
Abstract
A major component of green tea is (−)-epigallocatechin gallate (EGCG), which has strong antioxidant properties. Here, we investigated the effect of EGCG on neural stem cell (NSC) proliferation around the damaged area following traumatic brain injury (TBI). In this study, male Wistar rats that had access to normal drinking water, or water containing 0.1% (w/v) EGCG, ad libitum received TBI at 10 weeks of age. Immunohistochemistry revealed that the number of nestin-positive cells around the damaged area after TBI in the EGCG treatment group increased significantly compared with the normal water group (P < 0.05). However, the number of 8-hydroxy-2′-deoxyguanosine-, 4-hydroxy-2-nonenal-, single-stranded DNA (ssDNA)-positive cells and the level of peroxidation around the damaged area after TBI significantly decreased in the EGCG treatment group when compared with the water group (P < 0.05). Furthermore, in contrast to the EGCG group, almost all ssDNA-positive cells in the water group co-localized with NeuN and nestin-staining. Ex vivo studies revealed that spheres could only be isolated from injured brain tissue in the water group at 3 days following TBI. However, in the EGCG group, spheres could be isolated at both 3 and 7 days following TBI. A greater number of spheres could be isolated from the EGCG group, which differentiated into neurons and glia in culture without basic fibroblast growth factor. These results indicate that consumption of water containing EGCG pre- and post-TBI inhibits free radical-induced degradation of NSCs, which have the potential to differentiate into neurons and glia around the area of damage following TBI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Green tea is a widely consumed beverage that contains many biologically important polyphenols (Yu et al. 2010). The green tea polyphenols consist of four main components: (−)-epigallocatechin gallate (EGCG), (−)-epicatechin gallate, (−)-epigallocatechin and (−)-epicatechin (Yu et al. 2010). EGCG is the major constituent, accounting for more than 10% of the extract in dry weight (Yu et al. 2010). EGCG is abundant with phenolic hydroxyl groups on its aromatic rings, which confer its antioxidant and iron chelating activities. The importance of EGCG in enhancing cell resistance to oxidative stress goes beyond its simple scavenging and iron chelating activities, and its effects are most interesting in pathologies where oxidative stress and iron are involved (Weinreb et al. 2009). Numerous in vitro and in vivo studies in the last decade have shown that EGCG can prevent and/or reduce the deleterious effects of oxygen-derived free radicals, which are associated with several human diseases (Weinreb et al. 2009). Several lines of evidence suggest that oxidative stress, resulting in reactive oxygen species generation, either through an enzyme or metal catalyzed process, plays a pivotal role in clinical disorders, such as atherosclerosis, ischemia reperfusion injury, cancer, stroke and neurodegenerative disorders (Weinreb et al. 2009).
Brain ischemia has been reported to induce excitotoxicity in the central nervous system (CNS) (Kontos 1985). Excitotoxicity can be induced by excessive glutamate release from neurons that have been injured by ischemia. This overstimulation of glutamate receptors can result in increased Ca2+ influx into neuronal cells via these receptors (Kontos 1985). Increased intracellular Ca2+ concentrations lead to the production of free radicals, and ultimately to cell degeneration and death (Kontos 1985). In experimental focal ischemia in the rat brain, free radical production is increased by facilitation of the arachidonic acid cascade (Chan et al. 1985). Free radicals induce lipid peroxidation in cell membranes and initiate neuronal dysfunction and death (Hall and Braughler 1989; Clausen et al. 2004). Furthermore, the membrane peroxidation cascade proceeds automatically and induces edema, infarction and neuronal dysfunction in the brain after injury (Hall and Braughler 1989; Clausen et al. 2004). In addition, oxidative stress mediated by free radical production has been shown to contribute to neuronal dysfunction and death in a focal brain ischemia model (Hall and Braughler 1989; Clausen et al. 2004).
Traumatic brain injury (TBI) occurs as a result of a direct mechanical insult to the brain, and induces degeneration and death in the CNS (Chirumamilla et al. 2002; Rice et al. 2003). Following the initial mechanical insult, secondary effects including blood–brain barrier disruption, excitotoxic damage and free radical production are induced as a result of the circulatory disturbance caused by ischemia (Kawamata et al. 1995; Xiong et al. 1997; Gage 2000). Previous studies have shown that free radical production following TBI has deleterious effects on neural stem cell (NSC) populations (Itoh et al. 2007, 2009). However, in a recent study, we reported that the novel free radical scavenger, edaravone, which inhibits and absorbs free radicals, increases and protects NSC proliferation around the damaged region after TBI (Itoh et al. 2009).
The effects of EGCG on NSCs around the damaged area following TBI have not been investigated. In the present study, we hypothesized that EGCG treatment would inhibit free radical production by TBI, resulting in an increase in NSC number around the damaged area. Thus, in this study, we used the rat TBI model to investigate the effects of EGCG on NSCs around the damaged tissue using immunohistochemical and ex vivo techniques.
Materials and methods
Animals and (−)-epigallocatechin gallate (EGCG) treatment
Adult male Wistar rats (6 weeks of age, weighing 120–140 g) were housed at 22°C under a 12:12-h light–dark cycle and had free access to food. All experimental protocols conformed to the guidelines of the National Institutes of Health (Guide for the Care and Use of Laboratory Animals 1996), and were approved by the Institutional Animal Experimentation Committee of Kinki University School of Medicine.
Previously, we showed that EGCG (0.1% (w/v)) inhibited neuronal cell degeneration and death by free radicals following TBI (Itoh et al. 2011). Therefore, in this study we used the same concentration of EGCG. A 0.1% (w/v) EGCG (TEAVIGO; DSM Nutritional Products GmbH, Grenzach-Wylen, Germany) solution was prepared by dissolving the drug in drinking water (Lee et al. 2003).
A 6-week-old male Wistar rats were randomly divided into three groups: (1) water group (n = 18): rats had access to normal drinking water ad libitum from 6 to 10 weeks of age (n = 18), (2) EGCG treatment group (n = 18): rats had access to 0.1% (w/v) EGCG drinking water ad libitum from 6 to 10 weeks of age; and (3) Sham group (n = 18): aged-matched rats had access to normal drinking water ad libitum from 6 to 10 weeks of age and underwent a sham operation with no impact. Sham group rats were only used for the measurement of lipid peroxidation levels. The volume of normal drinking and 0.1% (w/v) EGCG drinking water, and the change in body weight of rats were measured every day in each group. There were no differences in volume of water consumed or changes in body weight.
Surgical procedures
When rats from each group reached 10 weeks of age (weighing 220–250 g) they were anesthetized by intraperitoneal pentobarbital (50 mg/kg) injection. The scalp was incised on the midline and the skull was exposed. A 2-mm hole was drilled (1 mm posterior, +1 mm right lateral to bregma) in the right parietal calvaria (Itoh et al. 2005, 2007). Brain injury above the dura mater was then inflicted with a pneumatic controlled injury device (Itoh et al. 2005, 2007) at an impact velocity of 4 m/s, with an impact tip diameter of 1 mm and a fixed impact deformation depth of 2 mm from the cerebral surface.
Immunohistochemistry
At 1, 3 and 7 days following TBI, seven rats in each of the water and EGCG groups were deeply anesthetized by an intraperitoneal pentobarbital (150 mg/kg) injection and then subjected to sequential intracardial perfusion with 300 mL of 0.1 M phosphate-buffered saline (PBS; pH 7.4–7.5), followed by 300 mL of 4% (w/v) paraformaldehyde (PFA) in PBS. The brains were removed and stored in PFA for 3 days, before the maximum size of the lesion was sliced into serial coronal sections (50-µm thick) using a microslicer (Dousaka EM, Kyoto, Japan). Each section was treated with 3% (v/v) H2O2 in Tris-buffered saline (TBS; 0.1 M Tris–HCl pH 7.5, 0.15 M NaCl) for 30 min to block endogenous peroxidase activity. Next, the sections were washed three times with TBS containing 0.1% (v/v) Triton X-100 (TBS-T), blocked with 3% (w/v) bovine serum albumin (BSA; Sigma, St. Louis, MO) in TBS-T (TBS-TB) for 30 min and incubated overnight at room temperature with the following primary antibodies in blocking solution: (1) monoclonal antibody against the rat NSC marker, nestin (1:1,000 in TBS-TB, BD Biosciences Pharmingen, San Diego, CA); (2) rabbit polyclonal antibody against the apoptosis marker, single-stranded DNA (ssDNA in TBS-TB, 1:1,000, DAKO, Glostrup, Denmark); (3) monoclonal antibody against DNA damage, 8-hydroxy-2′-deoxyguanosine (8-OHdG, 1:50 in TBS-TB, JaICA, Shizuoka, Japan); or (4) monoclonal antibody against the lipid peroxidation marker 4-hydroxy-2-nonenal (4-HNE, 1:50 in TBS-TB, JaICA, Shizuoka, Japan). Following extensive washing, the sections were further incubated with a HISTIFINE Rat-PO (mouse)-kit or HISTIFINE Rat-PO (rabbit)-kit (Nichirei, Osaka, Japan), comprising peroxidase-conjugated anti-mouse or anti-rabbit secondary antibody, respectively, for 60 min at room temperature. The HISTIFINE Rat-PO kit contained pre-absorbed rat serum and exhibited negligible non-specific binding of rat serum in injured rat tissues. Labeling was visualized using diaminobenzidine (DAB; Vector Peroxidase Substrate Kit; Vector Laboratories, Burlingame, CA) for 5 min, and the sections were counterstained with hematoxylin to quantify the number of nestin, 8-OHdG and 4-HNE-positive cells. Negative control staining was performed with normal mouse serum instead of primary antibodies following the procedure outlined above.
Quantification of nestin-, ssDNA-, 8-OHdG- and 4-HNE-positive cells
To determine the number of nestin-, ssDNA-, 8-OHdG- and 4-HNE-positive cells, all DAB-labeled cells within 500 µm of the edge of the damaged region (cortex), excluding white matter, following TBI were counted in three serial sections under a Nikon E 1000 M microscope (Nikon Corporation, Tokyo, Japan) using a 20× objective. The number of DAB labeled cells in three serial sections was averaged. An image of the measured area was captured at 20× magnification using a CCD camera (DS-2M; Nikon Corporation), and the measured area and volume in each image was traced and measured using ACT-2U image analysis software (Nikon Corporation). The number of DAB labeled cells was calculated from the average DAB labeled positive number and the volume. The numbers of nestin-, ssDNA-, 8-OHdG- and 4-HNE-positive cells were expressed as the numbers of positive cells/100 µm3.
Double immunofluorescence staining for ssDNA and nestin or NeuN
Three-day post-TBI sections were washed and blocked with 50 mM glycine in TBS for 2 h at 37°C to reduce non-specific fluorescence. The sections were washed with TBS-T, blocked with 3% (w/v) BSA in TBS-T and incubated with the anti-ssDNA antibody (1:300 in TBS-TB; DAKO) overnight at room temperature. Following extensive washing, the sections were further incubated with Alexa Flour 488 anti-rabbit IgG (1:300 in TBS-TB; BD Biosciences Pharmingen) for 80 min at room temperature. Next, the sections were washed extensively and incubated with a mouse monoclonal anti-NeuN antibody (1:300 in TBS-TB; Millipore, Billerica, MA, USA), a neuronal cell marker or a monoclonal anti-nestin antibody (1:300 in TBS-TB; BD Biosciences Pharmingen) overnight at room temperature. Following extensive washing, the sections were further incubated with Alexa Flour 555 anti-mouse IgG (1:300 in TBS-TB; BD Biosciences Pharmingen) for 80 min at room temperature. Subsequently, the sections were observed using a confocal laser scanning microscope (LSM5 PASCAL; Carl Zeiss Jena GmbH, Jena, Germany).
Isolation and culture of NSCs
NSCs were isolated and cultured according to a previously described method (Itoh et al. 2005). At 1, 3 and 7 days following TBI in the EGCG and water groups, cerebral cortex tissue 2 mm in diameter, measured from the center of the lesion, was separated by gross dissection under a dissecting microscope. Care was taken to remove and discard the meninges and blood vessels. One cerebral tissue sample from each of the five individual rats in six experimental groups was used for NSC culture. The tissue was then cut into small pieces and dissociated by incubation in Hanks’ Balanced Salt Solution (HBSS, Invitrogen, Carlsbad, CA) containing 0.1% (w/v) trypsin (Invitrogen) and 0.01% (w/v) DNase 1 (Roche, Indianapolis, IN, USA) at 37°C for 30 min. Subsequently, an equal volume of fetal calf serum (Invitrogen) was added to the tissue suspension and it was centrifuged at 1,300 rpm for 5 min. The supernatant was removed, HBSS was added, and cells were dissociated by trituration and centrifuged at 1,300 rpm for 5 min. The cells were plated as a single-cell suspension on ornithine- and fibronectin-coated 60-mm culture dishes in a plating medium (N2/DF) consisting of Dulbecco’s modified Eagle’s medium (Invitrogen)/F-12 medium (Invitrogen) supplemented with basic fibroblast growth factor (bFGF) (20 ng/mL, Roche), epidermal growth factor (EGF) (20 ng/mL, Roche), insulin (25 µg/mL, Roche), transferrin (100 µg/mL, Roche), and progesterone (100 µg/mL, Wako, Osaka, Japan), and maintained at 37°C in a humidified 5% CO2 atmosphere for 3 days. After 4 days, cells attached to the bottom of the culture dishes were desquamated by trituration, and collected and dissociated by trituration in N2/DF medium. Single cell suspensions were replated on untreated culture dishes, and after 10–14 days in culture, the spheres had formed. The number of spheres per culture dish was counted (n = 5), and sphere differentiation was investigated by growing cultures in media without bFGF and EGF. Cultures were maintained on ornithine-coated round cover glasses (1 cm2) in 24 well culture plates at 37°C in a humidified 5% CO2 atmosphere for 4 days.
Immunostaining of cultured cells
Immunostaining was performed on spheres and 4 day cultures that were fixed using 4% (w/v) PFA in PBS for 20 min. Spheres were stained with anti-rat nestin monoclonal antibody, anti monoclonal Tuj1 antibody (1:300 in TBS-TB; Chemicon, Berkeley, CA), anti polyclonal glial fibrillary acidic protein (GFAP) antibody (1:300 in TBS-TB; DAKO), or anti monoclonal O4 antibody (1:300 in TBS-TB; Chemicon). Tuj1 was used as a marker of neuronal cells, GFAP was as a marker of astrocytes, and O4 was used as a marker of oligodendrocytes. The secondary antibodies used were Alexa Flour 488 anti-mouse IgG, M (1:500 in TBS-TB; BD Biosciences Pharmingen) for nestin, and Tuj1 or O4 and Alexa Flour 555 anti-rabbit IgG (1:500 in TBS-TB; BD Biosciences Pharmingen) for GFAP. All antibodies were diluted in PBS containing 3% (w/v) BSA. Cells cultured for 4 days were used for fluorescence double labeling with Tuj1 and GFAP.
Measurement of lipid peroxidation
At 1, 3 and 7 days after TBI in the sham, water and EGCG groups, cerebral cortex tissue 2 mm in diameter, measured from the center of the lesion, was separated by gross dissection under a dissecting microscope for assays of malondialdehyde (MDA). The assay procedure for lipid peroxide in brain tissue was set up as follows: tissues were homogenized in 10 volumes (w/v) of cold homogenizing buffer (7.5 mmol/L sodium phosphate buffer (pH 7.0) containing 0.25 mol/L sucrose, 5 mmol/L ethylene glycol bis (2-aminoethyl ether)-tetraacetic acid, 25 μg/mL leupeptin, 25 μg/mL aprotinin) using a hand homogenizer. After treatment of the homogenates with ultrasonic waves at 4°C for 100 s and centrifugation at 20,000g at 4°C for 30 min, the resulting supernatants were collected. MDA measurement was performed using a BIOXYTECH MDA-586 (OxisResearch, Beverly Hills, CA) according to the manufacturer’s instructions. MDA has been identified as the product of lipid peroxidation that reacts with the thiobarbituric acid to give a red species absorbing at 535 nm.
Statistical analysis
Data were expressed as the mean ± SD. Statistical analysis was performed using one-way analysis of variance with Fisher’s post hoc test (Stat View®; SAS Institute Inc, Cary, NC, USA). Values of P < 0.05 were considered statistically significant.
Results
Quantification of nestin-immunopositive cells following TBI
At 1 day following TBI in the water groups, a few small nestin-positive cells were present around the damaged area. At 3 days following TBI in the water and at 1 and 3 days following TBI in the EGCG treatment groups, there were many larger nestin-positive cells around the damaged area, and these cells possessed a nestin-immunopositive cytoplasm and projections (Fig. 1a, b). Furthermore, there were more of these larger cells in the EGCG treatment group than in the water group. At 7 days following TBI in the water group, there were a few nestin-positive fibers among the cells around the damaged area (Fig. 1c). However, in the EGCG treatment group, there were many larger-sized nestin-positive cells that possessed a nestin-immunopositive cytoplasm and projections (Fig. 1d). The number of nestin-positive cells are shown in Fig. 1e.

Immunostaining for nestin around the damaged cerebral cortex following traumatic brain injury in the rat. At 3 days after injury in the water (a) and EGCG treatment (b) groups, abundant nestin immunoreactivity was present mainly in the cytoplasm and projections. At 7 days after injury in the water drinking group, nestin immunopositive elongating fibers are seen (c arrows), while in the EGCG treatment group, abundant nestin immunoreactivity was present mainly in the cytoplasm and projections (d). Scale bars 50 μm. Graph showing nestin immunopositive cell number around the damaged cerebral cortex following traumatic rat brain injury (e). The results are shown as the mean ± SD. **P < 0.01; n = 5. EGCG (−)-epigallocatechin gallate
At 1 day following TBI, the number of nestin-positive cells was 4.9 ± 3.8 and 48.3 ± 12.3 in the water and EGCG treatment groups, respectively. The number of nestin-positive cells in the EGCG treatment group showed a significant increase when compared with the number in the water group. However, the number of nestin-positive cells at 3 and 7 days following TBI in the EGCG treatment group (178.3 ± 16.9 and 62.3 ± 9.9, respectively) showed a significant increase when compared with the number in water group (73.0 ± 25.8 and 12.4 ± 3.3, respectively, P < 0.01).
Lipid peroxidation levels
At 1 and 3 days following TBI, the water group showed a significantly higher MDA level than the sham and EGCG treatment groups (Fig. 2, P < 0.05). However, at the same time following TBI, the EGCG treatment group showed a significantly higher MDA level than the sham group (Fig. 2, P < 0.05). At 7 days following TBI, there was no change in any of the groups.

Effect of EGCG treatment on lipid peroxide (MDA) formation in rat traumatic brain injured tissue. EGCG treatment significantly attenuated MDA production after traumatic brain injury. The results are shown as mean ± SD. *P < 0.05; **P < 0.01, n = 6. EGCG (−)-epigallocatechin gallate, MDA malondialdehyde
Quantification of 8-OHdG-immunopositive cells following TBI
At 1, 3 and 7 days following TBI in the water group, many 8-OHdG-positive cells were present around the damaged area and possessed 8-OHdG-immunopositive cytoplasm and projections morphologically similar to those of large neurons or nestin-positive cells (Fig. 3a).

Immunostaining for 8-OHdG around the damaged cerebral cortex following traumatic brain injury in the rat. At 3 days after injury in the water treatment group, there were many 8-OHdG-positive cells (a), but in the EGCG treatment group, there were only a few 8-OHdG-positive cells (b). Scale bars 50 µm. Graph showing the number of 8-OHdG immunopositive cells around the damaged cerebral cortex following traumatic brain injury in the rat (c). The results are shown as the mean ± SD. ***P < 0.001; n = 5. 8-OHdG 8-Hydroxy-2′-deoxyguanosine, EGCG (−)-epigallocatechin gallate
In contrast, at 1, 3 and 7 days following TBI in the EGCG treatment group, only a few 8-OHdG-immunopositive cells were present around the damaged area (Fig. 3b), with a significant reduction in the number of 8-OHdG-positive cells at 1, 3 and 7 days post-TBI in the EGCG treatment group (183.6 ± 53.6, 265.4 ± 59.9 and 87.8 ± 38.8, respectively) when compared with the water group (979.4 ± 107.6, 975.7 ± 136.9 and 988.5 ± 201.4, respectively; P < 0.001) (Fig. 3c).
Quantification of 4-HNE-immunopositive cells following TBI
At 1, 3 and 7 days following TBI in the water group, many 4-HNE-immunopositive cells were present around the damaged area, and possessed a 4-HNE-immunopositive cytoplasm and projections morphologically similar to those of large neurons or nestin-positive cells (Fig. 4a).

Immunostaining for 4-HNE around the damaged cerebral cortex following traumatic brain injury in the rat. At 3 days after injury in the water treatment group, there were many 4-HNE-positive cells (a), but in the EGCG treatment group, there were only a few 4-HNE-positive cells (b). Scale bars 50 μm. Graph showing the number of 4-HNE immunopositive cells around the damaged cerebral cortex following traumatic brain injury in the rat (c). The results are shown as the mean ± SD. ***P < 0.001; n = 5. 4-HNE 4-hydroxy-2-nonenal, EGCG (−)-epigallocatechin gallate
In contrast, at 1, 3 and 7 days following TBI in the EGCG treatment group, only a few 4-HNE-immunopositive cells were present around the damaged area (Fig. 4b), with a significant reduction in the number of 4-HNE-immunopositive cells at 1 and 3 days following TBI in the EGCG treatment group (116.6 ± 38.2 and 134.8 ± 53.0, respectively) when compared with the water group (488.1 ± 55.1 and 471.9 ± 48.8, respectively; P < 0.001) (Fig. 4c); there was no difference between the groups at 7 days. The number of 4-HNE-positive cells in the water group was significantly lower at 3 days when compared with 1 and 7 days after TBI (P < 0.001).
Quantification of ssDNA-immunopositive cells following TBI
At 1, 3 and 7 days following TBI in the water group, many ssDNA-immunopositive cells were present around the damaged area, and possessed an ssDNA-immunopositive cytoplasm morphologically similar to those of neurons or nestin-positive cells (Fig. 5a). In contrast, at 1, 3 and 7 days following TBI in the EGCG treatment group, only a few ssDNA-immunopositive cells were present around the damaged area (Fig. 5b), with a significant reduction in the number of ssDNA-immunopositive cells at 1, 3 and 7 days following TBI in the EGCG treatment group (28.0 ± 5.1, 108.8 ± 11.4 and 24.9 ± 4.7, respectively) as compared to the water group (72.7 ± 20.4, 276.7 ± 35.5 and 55.7 ± 18.7, respectively; P < 0.05) (Fig. 5c). The number of ssDNA-immunopositive cells at 3 days after TBI in the water group was markedly higher than at 1 and 7 days after TBI (P < 0.001).

Immunostaining for ssDNA around the damaged cerebral cortex following traumatic brain injury in the rat. At 3 days after injury in the water drinking group, abundant ssDNA immunoreactivity was present mainly in the nuclei (a), but in the EGCG treatment group, there were only a few ssDNA immunoreactive nuclei (b). Scale bars 50 µm. Graph showing ssDNA immunopositive cell number around the damaged cerebral cortex following traumatic rat brain injury (c). The results are shown as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001; n = 5. EGCG (−)-epigallocatechin gallate, ssDNA single-stranded DNA
Double immunofluorescence staining for ssDNA and nestin or NeuN
There were many ssDNA-positive cells in the water group (Fig. 6a, g) and a few ssDNA positive cells in the EGCG treatment group (Fig. 6d, j) around the damaged area at 3 days after TBI. In the water and EGCG groups, there were many NeuN-positive cells around the damaged area at 3 days after TBI (Fig. 6b, e). The majority of NeuN-positive cells were also immunopositive for ssDNA in the water group (Fig. 6c). However, after TBI in the EGCG treatment group, few NeuN-positive cells co-localized with ssDNA-positive cells (Fig. 6f).

Double immunofluorescence staining of ssDNA and NeuN or nestin around the damaged area at 3 days after traumatic brain injury. At 3 days after the injury, many ssDNA-immunopositive (a, g green) and NeuN-immunopositive (b red) or nestin-immunopositive (h red) cells were observed in the water group. The ssDNA-immunopositive cells were also immunopositive for NeuN (c yellow, arrows) or nestin (i arrows). However, in the EGCG treatment group, a few ssDNA-immunopositive (d, j green) and many NeuN- (e red) or nestin-immunopositive (k red) cells were observed. Double labeling of ssDNA-immunopositive cells with NeuN (f) or nestin (l) was not seen. EGCG (−)-epigallocatechin gallate, ssDNA single-stranded DNA
In addition, although there were nestin-positive cells and projections in the water (Fig. 6h) and EGCG treatment (Fig. 6k) groups at 3 days after TBI, the nestin-positive cells in the water group were smaller than those in the EGCG treatment group. Moreover, a subset of nestin-positive cells was positive for ssDNA-immunostaining in the water group (Fig. 6i). However, few nestin-positive cells co-localized with ssDNA-positive cells in the EGCG treatment group (Fig. 6l).
NSC isolation, counting and immunostaining
At 1 day following TBI in the water group, it was not possible to isolate spheres from around the damaged area of the cerebral cortex (Fig. 7a). However, 3 days after injury in the water, and at 1 and 3 days after injury in the EGCG treatment groups, spheres were isolated and cultured (Fig. 7b). Moreover, at 7 days after injury in the EGCG treatment group, but not the water group, spheres were isolated and cultured. Almost all aggregated cells in spheres isolated from the water and EGCG treatment groups were immunopositive for nestin (Fig. 7c). In addition, the spheres were not immunopositive for Tuj1, a marker of immature neuronal cells, or vimentin, a marker of immature glial cells (data not shown). At 1, 3 and 7 days following injury, a number of isolated and cultured spheres in the EGCG treatment group (8 ± 5, 32 ± 12 and 11 ± 6, respectively) were significantly increased when compared with the water group (0, 14 ± 6 and 0, respectively; P < 0.05) (Fig. 7d).

Photomicrographs of isolated spheres from the area surrounding the brain injury in the EGCG treatment group. No spheres were observed in cultures derived from 1-day tissue in the EGCG treatment group (a). In the EGCG treatment group, a few spheres were found in cultures derived from the 3-day tissue after 13 days of culture (b). Isolated spheres showing nestin immunoreactivity (c). Scale bars 50 μm. Graph showing the number of isolated and cultured spheres from the area surrounding the brain injury (d). The results are shown as mean ± SD. *P < 0.05, **P < 0.01; n = 5. EGCG (−)-epigallocatechin gallate
Differentiation of cultures
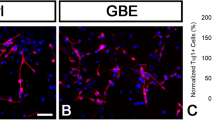
Spheres isolated from the water and EGCG treatment groups immediately attached to the bottom of the cell culture dishes. The number of fibers that elongated from the spheres increased with time. After 4 days in culture, many cells with elongated fibers had migrated from the spheres (Fig. 8a). Cells with O4 immunopositive labeling in the cytoplasm (Fig. 8b), and Tuj1 immunopositive labeling in the cytoplasm and elongated fibers were also present in cultures (Fig. 8c). In addition, there were also cells with GFAP immunopositive labeling in the cytoplasm (Fig. 8d). However, Tuji1 immunopositive labeling did not co-localize with GFAP immunopositive labeling (Fig. 8e).

Phase contrast and immunohistochemistry images of cells differentiated from neurospheres without bFGF and EGF after 4 days of culture in the EGCG treatment group. Phase contrast microscopic image showing cells differentiated from neurospheres in culture. Arrow indicates neurospheres (a). Differentiated cultures of neurospheres with Alexa Flour 488 immunostaining showing O4 immunoreactive cells (green b). Double labeling fluorescence immunostaining showing many Tuj1 immunoreactive cells (green c) and many GFAP immunoreactive cells (red d). A merged image of c and d is shown in e. Tuj1 and GFAP do not co-localize. Scale bars 50 μm. bFGF Basic fibroblast growth factor, EGCG (−)-epigallocatechin gallate, EGF epidermal growth factor, GFAP glial fibrillary acidic protein
Discussion
In this study, we found that 0.1% EGCG drinking, pre- and post-TBI, in inhibits TBI-induced increases in oxidative DNA damage, lipid peroxidation, lipid peroxidation levels, and neuronal and NSC apoptosis. Moreover, EGCG treatment increased the number of nestin-positive cells and NSCs around the damaged area after TBI.
MDA, which is produced by free radicals in glial and neuronal membranes via the breakdown of polyunsaturated fatty acids, serves as a convenient index for determining the extent of lipid peroxidation (Ates et al. 2007). It has been reported that in a rat ischemic model, MDA significantly increases in the brain after brain injury (Hong et al. 2000; Suzuki et al. 2004). Our previous study has shown that free radical production around damaged tissue after TBI increases, and significantly increases MDA levels (Itoh et al. 2009). Interestingly, EGCG prevented the production of free radicals in brain after rat ischemia, and significantly decreased the levels of MDA in the brain after brain injury (Hong et al. 2000; Suzuki et al. 2004). In this study, we monitored MDA levels in rats subjected to TBI that were treated with or without EGCG. MDA levels in the EGCG treatment group showed a significant decrease when compared with the water group. Furthermore, MDA in the EGCG treatment group did not show a significant increase compared with the sham group. These results indicate that EGCG eliminated and absorbed the free radicals produced by TBI around the damaged brain region.
The number of ssDNA-positive cells following TBI in the water group increased significantly when compared with the EGCG group. Furthermore, the number of 8-OHdG- and 4HNE-positive cells following TBI in the water group also decreased when compared with the EGCG treatment group. Previous studies provide strong evidence that the signaling pathways induced by free radicals can cause cellular damage and even cell death during brain injury. For example, the activation of mitochondrial pathways (Sugawara et al. 2002) and kinase signaling pathways (Irving and Bamford 2002) that can occur after a variety of injuries, including focal cerebral ischemia and TBI, have been demonstrated. The activation of caspase-3 has also been shown to be increased 3 days after an ischemic insult in hippocampal CA1 neurons, following which the release of cytochrome c from the mitochondria to the cytosol induces neural and glial cell degeneration and death (Sugawara et al. 2002). Significantly, EGCG inhibits the release of cytochrome c and blocks caspase activation in the cortex and hippocampus after an ischemic insult, and EGCG also blocks the production of free radicals (Weinreb et al. 2009). Therefore, EGCG inhibits neural and glial cell degeneration as well as death in the cortex and hippocampus that is mediated by free radical production during ischemia (Weinreb et al. 2009). The mitogen-activated protein kinase family is also known to be involved in ischemia; an increase in p38 activity has been observed after the onset of cerebral ischemia (Walton et al. 1998), and JNK (c-Jun N-terminal kinase) activation is also involved in free radical-mediated cell death (Tsuji et al. 2000). For example, spin-trapped α-butylnitrone protects the CA1 region of the hippocampus after induction of global ischemia in the gerbil, and the protective effect is associated with the inhibition of JNK and p38 activity (Tsuji et al. 2000). Furthermore, in a mouse Alzheimer’s model, β-amyloid protein significantly increases JNK and p38 activation in hippocampal neurons, and induces neuronal cell degeneration and death (Mandel et al. 2004; Kim et al. 2009). EGCG has been reported to significantly inhibit JNK and p38 activation after brain injury, and blocks induced free radical-mediated neural cell degeneration and death after brain injury, resulting in improvement of cerebral dysfunction (Mandel et al. 2004; Kim et al. 2009).
Bcl-2 inhibits the release of cytochrome c from the mitochondria to the cytosol and blocks the activation of capase-3, resulting in the inhibition of neuronal apoptotic cell death (Hockenbery et al. 1990). In a Bcl-2-overexpressing ischemic mouse model, the overexpression of Bcl-2 has been shown to be neuroprotective in the hippocampus after brain injury when compared with Bcl-2 deficient mice (Martinou et al. 1994; Hata et al. 1999). Interestingly, in mouse models of Alzheimer’s and Parkinson’s disease, EGCG increases the expression of Bcl-2, and EGCG decreases the expression of Bax, which induces apoptotic cell death, resulting in the prevention of neuronal and glial apoptotic cell death and the protection of neurons in the brain (Mandel et al. 2005; Weinreb et al. 2009). Furthermore, in a rat ischemic model, free radical production increased the expression of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) in neurons, and decreased the expression of Bcl-2, resulting in apoptotic neural cell death (Amemiya et al. 2005). EGCG has been shown to reduce the expression of NFκB induced by free radicals after ischemia, and EGCG after brain injury blocks neuronal apoptotic cell death (Lee et al. 2000). Moreover, it has been reported that free radicals react to T cells during neuroinflammation and activate NF-κB, resulting in T cell proliferation, an increase in tumor necrosis factor-α (TNF-α) synthesis, and induction of neuronal degeneration and cell death (Aktas et al. 2004; Herges et al. 2011). However, EGCG suppressed the activation of NF-kB by free radicals and inhibited neuronal degeneration and cell death by T cells during neuroinflammation, resulting in a neuroprotective effect (Aktas et al. 2004; Herges et al. 2011). The present results suggest that EGCG absorbs free radicals generated by TBI and/or inhibits their function, thereby preventing free radical-mediated apoptosis and cell death subsequent to TBI. In addition, there were many more NeuN- and nestin-positive cells that co-localized with ssDNA-positive cells in the water group than in EGCG group following TBI. These data demonstrated that EGCG inhibits apoptosis of neural cells and NSCs, which is mediated by free radicals following TBI.
In this study, nestin-positive cells were present at the early stages after TBI (1–7 days) in the water and EGCG groups. In both groups, the nestin-positive immunoreactive cells achieved their maximum number at 3 days following TBI. This result is consistent with our previous report (Itoh et al. 2005) as well as others where nestin-positive cells were increased around the damaged cerebral cortex at 1–4 days after cryoinjury, after which they decreased (Moon et al. 2004). In addition, the number of nestin-positive cells around the damaged cerebral cortex reached a maximum at 3 days after ablation of injury (Douen et al. 2004). Nestin-positive cells were present around the damaged cerebral cortex at 1–7 days following controlled cortical impact (CCI), and the number of nestin-positive cells reached a maximum at 4 days after CCI (Chen et al. 2003). These results indicate that the maximal numbers of nestin-positive cells were present around the damaged area at 3 or 4 days after injury. These data are consistent with our results described here.
In this study, the number of nestin-positive cells in the EGCG treatment group showed a significant increase when compared with the water group. It seems that the EGCG-meditated reduction in free radicals reduced the degeneration and death of nestin-positive cells, and allowed brain tissue and nestin-positive cells to proliferate around the damaged area following TBI.
At 1, 3 and 7 days following TBI in the water and EGCG treatment groups, the cerebral cortex, without white matter, was dissected from around the damaged area for the isolation of nestin-positive cells. At 3 days following TBI in the water and EGCG treatment groups, nestin-positive spheres could be isolated from around the damaged brain tissue, but the number of spheres in the EGCG treatment group increased significantly when compared with the water group. Furthermore, at 7 days following TBI, nestin-positive spheres could be isolated from around the damaged brain tissue in the EGCG treatment group, but not the water group. Our previous report showed the number of nestin-positive cells around the damaged area following TBI correlated with the number of spheres from brain tissue (Itoh et al. 2009). Therefore, these data may indicate that the number of nestin-positive cells around the damaged area correlate with the number of NSCs present.
Immunohistochemistry revealed that almost all aggregated cells in the spheres isolated from the water and EGCG groups were immunopositive for nestin, but not Tuj1 or vimentin. Neurospheres comprising NSCs may differentiate into neurons or glia following removal of bFGF or EGF, which are mitogenic factors in culture medium (Itoh et al. 2005). Neurospheres originating from rat brain differentiated into Tuj1-positive neurons and GFAP-positive astrocytes after 2–4 days of culture without bFGF in the culture medium (Yamamoto et al. 2001). In this study, the spheres differentiated into Tuj1-, GFAP- and O4-positive cells after 4 days of culture without bFGF in the culture medium. Recently, in the rat TBI model, we reported that neurospheres could be isolated from the damaged region at an early stage following rat TBI and had the ability to differentiate into neurons and glia (Itoh et al. 2005). We confirmed in this study that spheres isolated from the damaged region at an early stage following rat TBI were NSCs that could differentiate into neurons and glia.
Meanwhile, a recent study demonstrated that phosphatidylinositol 3-kinase (PI3K), a component of the PI3K-Akt pathway activated by free radicals, inhibited apoptotic cell death in cultured rat NSCs (Lin et al. 2004). Moreover, PI3K-Akt signaling has been shown to increase in vitro cultured neural cells treated with EGCG and to inhibit neuronal cell death induced by free radicals (Koh et al. 2004). Furthermore, our recent report has shown that radical scavenger administration after TBI protects nestin-positive cells, including NSCs, around the damaged area after TBI, and significantly increases the number of NSCs around the damaged area after injury (Itoh et al. 2009). These results indicate that EGCG absorbs free radicals induced by brain injury and protects nestin-positive cells, including NSCs, around the damaged area following TBI.
Recently, the absorption and pharmacokinetics of EGCG in various regions of the adult and fetal rat brain have been investigated following oral and intravenous administration. These studies revealed that EGCG could be detected in brain tissue, suggesting that it may potentially penetrate through the blood–brain barrier (BBB) (Weinreb et al. 2009). An in vitro model of the BBB demonstrated that various flavonoids and some metabolites were able to traverse the BBB and that the potential for permeation was consistent with compound lipophilicity (Weinreb et al. 2009). Therefore, we speculate that EGCG crosses the BBB and inhibits neuronal and NSC death by free radicals in the damaged area following TBI.
Reactive astrocytes have been identified by GFAP staining around the damaged area after brain injury (Seri et al. 2001; Picard-Riera et al. 2004). In this study, there were many GFAP-positive reactive astrocytes around the damaged area after TBI. Reactive astrocytes around the damaged area underwent blastogenesis following brain injury to give rise to NSCs that were immunopositive for nestin and could differentiate into neurons and glia (Seri et al. 2001; Picard-Riera et al. 2004). In the subventricular zone (SVZ) or the subgranular zone (SGZ) of the dentate gyrus-hilus, reactive astrocytes undergo blastogenesis following brain injury and differentiate into neural precursor cells that are immunopositive for both nestin and GFAP (Seri et al. 2001; Picard-Riera et al. 2004). In addition, we reported that cultured rat type I astrocytes underwent blastogenesis when cultured in the presence of bFGF to become nestin-positive NSCs, which could differentiate into neuronal and glial cells (Itoh et al. 2006). In contrast, it has been reported that NSCs migrate to the damaged area from the SVZ of the lateral ventricles or the SGZ interface following brain injury (Lois and Alvarez-Buylla 1994; Kuhn et al. 1996; Parent et al. 1997). Therefore, the occurrence of reactive astrocytes around the damaged area following brain injury appears to be important. However, it is unclear whether NSCs migrate to the damaged area from the SVZ and SGZ, or whether astrocytes undergo blastogenesis following injury and differentiate into nestin-positive cells. Our experiments presented here do not answer this question and so further investigations are required.
This study may be potentially important for those people who have received TBI through traffic accidents, war and other events. Thus, clinical trials in humans using EGCG must be performed because drinking green tea may improve cerebral dysfunction after TBI. EGCG (0.1% (w/v) in drinking water significantly decreased DNA damage, lipid peroxidation levels and neuronal cell and NSC apoptosis around the damaged area following TBI at 1, 4 and 7 days. Moreover, the number of nestin-positive cells (NSC maker) after TBI in the EGCG treatment group increased significantly, and a greater number of neurospheres could be isolated from brain injured tissue in the EGCG group at both 3 and 7 days following TBI. It will be interesting to investigate not only the sustained effect of EGCG treatment on NSCs around the damaged area post-TBI but also the effect of pretreatment, studies we aim to perform in the future.
From our current study, we propose that EGCG treatment before and after TBI eliminated and/or absorbed free radicals (such as O2 − and ·OH) induced by brain injury. Apoptosis and cell death of neuronal cells and NSCs induced by free radical production were inhibited, thus protecting nestin-positive cells, including NSCs, around the damaged area in the early phase following TBI.
References
Aktas O, Prozorovski T, Smorodchenko A, Savaskan NE, Lauster R, Kloetzel PM, Infante-Duarte C, Brocke S, Zipp F (2004) Green tea epigallocatechin-3-gallate mediates T cellular NF-kappa B inhibition and exerts neuroprotection in autoimmune encephalomyelitis. J Immunol 173:5794–5800
Amemiya S, Kamiya T, Nito C, Inaba T, Kato K, Ueda M, Shimazaki K, Katayama Y (2005) Anti-apoptotic and neuroprotective effects of edaravone following transient focal ischemia in rats. Eur J Pharmacol 516:125–130
Ates O, Cayli S, Altinoz E, Gurses I, Yucel N, Sener M, Kocak A, Yologlu S (2007) Neuroprotection by resveratrol against traumatic brain injury in rats. Mol Cell Biochem 294:137–144
Chan PH, Fishman RA, Longar S, Chen S, Yu A (1985) Cellular and molecular effects of polyunsaturated fatty acids in brain ischemia and injury. Prog Brain Res 63:227–235
Chen S, Pickard JD, Harris NG (2003) Time course of cellular pathology after controlled cortical impact injury. Exp Neurol 182:87–102
Chirumamilla S, Sun D, Bullock MR, Colello RJ (2002) Traumatic brain injury induced cell proliferation in the adult mammalian central nervous system. J Neurotrauma 19:693–703
Clausen F, Lundqvist H, Ekmark S, Lewen A, Ebendal T, Hillered L (2004) Oxygen free radical-dependent activation of extracellular signal-regulated kinase mediates apoptosis-like cell death after traumatic brain injury. J Neurotrauma 21:1168–1182
Douen AG, Dong L, Vanance S, Munger R, Hogan MJ, Thompson CS, Hakim AM (2004) Regulation of nestin expression after cortical ablation in adult rat brain. Brain Res 1008:139–146
Gage FH (2000) Mammalian neural stem cells. Science 287:1433–1438
Hall ED, Braughler JM (1989) Central nervous system trauma and stroke. II. Physiological and pharmacological evidence for involvement of oxygen radicals and lipid peroxidation. Free Radic Biol Med 6:303–313
Hata R, Gillardon F, Michaelidis TM, Hossmann KA (1999) Targeted disruption of the bcl-2 gene in mice exacerbates focal ischemic brain injury. Metab Brain Dis 14:117–124
Herges K, Millward JM, Hentschel N, Infante-Duarte C, Aktas O, Zipp F (2011) Neuroprotective effect of combination therapy of glatiramer acetate and epigallocatechin-3-gallate in neuroinflammation. PLoS One 6:e25456
Hockenbery D, Nunez G, Milliman C, Schreiber RD, Korsmeyer SJ (1990) Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 348:334–336
Hong JT, Ryu SR, Kim HJ, Lee JK, Lee SH, Kim DB, Yun YP, Ryu JH, Lee BM, Kim PY (2000) Neuroprotective effect of green tea extract in experimental ischemia–reperfusion brain injury. Brain Res Bull 53:743–749
Irving EA, Bamford M (2002) Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab 22:631–647
Itoh T, Satou T, Hashimoto S, Ito H (2005) Isolation of neural stem cells from damaged rat cerebral cortex after TBI. Neuroreport 16:1687–1691
Itoh T, Satou T, Nishida S, Hashimoto S, Ito H (2006) Cultured rat astrocytes give rise to neural stem cells. Neurochem Res 31:1381–1387
Itoh T, Satou T, Hashimoto S, Ito H (2007) Immature and mature neurons coexist among glial scars after rat traumatic brain injury. Neurol Res 29:734–742
Itoh T, Satou T, Nishida S, Tsubaki M, Hashimoto S, Ito H (2009) The novel free radical scavenger, edaravone, increases neural stem cell number around the area of damage following rat traumatic brain injury. Neurotoxic Res 16:378–389
Itoh T, Imano M, Nishida S, Tsubaki M, Hashimoto S, Ito A, Satou T (2011) (−)-Epigallocatechin-3-gallate protects against neuronal cell death and improves cerebral function after traumatic brain injury in rats. Neuromolecular Med 13:300–309
Kawamata T, Katayama Y, Hovda DA, Yoshino A, Becker DP (1995) Lactate accumulation following concussive brain injury: the role of ionic fluxes induced by excitatory amino acids. Brain Res 674:196–204
Kim CY, Lee C, Park GH, Jang JH (2009) Neuroprotective effect of epigallocatechin-3-gallate against beta-amyloid-induced oxidative and nitrosative cell death via augmentation of antioxidant defense capacity. Arch Pharm Res 32:869–881
Koh SH, Kim SH, Kwon H, Kim JG, Kim JH, Yang KH, Kim J, Kim SU, Yu HJ, Do BR, Kim KS, Jung HK (2004) Phosphatidylinositol-3 kinase/Akt and GSK-3 mediated cytoprotective effect of epigallocatechin gallate on oxidative stress-injured neuronal-differentiated N18D3 cells. Neurotoxicology 25:793–802
Kontos HA (1985) George E. Brown memorial lecture. Oxygen radicals in cerebral vascular injury. Circ Res 57:508–516
Kuhn HG, Dickinson-Anson H, Gage FH (1996) Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci 16:2027–2033
Lee S, Suh S, Kim S (2000) Protective effects of the green tea polyphenol (−)-epigallocatechin gallate against hippocampal neuronal damage after transient global ischemia in gerbils. Neurosci Lett 287:191–194
Lee SY, Kim CY, Lee JJ, Jung JG, Lee SR (2003) Effects of delayed administration of (−)-epigallocatechin gallate, a green tea polyphenol on the changes in polyamine levels and neuronal damage after transient forebrain ischemia in gerbils. Brain Res Bull 61:399–406
Lin HJ, Wang X, Shaffer KM, Sasaki CY, Ma W (2004) Characterization of H2O2-induced acute apoptosis in cultured neural stem/progenitor cells. FEBS Lett 570:102–106
Lois C, Alvarez-Buylla A (1994) Long-distance neuronal migration in the adult mammalian brain. Science 264:1145–1148
Mandel S, Weinreb O, Amit T, Youdim MB (2004) Cell signaling pathways in the neuroprotective actions of the green tea polyphenol (−)-epigallocatechin-3-gallate: implications for neurodegenerative diseases. J Neurochem 88:1555–1569
Mandel SA, Avramovich-Tirosh Y, Reznichenko L, Zheng H, Weinreb O, Amit T, Youdim MB (2005) Multifunctional activities of green tea catechins in neuroprotection. Modulation of cell survival genes, iron-dependent oxidative stress and PKC signaling pathway. Neurosignals 14:46–60
Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C et al (1994) Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron 13:1017–1030
Moon C, Ahn M, Kim S, Jin JK, Sim KB, Kim HM, Lee MY, Shin T (2004) Temporal patterns of the embryonic intermediate filaments nestin and vimentin expression in the cerebral cortex of adult rats after cryoinjury. Brain Res 1028:238–242
Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH (1997) Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci 17:3727–3738
Picard-Riera N, Nait-Oumesmar B, Baron-Van EA (2004) Endogenous adult neural stem cells: limits and potential to repair the injured central nervous system. J Neurosci Res 76:223–231
Rice AC, Khaldi A, Harvey HB, Salman NJ, White F, Fillmore H, Bullock MR (2003) Proliferation and neuronal differentiation of mitotically active cells following traumatic brain injury. Exp Neurol 183:406–417
Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A (2001) Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci 21:7153–7160
Sugawara T, Noshita N, Lewen A, Gasche Y, Ferrand-Drake M, Fujimura M, Morita-Fujimura Y, Chan PH (2002) Overexpression of copper/zinc superoxide dismutase in transgenic rats protects vulnerable neurons against ischemic damage by blocking the mitochondrial pathway of caspase activation. J Neurosci 22:209–217
Suzuki M, Tabuchi M, Ikeda M, Umegaki K, Tomita T (2004) Protective effects of green tea catechins on cerebral ischemic damage. Med Sci Monit 10:BR166–BR174
Tsuji M, Inanami O, Kuwabara M (2000) Neuroprotective effect of alpha-phenyl-N-tert-butylnitrone in gerbil hippocampus is mediated by the mitogen-activated protein kinase pathway and heat shock proteins. Neurosci Lett 282:41–44
Walton KM, DiRocco R, Bartlett BA, Koury E, Marcy VR, Jarvis B, Schaefer EM, Bhat RV (1998) Activation of p38MAPK in microglia after ischemia. J Neurochem 70:1764–1767
Weinreb O, Amit T, Mandel S, Youdim MB (2009) Neuroprotective molecular mechanisms of (−)-epigallocatechin-3-gallate: a reflective outcome of its antioxidant, iron chelating and neuritogenic properties. Genes Nutr 4:283–296
Xiong Y, Gu Q, Peterson PL, Muizelaar JP, Lee CP (1997) Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J Neurotrauma 14:23–34
Yamamoto S, Yamamoto N, Kitamura T, Nakamura K, Nakafuku M (2001) Proliferation of parenchymal neural progenitors in response to injury in the adult rat spinal cord. Exp Neurol 172:115–127
Yu J, Jia Y, Guo Y, Chang G, Duan W, Sun M, Li B, Li C (2010) Epigallocatechin-3-gallate protects motor neurons and regulates glutamate level. FEBS Lett 584:2921–2925
Acknowledgments
This work was supported by the Grant-in-Aid for Scientific Research (21500803). The authors thank Mari Yachi for technical assistance.
Conflict of interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Itoh, T., Imano, M., Nishida, S. et al. (−)-Epigallocatechin-3-gallate increases the number of neural stem cells around the damaged area after rat traumatic brain injury. J Neural Transm 119, 877–890 (2012). https://doi.org/10.1007/s00702-011-0764-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-011-0764-9