. Methionine-homocysteine cycle: 5-methyl-THF is the methyl group donor to transform homocysteine into methionine by MS and vitamin B12. Methionine is transformed by MAT into SAM, which is the methylgroup donor for many acceptors like phospholipide, myelin, DNA, RNA, protein, enzymes (PP2A), choline, acetylcholine, catecholamine, melatonin. SAM is transformed to SAH when the acceptor is methylated by SAMdepMT. Accumulation of SAH which has an equilibrium to homocysteine strongly inhibits further methylation. SAH is hydrolyzed to homocysteine by SAHH. Lack of two major routes of homocysteine degradation in brain tissue (betaine-mediated conversion and transsulfuration into cystathionine and cysteine). FR Folatreductase, DHFR dihydrofolate reductase, MTHFD1 cytoplasmatic trifunctional enzyme: methylene-THF dehydrogenase (NADP + dependent)1 with FTHFS and MTHFC function: cFTHFS/MTHFD1 cytoplasmic formyl-THF synthase function of MTHFD1, cMTHFC/MTHFD1 cytoplasmic methenyl-THF cyclohydrolase function of MTHFD1; MTHFD2 mitochondrial bifunctional enzyme: methylene-THF dehydrogenase (NADP + dependent) 2 with MTHFC function: mMTHFC/MTHFD2 mitochondrial methenyl-THF cyclohydrolase function of MTHFD2, mFTHFS mitochondrial formyl-THF synthase, fMet-tRNA formyl-methionine-tRNA, mSHMT mitochondrial serine hydroxymethyltransferase, cSHMT cytoplasmic serine hydroxy methyltransferase, MTHFR methylene THF reductase, MS methionine synthase, hcy homocysteine, MAT methionine adenosyltransferase, SAM S-adenosy-methionine, SAMdepMT SAM-dependent methyltransferase; SAH S-adenosyl-homocysteine, SAHH SAH hydrolase, dUMP deoxy-uridine monophophate, dTMP deoxy thymidine monophosphate, TS thymidilate synthase, acceptor: phospholipide, myelin, DNA, RNA, protein, enzymes: (e.g.: PP2A), choline, acetylcholine, catecholamine, melatonin. Gray background indicates CH3 carrier, rectangle box bioactive THF derivative
Whether folate supplementation can delay or prevent Alzheimer’s disease is a matter of debate (Malouf and Grimley 2008). Epidemiologic studies found low folate or high homocysteine being a risk factor for later cognitive decline or AD (Fischer et al. 2008; Seshadri et al. 2002). However, reviews and meta-analyses of randomized controlled trials (RCT) on folate and cognition have provided inconclusive evidence (Dangour et al. 2010). Several weaknesses are running like a red thread through most of these studies: (1) heterogeneity of the subjects concerning age, recruitment and baseline cognition. (2) Different methods of assessment of cognition or dementia. (3) Different methods of determination of folate levels (food questionnaires, supplemental use and laboratory analyses). (4) Lack of a standardized threshold level and inconsistent definitions of “low” folate or “high” homocysteine levels with heterogeneous study entry criteria. (5) “Misleading” folate levels due to short-term use of multivitamins without reporting. (6) Mandatory folic acid fortification. (7) Short follow-up.
This review aims on the pathophysiologic connections between folate deficiency and different hypothesized AD pathomechanisms and on selected papers fulfilling the following criteria: longitudinal/observational studies or RCT on the impact of folate on cognition or AD incidence after a follow-up of at least 2 (RCT) or 4 and more (observational) years.
Folate metabolism (Fig. 2)
Folate is indispensable for purines syntheses and transformation of uracil into thymine (Herbig et al. 2002), thus maintaining DNA replication, repair and mtDNA integrity (Fenech 2010). Tetrahydrofolate (THF) transformation cycle and folate/vit B12-dependent methionine-methylation cycle necessary for methyl group donation to many acceptors are shown in Fig. 2. The impact of polymorphism of multiple folate metabolizing enzymes on AD is controversial (Friso et al. 2002; Kageyama et al. 2008; Hua et al. 2011; Naj et al. 2010).
Pathomechanism connecting folate deficiency and Alzheimer’s dementia
Folate deficiency impairs DNA and mitochondrial DNA (mtDNA) causing oxidative stress and ROS generation—an accepted early hit in the AD pathogenesis (Querfurth and LaFerla 2010) followed by neuronal impairment and cell death in AD involved brain areas (Kruman et al. 2002, 2005). Folate deficiency causes hypomethylation of enzymes and promoter regions of genes reportedly involved in AD pathogenesis (Fuso and Scarpa 2011). Folate deficiency is accompanied by increase of cytosolic Ca++ and of homocysteine (Mattson and Shea 2003) which is a known risk factor for myocardial infarction, stroke and cognitive decline (Selhub 2008; Seshadri et al. 2002).
Folate deficiency and DNA
Lack of methyl group donation precludes conversion of dUMP (deoxy-uridine monophosphate) into dTMP (deoxy thymidine monophosphate) leading to uracil misincorporation into DNA causing double strand breaks prone to errors eventually leading to DNA damage and apoptosis (Fenech 2010; Quinlivan et al. 2005). Impaired DNA repair was found in folate-deficient hippocampal pyramidal neurons (region CA1-3) and proliferation of adult hippocampal progenitors, synaptic plasticity and neuronal function is inhibited by folate deficiency in synergy with homocysteine (Ho et al. 2001, 2003; Kruman et al. 2002, 2005; Mattson and Shea 2003).
Folate deficiency has the same detrimental impact on mtDNA leading to reduced ATP generation and oxidative stress, with production of reactive oxygen species (ROS), dysregulation of calcium homeostasis, reduction of mitochondrial number and eventually apoptosis of the neuron (Fenech 2010; Ho et al. 2003; Ross 2005).
Folate deficiency and oxidative stress
In the brains of AD patients, ROS are responsible for damage of nuclear and mitochondrial DNA, protein oxidation, lipid peroxidation and β-amyloid aggregation (Christen 2000). APOE and folate have antioxidative capacities (Chan and Shea 2006a, b; Chan et al. 2009; Shea et al. 2002; Stanger and Wonisch 2012). Folate deprivation was shown to induce increase of ROS directly and via homocysteine (Ho et al. 2003; Mattson and Shea 2003). Tetrahydrofolate (THF) is very susceptible to oxidation leading to further intracellular and mitochondrial depletion of folate (Fuchs et al. 2001). Folate supplementation enhances antioxidant status and memory (Singh et al. 2011), prevents β-amyloid-induced oxidative stress and neurotoxicity (Ho et al. 2003) and enhances growth and repair mechanisms even in the adult central nervous system (Iskandar et al. 2004).
Folate deficiency and homocysteine
Folate or B12 deficiency increases homocysteine—a cytotoxic, atherogenic, non-proteinogenic amino acid—which induces Ca++ influx via NMDA channels and increases β-amyloid-induced Ca++ influx, potentiates glutamate excitotoxicity, induces oxidative stress and increases ROS-induced β-amyloid formation, increases hyperphosphorylated tau (Ho et al. 2003; Mattson and Shea 2003) and sensitizes hippocampal neurons to β-amyloid toxicity and apoptosis. During excessive oxidative stress, however, irreversible degradation with paradox reduction of plasma homocysteine by transsulfuration pathway in liver or gut (Fig. 2) occurs (Rogers et al. 2007). Lack of betaine-mediated conversion and irreversible transsulfuration of homocysteine in brain tissue aggravates local toxicity (Shea and Rogers 2002).
Folate deficiency and methyl group donation via methionine cycle
High homocysteine increases S-adenosyl homocysteine (SAH), which is a potent inhibitor of all methylation reactions (James et al. 2002). Genomic CpG dinucleotide methylation—regulating DNA stability, structure, and gene expression via promoter regions (Fenech 2010)—inversely correlates with homocysteine levels and directly with folate status (Friso et al. 2002). SAH/homocysteine-dependent DNA hypomethylation—which is an activating input on the promoter region—has already been demonstrated for APP (amyloid precursor protein) gene, PSEN1 (gene encoding presenilin 1) and BACE 1 (gene encoding beta site APP cleaving enzyme 1) leading to increased formation of β-amyloid (Fuso et al. 2005; Fuso and Scarpa 2011; Lin et al. 2009; West et al. 1995), but results are controversial (Brohede et al. 2010; Wang et al. 2008; Jung et al. 2011). Reduced methylation impedes protein phosphatase 2A (PP2A) leading to hyperphosphorylation of tau and APP (Sontag et al. 2007, 2008) rendering it more susceptible to β cleavage.
Observational and interventional studies
Folic acid supplementation in atherosclerotic diseases has generally fallen short of expectations (Maron and Loscalzo 2009). Interventional studies on folic acid with and without B12 for preventing dementia or cognitive decline in healthy or demented persons seemingly are not doing better (Malouf and Grimley 2008; Shea et al. 2012). Heterogeneity of methods in these studies almost precludes final comparisons and conclusions. In addition, it is not surprising that interventional studies with follow-up periods of weeks are failing when the disease triggered by a suspected deficiency takes decades or probably the whole life to evolve (Lahiri and Maloney 2010).
Recent mandatory folic acid fortification or short-term use of multivitamins without reporting produce “misleading” folate, B12 and—to a much lesser extent—homocysteine levels (Jacques et al. 1999) not representative for the vitamin exposure during the past. In the VITA study (longitudinal, community-based birth cohort study) about 5–10 % of those without vitamin history at baseline supposedly did not report about it (Hinterberger, personal communication). Therefore, levels of folate and B12 might not depict the real, long-lasting, possibly deficient levels but are probably only a short flashlight of 1 day or 1 week. Sporadic supplementation might be explained by increasing concerns of older people about their decreasing health and memory conditions.
Population-based studies with observation periods of ≥4 years (Table 1)
Studies using food frequency questionnaires (FFQ)
Five studies analysed elder subjects free of dementia at baseline using food frequency questionnaires (FFQ) to quantify impact of folate and B12 intake on incident AD and/or dementia or cognitive decline after 4–9 years (Table 1). Corrada et al. (2005) and Luchsinger et al. (2007) found a reduced risk of AD with higher total (dietary and supplementary) folate intake, but not with B12. Both studies started long before the start of mandatory folic acid fortification and ended 1 (Corrada et al. 2005) or 2 years afterward (Luchsinger et al. 2007).
Nelson et al. (2009), however, did not find any association with incident AD, but CCMS started 1995 2 to 3 years before mandatory folic acid fortification possibly beneficially changed folate and homocysteine levels (Yetley and Johnson 2011) during the following 6 years thus confounding former low folate and high homocysteine exposure. The problem of “misleading” folate might apply to CCMS, too. The rate of multivitamin users dramatically increases with increasing folate quintiles from lowest (1.7 %) to highest (95 %) without differentiation between long-term exposure and only short-term use. The AD incidence in CCMS seems to be rather small compared to epidemiologic data (Ferri et al. 2005). Only 5.8 % (212 out of 3,634) of a population with a median age of 75 years and a follow-up of 9 years. The MMSE screening policy and drop outs might have underestimated the AD incidence. There remains, however, the possibility of reduced AD incidence because of advantageous impact of fortification. In a comparable study, Morris et al. (2006) reported about 161/1,041 incident AD (15.5 %) after 3.9 years. Dietary intake of folate or B12 again was not associated with AD incidence, but like CCMS this study crossed the fortification start, too, and had a high rate of supplementation users.
However, in contrast to the studies above Morris et al. (2005) from Chicago reported fastest cognitive decline in the highest total folate intake quintile compared to the lowest but reduced cognitive decline in the oldest subjects with high total B12 intake. There might be several explanations to these results: (1) the highest quintile of total folate intake also presented with exceedingly good cognitive performance at baseline. A greater regression to the mean from maximum is not surprising and possibly with less clinical relevance than a smaller decline from a worse starting point. (2) Again in the highest folate quintile there are 96.9 % multivitamin users, compared to 3.1 % in the lowest quintile with the uncertainty of duration of the supplementation. (3) No increased AD incidence was reported as mentioned above (Morris et al. 2006).
In part, similar results were reported by Morris et al. (2007, 2010) from Boston in a cross-sectional study using serum values instead of FFQ. They found worse cognitive performance in subjects with fortification-induced high levels of folate and unmetabolized free folic acid but only in subjects with concomitant low B12. Those with high folate but normal B12 levels, however, showed better cognitive performance. In the elderly, B12 deficiency (<148 pmol/L) was reported to occur in about 6 %, marginal depletion (148–221 pmol/L) in more than 20 % (Allen 2009) probably due to gastritis-induced worse B12 uptake (Selhub et al. 2000). Therefore, some experts have already suggested mandatory B12 fortification (Refsum and Smith 2008; Selhub and Paul 2011) to protect the B12-deficient elderly from the harm of elevated folate intake.
Studies using serum/plasma/erythrocyte levels of folate
Nine studies analysed influence of baseline folate, B12 and homocysteine levels on incident AD/dementia or cognitive decline after 4–8 years (Table 1). Three studies reported low folate (Fischer et al. 2008, personal communication; Kado et al. 2005; Ravaglia et al. 2005), three reported B12 (Elias et al. 2005; Haan et al. 2007; Hooshmand et al. 2010) and six reported homocysteine as independent risk factor for cognitive decline, dementia or AD (Elias et al. 2005; Haan et al. 2007; Kivipelto et al. 2009; Nurk et al. 2005, 2010; Ravaglia et al. 2005; Seshadri et al. 2002). Only three studies crossed the fortification start before closure and five took place in unfortified areas (Table 1).
Kado et al. (2005) reported worse cognitive outcome in subjects with excellent physical and cognitive performance when in the lowest folate quartile at baseline.
Elias et al. (2005) showed an association of higher homocysteine and low B12 with worse cognitive performance after 7.6 years only in the oldest of three age groups (60–82 years) demonstrating the long time necessary for high homocysteine to produce clinically measurable cerebral damage. Folate showed no association. However, folate plasma levels significantly increased from the youngest to the oldest age group with increasing SD and again might be a hint at confounding short-term supplementation.
VITA study (Fischer et al. 2008, personal communication) found folate being an independent risk factor for AD in multiple adjusted models, also including homocysteine and B12.
Haan et al. (2007) demonstrated that the risk of homocysteine-induced dementia or cognitive decline may be modified by plasma vitamin B12. No impact of red cell folate could be demonstrated probably because the study started after the beginning of the folate fortification.
Hooshmand et al. (2010) reported only holotranscobalamin (active form of B12) but not folate being an independent risk factor for incident AD. Homocysteine lost its significance when holotranscobalamin entered the model but remained significant in the below median group of holotranscobalamin.
Kivipelto et al. (2009) reported a doubled risk of AD in the highest homocysteine quartile, and a reduced risk of dementia and AD in the third holotranscobalamin quartile. The U-shaped association probably might be due to infrequent and unreported supplement use or parenteral administration. Only 15/228 subjects reported supplementation with B12 and folic acid and were obviously excluded from analysis. But only 6.6 % supplemented subjects in a 80-year-old study group seems to be less than reported from FFQ studies (Table 1) or our own experience (VITA study: 80-year old: folate: 24.9 %, B12: 25.7 %).
Nurk et al. (2005) reported significantly increased risk of memory deficits for the highest homocysteine quintile at baseline. Folate association was lost when controlled for homocysteine.
RCT with at least 2 years of intervention
Clarke et al. (2010) on behalf of the B vitamin treatment trialists’ collaboration reported in the meta-analysis no benefit of B vitamins (folate, B6, B12) in vascular diseases but also no adverse effects especially on cancer or survival which is important information about the general safety of B vitamin trials.
Only 9 RCTs with folic acid supplementation with or without B12 and B6 lasting 2–5.4 years with different inclusion criteria have been reported up to now (Table 2). Only one was confounded by folic acid fortification (Kang et al. 2008).
Two population-based studies without exclusion of cognitively impaired subjects found cognitive benefits in the vitamin group of moderately hyperhomocysteinemic subjects (Durga et al. 2007) or in the subgroup with low dietary intake of B vitamins (Kang et al. 2008).
Durga chose well-considered exclusion criteria: normal homocysteine ≤13 or excessive homocysteine ≥26 μmol/L with concomitant low B12 levels (≤200 pmol/L). Mild cognitive impaired (MCI) or demented subjects were neither diagnosed nor excluded from analysis, but only 7 subjects had a dementia compatible MMSE (0–30; Morris et al. 1989) below 24, 6 of them accidentally randomised into the placebo group. Supplementation of 818 subjects with folic acid for 3 years significantly improved 3 year change of cognitive function.
Kang et al. (2008) reported about possible cognitive benefits of supplementation among women with a low dietary intake of B vitamins but no effects in the total cohort. This might be explained by increased background dietary intakes due to mandatory folic acid fortification starting just 3 months earlier.
Two other population-based studies in high-functioning subjects found no effect after 2 years of supplementation (MacMahon et al. 2006; Ford et al. 2010).
MacMahon’s study group was small and the test results were excellent at baseline and at follow-ups, making it impossible to find any difference within only 2 years (Clarke 2006).
Ford et al. (2010) did not find any cognitive benefit of 2 years of supplementation in older hypertensive men after 8 years of follow-up. However, there was a small immediate benefit after 1 and 2 years of active supplementation in two subtests.
Walker et al. (2012) reported significantly improved cognitive performance after 2 years of supplementation in elderly, psychological distressed, but not severely depressed subjects. These results are all the more remarkable since subjects with folate or B12 deficiency at baseline, probably benefitting most, were excluded.
Kwok et al. (2011) found a cognitive benefit after 2 years of supplementation in a small sample of mild to moderate AD/VD in one subtest only in the subgroup with elevated homocysteine.
de Jager et al. (2011) reported 2 years of supplementation slowing down cognitive and clinical decline in MCI, in particular in those with elevated homocysteine. Smith et al. (2010) found in the same study group that 2 years of supplementation slowed down the rate of brain atrophy. This has been already suggested by Snowdon et al. (2000) and has been confirmed by Blasko et al. (2012) who found fewer converters from MCI to AD in those supplemented with folic acid and B12.
Conclusion concerning the studies
Eleven out of 14 epidemiologic studies with observation periods between 4 and 9.3 years reported significant, independent and beneficial impact of at least 1 of the 3 players—folate/B12/homocysteine—on cognition. Six out of seven RCTs with B vitamin intervention periods between 2 and 5.4 years reported about cognitive benefits of the supplemented groups (cognitively healthy or MCI or AD) mainly for those subjects with high homocysteine levels or low folate intake at baseline. Only one study found no difference. These are encouraging results. It is of utmost importance to design further studies avoiding the general pitfalls of short follow-up, heterogeneous study entry criteria, and of folate/B12/homocysteine levels not representative for midlife exposure due to short-term supplementation or fortification. Deduced from these results, we suggest that serum levels of folate and B12 should exceed low normal values (folate >14 nmol/L; B12 >220 pmol/L) at any age and homocysteine should not exceed 13 μmol/L by means of (1) Mediterranean (“Cretan”) diet rich in vegetables and fruits for adequate folate intake (Morris and Tangney 2007) in countries without mandatory folate—fortification of cereal—grain products, (2) dairy products for B12 intake (Allen 2009), (3) voluntarily fortified breakfast cereals for both or (4) low dose supplementation (folic acid ≤0.4 mg/day, B12 ≥0.003 mg/day). An artificial folic acid load exceeding 1 mg/d in concomitant B12 deficiency (vegans, age beyond 60 years) must be avoided until proved to be safe. Mandatory folate fortification has already started 1998 in several countries and rapidly reduced neural tube defects. It has also offered the unique opportunity of a huge field study hopefully finding an AD incidence lower than historical controls when the mid-fourth generation of 1998 reaches the age of sporadic AD.
References
Allen LH (2009) How common is vitamin B12 deficiency? Am J Clin Nutr 89 (suppl):693S–696S
Appling DR (1991) Compartmentation of folate-mediated one-carbon metabolism in eukaryotes. FASEB J 5:2645–2651
Blasko I, Hinterberger M, Kemmler G, Jungwirth S, Krampla W, Leitha T, Tragl KH, Fischer P (2012) Conversion from mild cognitive impairment to dementia: influence of folic acid and vitamin B12 use in the VITA cohort. J Nutr Health Aging (in press) (accepted 2011-10-11)
Bostom AG, Selhub J, Jacques PF, Rosenberg IH (2001) Power shortage: clinical trials testing the “homocysteine hypothesis” against a background of folic acid-fortified cereal grain flour. Ann Int Med 135:133–137
Brohede J, Rinde M, Winblad B, Graff C (2010) A DNA methylation study of the amyloid precursor protein gene in several brain regions from patients with familial Alzheimer disease. J Neurogenet 24:179–181
Chan A, Shea TB (2006a) Dietary and genetically induced oxidative stress alters tau-phosphorylation: influence of folate and apolipoprotein E deficiency. J Alzheimers Dis 9:399–406
Chan A, Shea TB (2006b) Supplementation with apple juice attenuates presenilin-1 over expression during dietary and genetically induced oxidative stress. J Alzheimers Dis 10:353–358
Chan A, Tchantchou F, Rogers EJ, Shea TB (2009) Dietary deficiency increases presenilin expression, gamma-secretase activity, and Abeta levels: potentiation by ApoE genotype and alleviation by S-adenosyl methionine. J Neurochem 110:831–836
Clarke R (2006) Vitamin B12, folic acid, and the prevention of dementia. N Engl J Med 354:2817–2819
Clarke R, Halsey J, Lewington S, Lonn E, Armitage J, Manson JE, Bonaa KH, Spence D, Nygard O, J amison R, Gaziano JM, Guarino P, Bennet D, Mir F, Peto R, Collins R, For the B-Vitamin treatment trialists'collaboration (2010) Effect of lowering homocysteine levels with B vitamins on cardiovascular disease, cancer, and cause-specific mortality. Meta-analysis of 8 randomized trials involving 37 485 individuals. Arch Intern Med 170:1622–1631
Corrada MM, Kawas CH, Hallfrisch J, Muller D, Brookmeyer E (2005) Reduced risk of Alzheimer’s disease with high folate intake: The Baltimore Longitudinal Study of Aging. Alzheimer’s Dement 1:11–18
Dangour AD, Whitehouse PJ, Rafferty K, Mitchell SA, Smith L, Hawkesworth S, Vellas B (2010) B-vitamins and fatty acids in the prevention and treatment of Alzheimer`s disease and dementia: a systematic review. J Alzheimer Dis 22:205–224
de Jager CA, Oulhay A, Jacoby R, Refsum H, Smith DA (2011) Cognitive and clinical outcomes of homocysteine-lowering B-vitamin treatment in mild cognitive impairment: a randomized controlled trial. Int J Geriatr Psychiatry. doi:10.1002/gps.2758
Dietrich M, Brown CJ, Block G (2005) The effect of folate fortification of cereal-grain products on blood folate status, dietary folate intake, and dietary folate sources among adult non-supplement users in the United States. J Am Coll Nutr 24:266–274
Durga J, van Boxtel MP, Schouten EG, Kok FJ, Jolles J, Katan MB, Verhoef P (2007) Effect of 3-year folic acid supplementation on cognitive function in older adults in the FACIT trial: a randomised, double blind, controlled trial. Lancet 369:208–216
Elias MF, Sullivan LM, D′Agostino RB, Elias PK, Jacques PF, Selhub J, Seshadri S, Au R, Beiser A, Wolf PA (2005) Homocysteine and cognitive performance in the Framingham Offspring Study: age is important. Am J Epidemiol 162:644–653
Fenech M (2010) Folate, DNA damage and the aging brain. Mech Ageing Develop 131:236–241
Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M, For Alzheimer′s Disease International (2005) Global prevalence of dementia: a Delphi consensus study. Lancet 366:2112–2117
Fischer P, Zehetmayer S, Jungwirth S, Weissgram S, Krampla W, Hinterberger M, Torma S, Rainer M, Huber K, Hoenigschnabl S, Gelpi E, Bauer K, Leitha T, Bauer P, Tragl KH (2008) Risk factors for Alzheimer dementia in a community-based birth cohort at the age of 75 years. Dement Geriatr Cogn Disord 25:501–507
Fleming JL, Phiel CJ, Toland AE (2011) The role for oxidative stress in aberrant DNA methylation in Alzheimer’s disease. Current Alzheimer Res 8 (Epub ahead of print)
Ford AH, Flicker L, Alfonso H, Thomas J, Clarnette R, Martins R, Almeida OP (2010) Vitamin B12, B6, and folic acid for cognition in older men. Neurology 75:1540–1547
Friso S, Choi SW, Girelli D, Mason JB, Dolnikowski GG, Babley PJ, Olivieri O, Jacques PF, Rosenberg IH, Corrocher R, Selhub J (2002) A common mutation in the 5,10 methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. PNAS 99:5606–5611
Fuchs D, Jaeger M, Widner B, Wirleitner B, Artner-Dworzak E, Leblhuber F (2001) Is homocysteinemia due to oxidative depletion of folate rather than to insufficient dietary intake? Clin Chem Lab Med 39:691–694
Fuso A, Scarpa S (2011) One-carbon metabolism and Alzheimer′s disease: is it all a methylation matter? Neurobiol Aging 32:1192–1195
Fuso A, Seminara L, Cavallaro RA, Anselmi F, Scarpa S (2005) S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and β-amyloid production. Mol Cell Neurosci 28:195–204
Haan MN, Miller JW, Aiello AE, Whitmer RA, Jagust WJ, Mungas DM, Allen LH, Green R (2007) Homocysteine, B vitamins, and the incidence of dementia and cognitive impairment: results from the Sacramento Area Latino Study on Aging. Am J Clin Nutr 85:511–517
Herbig K, Chiang EP, Lee LR, Hills J, Shane B, Stover PJ (2002) Cytoplasmic serine hydroxymethyltransferase mediates competition between folate dependent deoxyribonucleotide and S-adenolsymethionine biosyntheses. J Biol Chem 277:38381–38389
Ho PI, Collins SC, Dhitavat S, Ortiz D, Ashline D, Rogers E, Shea TB (2001) Homocysteine potentiates beta-amyloid neurotoxicity: role of oxidative stress. J Neurochem 78:249–253
Ho PI, Ashline D, Dhitavat S, Ortiz D, Collins SC, Shea TB, Rogers E (2003) Folate deprivation induces neurodegeneration: roles of oxidative stress and increased homocysteine. Neurobiol Dis 14:32–42
Hooshmand B, Solomon A, Kareholt I, Leiviskä J, Rusanen M, Ahtiluoto S, Winblad B, Laatikainen T, Soininen H, Kivipelto M (2010) Homocysteine and holotranscobalamin and the risk of Alzheimer disease. A longitudinal study. Neurology 75:1408–1414
Hua Y, Zhao H, Kong Y, Ye M (2011) Association between the MTHFR gene and Alzheimer’s disease: a metaanalysis. Int J Neurosci 121:462–471
Iskandar BJ, Nelson AN, Resnick D, Skene P, Gao P, Johnson C, Cook TD, Hariharan N (2004) Folic acid supplementation enhances repair of the adult central nervous system. Ann Neurol 56:221–227
Jacques PF, Selhub J, Bostom AG, Wilson PW, Rosenberg IH (1999) The effect of folic acid fortification on plasma folate and total homocysteine concentrations. NEJM 340:1449–1454
James SJ, Melnyk S, Pogribna M, Pogribny IP, Caudill MA (2002) Elevation in S-adenosylhomocysteine and DNA hypomethylation: potential epigenetic mechanism for homocysteine-related pathology. J Nutr 132:2361S–2366S
Jung AY, Smulders Y, Verhoef P, Kok FJ, Blom H, Kok RM, Kampman E, Durga J (2011) No effect of folic acid supplementation on global DNA methylation in men and women with moderately elevated homocysteine. PLoS ONE 6(9):e24976. doi:10.1371/journal.pone.0024976
Kado DM, Karlamangla AS, Huang MH, Troen A, Rowe JW, Selhub J, Seeman TE (2005) Homocysteine versus the vitamins folate, B6, and B12 as predictors of cognitive function and decline in older high functioning adults: MacArthur studies of successful aging. Am J Med 118:161–167
Kageyama M, Hiraoka M, Kagawa Y (2008) Relationsship between genetic polymorphism, serum folate and homocysteine in Alzheimer′s disease. Asia Pac J Public Health 20 Suppl:111–117
Kang JH, Cook N, Manson JA, Buring JE, Albert CM, Grodstein F (2008) A trial of B vitamins and cognitive function among women at high risk of cardiovascular disease. Am J Clin Nutr 88:1602–1610
Kivipelto M, Annerbo S, Hultdin J, Bäckman L, Viitanen M, Fratiglioni L, Lökk J (2009) Homocysteine and holotranscobalamin and the risk of dementia and Alzheimer disease: a prospective study. Eur J Neurol 16:808–813
Kruman II, Kumaravel TS, Lohani A, Pedersen WA, Cutler RG, Kruman Y, Haughey N, Lee J, Evans M, Mattson MP (2002) Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J Neurosci 22:1752–1762
Kruman II, Mouton PR, Emokpae R Jr, Cutler RC, Mattson MP (2005) Folate deficiency inhibits proliferation of adult hippocampal progenitors. NeuroReport 16:1055–1059
Kwok T, Lee J, Law CB, Pan PC, Yung CY, Choi KC, Lam LC (2011) A randomized placebo controlled trial of homocysteine lowering to reduce cognitive decline in older demented people. Clin Nutr 30:297–302
Lahiri DK, Maloney B (2010) The “LEARn” (Latent Early Life Associated Regulation) model integrates environmental risk factors and the developmental basis of Alzheimer’s disease, and proposes remedial steps. Exp Gerontol 45:291–296
Lin HC, Hsieh HM, Chen YH, Hu ML (2009) S-Adenosylhomocysteine increases beta-amyloid formation in BV-2 microglial cells by increased expressions of beta-amyloid precursor protein and presenilin 1 and by hypomethylation of these gene promotors. Neurotoxicology 30:622–627
Luchsinger JA, Tang MX, Miller J, Green R, Mayeux R (2007) Relation of higher folate intake to lower risk of Alzheimer disease in the elderly. Arch Neurol 64:86–92
MacFarlane AJ, Perrs CA, Girnary HH, Gao D, Allen RH, Stabler SP, Shane B, Stover PJ (2009) Mthfd1 is an essential gene in mice and alters biomarkers of impaired one-carbon metabolism. J Biol Chem 284:1533–1539
MacMahon JA, Green TJ, Skeaff CM, Knight RG, Mann JI, Williams SM (2006) A controlled trial of homocysteine lowering and cognitive performance. N Engl J Med 354:2764–2772
Malouf R, Grimley EJ (2008) Folic acid with or without vitamin B12 for the prevention and treatment of healthy elderly and demented people. Cochrane Database Syst Rev 4:CD004514. doi:10.1002/14651858.CD004514.pub2
Maron BA, Loscalzo J (2009) The treatment of hyperhomocysteinemia. Annu Rev Med 60:39–54. doi:10.1146/annurev.med.60.041807.123308
Mattson MP, Shea TB (2003) Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci 26(3):137–146
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34:939–944
Morris JC (1993) The clinical dementia rating (CDR) current version and scoring rules. Neurology 43:2412–2414
Morris MC, Tangney CC (2007) Is dietary intake of folate too low? Lancet 369:166–167
Morris JC, Heyman A, Mohs RC, Hughes JP, van Belle G, Fillenbaum G, Mellits ED, Clark C (1989) CERAD investigators: The Consortium to Establish a Registry for Alzheimer’s disease (CERAD). I. Clinical and neuropsychological assessment of Alzheimer’s disease. Neurology 39:1159–1165
Morris MC, Evans DA, Bienias JL, Tangney CC, Hebert LE, Scherr PA, Schneider JA (2005) Dietary folate and vitamin B12 intake and cognitive decline among community dwelling older persons. Arch Neurol 62:641–645
Morris MC, Evans DA, Schneider JA, Tangney CC, Bienias JL, Aggarwal NT (2006) Dietary folate and vitamin B12 and B6 not associated with incident Alzheimer’s disease. J Alzheimers Dis 9:435–443
Morris MS, Jaques PF, Rosenberg IH, Selhub J (2007) Folate and vitamin B12 status in relation to anemia, macrocytosis, and cognitive impairment in older Americans in the age of folic acid fortification. Am J Clin Nutr 85:193–200
Morris MS, Jacques PF, Rosenberg IH, Selhub J (2010) Circulating unmetabolized folic acid and 5-methyltetrahydrofolate in relation to anemia, macrocytosis and cognitive test performance in American seniors. Am J Clin Nutr 91:1733–1744
Naj AC, Beecham GW, Martin ER, Gallins PJ, Powell EH, Konidari I, Whitehead PL, Cai G, Haroutunian V, Scott WK, Vance JM, Slifer MA, Gwirtsman HE, Gilbert JR, Haines JL, Buxbaum JD, Pericak-Vance MA (2010) Dementia revealed: novel chromosome 6 locus for late onset Alzheimer Disease provides genetic evidence for folate pathway abnormalities. PLoS Genet 6(9):e1001130. doi:10.1371/journal.pgen.1001130
Nelson C, Wengreen HJ, Munger RG, Corcoran CD, The Cache County Investigators (2009) Dietary folate, vitamin B-12, vitamin B-6 and incident Alzheimer’s disease: The Cache County Memory, Health and Aging Study. J Nutr Health Aging 13:899–905
Nurk E, Refsum H, Tell GS, Engedal K, Vollset SE, Ueland PM, Nygaard HA, Smith AD (2005) Plasma total homocysteine and memory in the elderly: The Hordaland homocysteine study. Ann Neurol 58:847–857
Petersen RC, Roberts RO, Knopman DS, Boeve BF, Geda YE (2009) Mild cognitive impairment:ten years later. Arch Neurol 66:1447–1455
Pfeiffer CM, Caudill SP, Gunter EW, Osterloh J, Sampson EJ (2005) Biochemical indicators of B vitamin status in the US population after folic acid fortification:results from the National Health and Nutrition Examination Survey 1999–2000. Am J Clin Nutr 82:442–450
Pierrot N, Ferrao Santos S, Feyt C, Morel M, Jean-Pierre Brion JP, Octave JN (2006) Calcium mediated transient phosphorylation of Tau and amyloid precursor protein followed by intraneuronal amyloid β accumulation. J Biol Chem 281:39907–39914
Querfurth HW, LaFerla FM (2010) Alzheimer’s disease. NEJM 362:329–344
Quinlivan EP, Davis SR, Shelnutt KP, Henderson GN, Ghandour H, Shane B, Selhub J, Bailey LB, Stacpoole PW, Gregory JF (2005) Methylenetetrahydrofolate reductase 677 C → T polymorphism and folate status affect one-carbon incorporation into human DNA deoxynucleosides. J Nutr 135:389–396
Ravaglia G, Forti P, Maioli F, Martelli M, Servadei L, Brunetti N, Porcellini E, Licastro F (2005) Homocysteine and folate as risk factors for dementia and Alzheimer disease. Am J Clin Nutr 82:636–643
Refsum H, Smith AD (2008) Are we ready for mandatory fortification with vitamin B12? Am J Clin Nutr 88:253–254
Rogers EJ, Chen SS, Chan A (2007) Folate deficiency and plasma homocysteine during increased oxidative stress. NEJM 357:421–422
Roman GC, Tatemichi TK, Erkinjuntti T, Cummings JL, Masdeu JC, Garcia JH, Amaducci L, Orgogozo JM, Brun A, Hofman A, Moody DM, O’Brien MD, Yamaguchi T, Grafman J, Drayer BP, Bennett DA, Fisher M, Ogata J, Kokmen E, Bermejo F, Wolf PA, Gorelick PB, Bick KL, Pajeau AK, Bell MA, DeCarli C, Culebras A, Korczyn AD, Bogousslavsky J, Hartmann A, Scheinberg P (1993) Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology 43:250–260
Ross CM (2005) Folate, mitochondria, ROS, and the aging brain. Am J Med 118:1174–1178
Selhub J (2008) Public health significance of elevated homocysteine. Food Nutr Bull 29(2 Suppl):S116–S125
Selhub J, Paul L (2011) Folic acid fortification: why not vitamin B12 also? BioFactors 37:269–271
Selhub J, Bagley LC, Miller J, Rosenberg IH (2000) B vitamins, homocysteine and neurocognitive function in the elderly. Am J Clin Nutr 71:614S–620S
Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D′Agostino RB, Wislon PWF, Wolf PA (2002) Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl J Med 346:476–483
Shea TB, Rogers E (2002) Homocysteine and dementia. N Engl J Med 346:2007–2008
Shea TB, Rogers E, Ashline D, Ortiz D, Sheu MS (2002) Apolipoprotein E deficiency promotes increased oxidative stress and compensatory increases in antioxidants in brain tissue. Free Radic Biol Med 33:1115–1120
Shea TB, Rogers E, Remington R. (2012) Nutrition and dementia: Are we asking the right questions? J Alzheimers Dis 29. doi:10.3233/JAD-2012-112231 (Epub ahead of print)
Singh R, Kanwar SS, Sood PK, Nehru B (2011) Beneficial effects of folic acid on enhancement of memory and antioxidant status in aged rat brain. Cell Mol Neurobiol 31:83–91
Smith AD, Smith SM, deJager CA, Whitbread P, Johnston C, Agacinski G, Oulhaj A, Bradley KM, Jacoby R, Refsum H (2010) Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: a randomized controlled trial. PLoS ONE 5(9):e12244. doi:10.1371/journal.pone.0012244
Snowdon DA, Tully CL, Smith CD, Riley KP, Markesbery WR (2000) Serum folate and the severity of atrophy of the neocortex in Alzheimer disease: findings from the Nun Study. Am J Clin Nutr 71:993–998
Sontag E, Nunbhakdi-Craig V, Sontag JM, Arrastia RD, Ogris E, Dayal S, Lentz SR, Arning E, Bottiglieri T (2007) Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J Neurosci 27:2751–2759
Sontag JM, Nunbhakdi-Craig V, Montgomery L, Arning E, Bottiglieri T, Sontag E (2008) Folate deficiency induces in vitro and mouse brain region-specific downregulation of leucine carboxyl methyltransferase 1 and protein phosphatase 2A Bα subunit expression that correlate with enhanced tau phosphorylation. J Neurosci 28:11477–11487
Stanger O, Wonisch W (2012) Enzymatic and non-enzymatic antioxidative effects of folic acid and its reduced derivates. Subcell Biochem 56:131–161
Ulrey CL, Liu L, Andrews LG, Tollefsbol TO (2005) The impact of metabolism on DNA methylation. Hum Mol Genet 14(1):R139–R147
Walker JG, Batterham PJ, Mackinnon AJ, Jorm AF, Hickie I, Fenech M, Kljakovic M, Crisp D, Christensen H (2012) Oral folic acid and vitamin B-12 supplementation to prevent cognitive decline in community dwelling older adults with depressive symptoms—the Beyond Ageing Project: a randomized controlled trial. Am J Clin Nutr 95:194–203
Wang S-C, Oelze B, Schumacher A (2008) Age-specific epigenetic drift in late onset Alzheimer’s disease. PLoS ONE 3(7):e2698. doi:10.1371/journal.pone.0002698
West RL, Lee JM, Maroun LE (1995) Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J Mol Neurosci 6:141–146
Yetley EA, Johnson CL (2011) Folate and vitamin B12 biomarkers in NHANES: history of their measurement and use. Am J Clin Nutr 94(suppl):1S–10S
Yves Christen (2000) Oxidative stress and Azheimer disease. Am J Clin Nutr 71(Suppl):621S–629S
Acknowledgments
The Vienna Transdanube Aging (VITA) study is supported by the Ludwig Boltzmann Society, Vienna-Austria, and is carried out at the Ludwig Boltzmann Institute of Aging Research which is located at the Donauspital, Vienna-Austria.
Conflict of interest
Hinterberger and Fischer declare that they have no conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hinterberger, M., Fischer, P. Folate and Alzheimer: when time matters. J Neural Transm 120, 211–224 (2013). https://doi.org/10.1007/s00702-012-0822-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-012-0822-y