
Abstract
Huntington’s disease (HD) is a progressive neurodegenerative disorder, the pathomechanism of which is not yet fully understood. Excitotoxicity is known to be involved in the development of HD and antiglutamatergic agents may, therefore, have beneficial neuroprotective effects. One of these agents is the tryptophan metabolite kynurenic acid (KYNA), which is an endogenous NMDA receptor antagonist. However, its pharmacological properties rule out its systemic administration in CNS disorders. We have tested a novel KYNA analogue, N-(2-N,N-dimethylaminoethyl)-4-oxo-1H-quinoline-2-carboxamide hydrochloride, in the N171-82Q transgenic mouse model of HD. The analogue exhibited several significant effects: it prolonged the survival of the transgenic mice, ameliorated their hypolocomotion, prevented the loss of weight and completely prevented the atrophy of the striatal neurons. The beneficial effects of this KYNA analogue are probably explained by its complex anti-excitotoxic activity. As it did not induce any appreciable side-effect at the protective dose applied in a chronic dosing regime in this mouse model, it appears worthy of further thorough investigations with a view to eventual clinical trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glutamate-induced excitotoxicity seems to play an important role in the development of Huntington’s disease (HD; Coyle and Schwarcz 1976; McGeer and McGeer 1976; reviewed by DiFiglia 1990), which can be influenced to a great extent by the metabolites of the kynurenine pathway, the main pathway of the tryptophan metabolism (reviewed by Zádori et al. 2009a; Schwarcz et al. 2010). The design and preclinical testing of synthetic drugs targeting this altered metabolic route is therefore particularly important.
Huntington’s disease is an autosomal dominantly inherited progressive neurodegenerative disorder. Characteristically, it is manifested in cognitive, psychiatric and motor symptoms in middle-aged people and ends in death after a course of 15–20 years (reviewed by Gárdián and Vécsei 2004; Walker 2007). With regard to the motor symptoms, in addition to the loss of coordination of voluntary movements and the appearance of involuntary movements, a progressive gait impairment also develops, manifested in brady- and hypokinesia (Thompson et al. 1988; van Vugt et al. 1996). This alteration is particularly important, because it can lead to the loss of independence in motion, resulting in the constant need of care.
One of the neurochemical features of HD is the imbalance of the tryptophan metabolism in the striatum in different stages of the disease. A relative decrease in kynurenic acid (KYNA; Fig. 1a) level and a reduced activity of kynurenine aminotransferase (KAT), the enzyme responsible for KYNA biosynthesis, have been demonstrated in the striatum of HD patients (Beal et al. 1990; Jauch et al. 1995; Guidetti et al. 2004). These alterations have been suggested to be related to the development of the disease. A severe reduction in KAT-I immunoreactivity has also been revealed in the rat striatum after chronic 3-nitropropionic acid treatment (Csillik et al. 2002). Mice with the genomic deletion of another KYNA-producing enzyme, KAT-II, displayed an increased neuronal vulnerability to the intrastriatal administration of quinolinic acid (QUIN), which has been postulated to be the possible endogenous neurotoxin in HD (reviewed by Schwarcz et al. 2010). This finding suggests the importance of KYNA in controlling the neurotoxicity of QUIN (Sapko et al. 2006). This is supported by the fact that an increase in KYNA concentration proves protective in the QUIN model (Harris et al. 1998).

The chemical structures of kynurenic acid (a) and the novel analogue, N-(2-N,N-dimethylaminoethyl)-4-oxo-1H-quinoline-2-carboxamide hydrochloride (b)
Transgenic (tg) mouse models of HD replicate the disturbance in the tryptophan metabolism rather well (Guidetti et al. 2006). Although the neuroprotective KYNA shows unaltered levels in these models, the significant elevation in the concentrations of neurotoxic kynurenine pathway compounds leads to a shift in the metabolism resulting in relative KYNA deficiency. These findings raise the possibility that increasing KYNA effect would be beneficial from a therapeutic aspect. However, the systemic administration of KYNA does not seem a good therapeutic approach for several reasons. In higher doses, its solubility is a limiting factor, it penetrates the blood–brain barrier poorly (Fukui et al. 1991), and it undergoes a rapid clearance from the brain and the body, this clearance being mediated by organic anion transporters (Bahn et al. 2005). To overcome these disadvantages, an amide analogue bearing a water-soluble side-chain, 2-N,N-dimethylaminoethylamine, in the amide moiety, has been synthesised (Fig. 1b). This analogue has already been proved to be beneficial in migraine models (Vámos et al. 2009b, 2010).
Our aim was to test how this novel KYNA analogue, N-(2-N,N-dimethylaminoethyl)-4-oxo-1H-quinoline-2-carboxamide hydrochloride, affects longevity and the behavioural and histopathological properties of the N171-82Q tg mouse model of HD, which model seems to be appropriate for the modelling of the human disease (Schilling et al. 1999). The intracerebroventricular injection of KYNA per se reduces exploratory activity and induces ataxia and stereotypy in rats (mainly at higher doses; Vécsei and Beal 1991), and KYNA has also been shown to be an anxiolytic (Schmitt et al. 1990). We therefore also investigated the possible side-effects of the KYNA analogue on these aspects of the behaviour of wild-type (wt) mice. We additionally tested the stereological differences in the striatum between the wt and N171-82Q tg mice, using unbiased design-based stereological procedures.
Materials and methods
Materials
The KYNA analogue N-(2-N,N-dimethylaminoethyl)-4-oxo-1H-quinoline-2-carboxamide hydrochloride was synthesised starting from KYNA. The synthetic procedure involved an amidation reaction between KYNA and N,N-dimethylethylenediamine in dimethyl-formamide in the presence of 1-hydroxy-benzotriazole by using N,N′-diisopropyl-carbodiimide as coupling reagent, followed by chromatographic purification.
Animals
N171-82Q, also known as B6C3-Tg(HD82Gln)81Dbo/J, tg mice (Jackson Laboratories, Bar Harbor, ME, USA) were maintained on the B6C3F1 wt background in our laboratory and the genotype was identified by PCR from tail biopsies. The mice were housed in cages (at most 5 per cage), with free access to food and water, under standard conditions. Male and female mice were distributed equally between the experimental groups. All animal experiments were carried out in accordance with the European Communities Council Directive (86/609/EEC) and were approved in advance by the local animal care committee.
Treatments
We tested three groups of mice: the control wt mice (n = 10 for survival, n = 8 for open-field test and n = 7 for anatomy and immunohistochemistry), the control N171-82Q tg mice (n = 10 for survival, n = 8 for open-field test and n = 7 for anatomy and immunohistochemistry) and the KYNA analogue-treated N171-82Q tg mice (n = 10 for survival, n = 9 for open-field test and n = 7 for anatomy and immunohistochemistry). The treated N171-82Q tg mice received intraperitoneal injections of the KYNA analogue (100 mg/kg/day, in a volume of 5 ml/kg, dissolved in distilled water, the pH of which was adjusted to 6.5 with NaOH) while the control wt mice and the control N171-82Q tg mice received 0.1 M phosphate-buffered saline (PBS; in a volume of 5 ml/kg) at the same time each day, 5 days per week, from 7 weeks of age.
Survival
The N171-82Q tg mice were treated with the KYNA analogue (n = 10), or PBS (n = 10), according to the regime detailed above, until death occurred. Wt littermates treated with 0.1 M PBS served as controls (n = 10).
Open-field test
Since N171-82Q tg mice display low locomotor activity and exploratory behaviour (Klivényi et al. 2006), we also tested the effects of the KYNA analogue in the open-field paradigm. The treatment regime was the same as in case of the survival time analysis (control wt mice: n = 8, control N171-82Q tg mice: n = 8, treated N171-82Q tg mice: n = 9). We examined the spontaneous locomotor activity and the exploration activity of the mice once a week, 2 h after the treatment, on the same day each week. The tests were performed at the same time of day to avoid possible changes due to the diurnal rhythm. Each mouse was placed in the centre of a box (48 × 48 × 40 cm) and its behaviour was recorded for 5 min with the aid of Conducta 1.0 software (Experimetria Ltd., Budapest, Hungary). The ambulation distance, the mean velocity during ambulation, the duration of immobility and the number of rearings were evaluated for 9 weeks (between 7 and 15 weeks of age). After calculation of the averages for the first, second and third 3 weeks in each group, the three experimental groups were compared. With regard to the locomotor activity of these mice, we report here only the ambulation distance data: the mean velocity during ambulation and the duration of immobility are dependent variables of the ambulation distance and revealed identical results (data not shown).
Testing for possible behavioural side-effects of the KYNA analogue in wt mice
To test for possible behavioural side-effects of the KYNA analogue, another population of wt B6C3F1 mice were divided into two groups (n = 8 in each group). These mice received either the KYNA analogue or 0.1 M PBS for 9 weeks according to the treatment regime detailed above.
First, open-field tests were performed once a week and were evaluated similarly as in the former experiment.
Secondly, we used the elevated plus maze paradigm. This method was applied in the last week of treatment in order to test the influence of the drug administration on the anxiety-like behaviour of these mice. Two hours after the intraperitoneal injection, the mice were placed in the central area of the device (height: 40 cm; length of arms: 30 cm; width of arms: 10 cm; height of walls: 10 cm) facing an open arm opposite the two observers. The numbers of open and closed arm entries and the times spent in these arms were recorded for 5 min.
Thirdly, we applied the conventional stereotypy rating scale (SRS) method (McNamara et al. 2003), also in the last week of treatment, 2 h after the daily injection, to examine the possible stereotypic behaviour-inducing effects of the KYNA analogue. They were removed from their home cages and placed individually in a clean cage for observation. The animals were assessed over a 30-s period and were scored in accordance with the nature of their activity: 0 = asleep or inactive, 1 = episodes of normal activity, 2 = discontinuous activity with bursts of prominent sniffing or rearing, 3 = continuous stereotyped activity, such as sniffing or rearing along a fixed path, 4 = stereotyped sniffing or rearing fixated in one location, 5 = stereotyped behaviour with bursts of licking or gnawing, 6 = continuous licking or gnawing. The examination was repeated on a further two occasions at 20-min intervals, and the scores were averaged.
Anatomy and immunohistochemistry
The body weight of the mice was measured once a week on the same day each week, at the same time of the day, from 7 weeks of age (n = 7 in each group). When the animals reached 16 weeks of age, they were deeply anesthetized with isoflurane (Forane®; Abott Laboratories Hungary Ltd., Budapest, Hungary) and perfused transcardially with 0.9% (wt/vol) NaCl solution, followed by 4% (wt/vol) paraformaldehyde. The perfused brains were removed, weighed, post-fixed with 4% (wt/vol) paraformaldehyde for 24 h and transferred into 0.1 M phosphate buffer (PB; pH 7.4) containing 0.05% (wt/vol) azide. Blocks containing the whole striatum were dissected and incised in the middle, so that the hemispheres could be handled separately. The whole striatum was sectioned on a vibratome at 60-μm thickness and the sections were collected in a systematic random fashion in seven vials. In this way, the sections with a distance of 420 μm (between the upper surfaces) were put into the same glass vials. The sampling was followed by washing in 0.1 M PB and incubation in 10 and 30% (wt/vol) sucrose for cryoprotection. After freeze–thawing, the sections of the left hemisphere were washed repeatedly in 0.1 M PB, and were processed for immunostaining. Following rinsing in 0.05 M Tris-buffered saline (TBS; pH 7.4), the free floating sections were incubated in 1% (wt/wt) hydrogen peroxide in 0.05 M TBS to suppress endogenous peroxidase activity. After washing in 0.05 M TBS and incubation in 2% (vol/vol) normal horse serum (Vector Laboratories Inc., Burlingame, CA, USA) diluted in 0.05 M TBS, sections were incubated in solutions of either anti-NeuN (clone 60; mouse monoclonal antibody; catalogue number MAB377; lot number LV1616015; 1:5,000; Millipore, Bilerica, MA, USA) or anti-huntingtin (clone mEM48, recognising the first 256 amino acids of the human huntingtin protein; mouse monoclonal antibody; catalogue number MAB5374; lot number LV1541616; 1:200; Millipore, Bilerica, MA, USA) primary antibodies diluted in 0.05 M TBS for 35 h at room temperature. After subsequent extensive washes in 0.05 M TBS, the sections were incubated overnight at 4°C with biotinylated donkey anti-mouse (1:1,000; Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA) secondary antibody, diluted in 0.05 M TBS, followed by avidin-biotinylated horseradish peroxidase complex (Elite ABC, 1:333; Vector Laboratories Inc., Burlingame, CA, USA) diluted in 0.05 M TBS for 3 h at room temperature. The immunoperoxidase reaction was developed by using 3′,3′-diaminobenzidine (Sigma-Aldrich Hungary Ltd., Budapest, Hungary) as a chromogen. The sections were treated with osmium tetroxide in PB, then dehydrated in an ascending alcohol series and acetonitrile and embedded in Durcupan (ACM; Fluka, Buchs, Schwitzerland).
Stereology
We used the unbiased design-based stereology method (reviewed by West 2002) to estimate the number and size of the neurons in the striatum. The complete penetration of the antibodies was verified by using the z-axis histograms in the sections, and the osmication procedure ensured the clear identification of cell and nucleus contours, confirming that these samples were suitable for accurate stereological analyses (Fig. 4a–f). The contours of a striatum were traced in each serial section (n = 8–9) according to the online-available Allen Brain Atlas: Mouse Brain (Allen Institute for Brain Science, Seattle, WA, USA; http://www.alleninstitute.org), using a 10× objective of a Zeiss Axioskop 2 (Carl Zeiss MicroImaging Ltd., Göttingen, Germany) and the Stereo Investigator software (MBF Bioscience, Williston, VT, USA). Subsequently, we utilised a 100× objective with oil immersion (numeric aperture: 1.4) for quantification of the anti-NeuN-stained neurons (grid size: 500 × 500 μm) and anti-huntingtin-stained nuclei (grid size: 350 × 350 μm) with the optical fractionator method, applying a counting frame of 40 × 40 μm. As a counting rule, we made use of the unbiased counting brick rule with a brick height of 15 μm and guard zones of 4 μm. On average, we counted ~623 ± 12 anti-NeuN-stained neurons and ~267 ± 49 anti-huntingtin-stained nuclei in each animal in order to estimate the total number of immunoreactive neurons in its striatum, and calculated the radii of ~87 ± 2 neurons per mouse in order to estimate the average volume of its neurons in its striatum.
For estimation of the striatal volume, we used the following formula:
where A i is the area of the left striatum in the consecutive sections, and ssf is the section sampling fraction. 60 is the original section thickness, and the product is multiplied by 2 to calculate the striatal volume for the 2 hemispheres.
The total numbers of anti-NeuN-stained neurons and anti-huntingtin-stained nuclei in the whole striatum were estimated by using the optical fractionator method and the following formula:
where ssf is the section sampling fraction, asf is the area sampling fraction and hsf is the height sampling fraction. The product is likewise multiplied by 2 to calculate the total number of cells in the whole striatum of the two hemispheres.
The isotropic orientation of the striatal neurons allowed the use of the nucleator probe in our samples to estimate neuronal volumes (Schmitz et al. 1999). Here, the systematic random selection of neurons was ensured by the selection of the first neurons coming into focus in each counting brick. The Stereo Investigator software calculated the neuronal volume via the following formula, where r is the average radius of a neuron:
Statistics
All statistical analyses were performed with the help of the Statistica software (StatSoft Inc., 2008, STATISTICA version 8.0; http://www.statsoft.com). Survival data were analysed by using Kaplan–Meier survival curves and the Mantel-Cox log rank test. In the cases of behavioural tests and anatomical analyses, we first checked the distribution of data populations with the Shapiro–Wilk W test. If the data showed a non-Gaussian distribution, we used nonparametric statistics: the Mann–Whitney U test when two groups were compared, and the Kruskal–Wallis test when there were more than two groups. If the data were distributed normally, we used parametric statistics: two groups were compared with the independent t test, and multiple groups of data were compared by using one-way or repeated measures of ANOVA followed by post hoc comparisons. The null hypothesis was rejected when the P level was <0.05, and in such cases the differences were considered significant. In the event of Gaussian or of non-Gaussian distributions, data were plotted as means (±SEM) or medians (and interquartile range) in the graphs, respectively.
Results
Survival
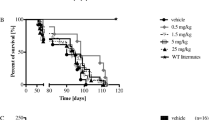
The novel KYNA analogue, administered intraperitoneally at a dose of 100 mg/kg, significantly increased the mean survival time of the N171-82Q tg mice from 111.3 ± 6.6 days to 145.5 ± 12.0 days, that is by 30.7% (P = 0.016, Mantel-Cox log rank test; Fig. 2).

Kaplan-Meier survival curves of the 2 groups of N171-82Q tg mice (n = 10 in each group). One group received PBS, while the other group was treated with the novel kynurenic acid analogue. The treatment with the analogue significantly increased the survival time relative to the controls (P < 0.05, Mantel-Cox log rank test; tg transgenic, KYNA kynurenic acid)
Behavioural tests
Through use of the open-field paradigm, we tested the control wt mice, the N171-82Q tg mice and the KYNA analogue-treated N171-82Q tg mice. We first assessed the locomotor activity of these animals by measuring their ambulation distance in the open-field during the test period for 9 consecutive weeks. During the tests in the first 3 weeks, the N171-82Q tg mice covered a 19.9% shorter distance than the wt mice, this difference proving significant (P = 0.0046, Fischer’s LSD post hoc test), and the KYNA analogue had no effect. During the second 3 weeks, we could already detect some positive changes in the behaviour of the treated N171-82Q tg mice, because a significant difference could no longer be detected between the wt and the treated N171-82Q tg mice. During the third 3 weeks, the KYNA analogue restored the locomotor activity of the treated N171-82Q tg mice to the wt level and their performance became significantly different from that of the non-treated N171-82Q tg mice group (P = 0.0094, Fischer’s LSD post hoc test; Fig. 3a). In the independent control experiments, the KYNA analogue itself did not influence the spontaneous locomotor activity of the wt mice (Fig. 3c).

The effects of the kynurenic acid analogue on the examined behavioural parameters in N171-82Q tg and wt mice. a The analogue significantly ameliorated the characteristic hypolocomotion in the N171-82Q tg mice, examined via the ambulation distance in open-field tests, while c it did not have any effect on the spontaneous locomotor activity of the wt mice. b With regard to the exploratory activity, the analogue did not influence the low rearing count characteristic of the N171-82Q tg animals and d did not exert any significant effect in the wt mice either. e The results of the elevated plus maze test, examined via the percentage of time spent in the closed arms and f the results of the SRS did not reveal any significant difference between the analogue-treated and control wt animals. (wt mice: n = 8; N171-82Q tg control mice: week 7–12: n = 8, week 13–15: n = 7; N171-82Q tg treated mice: week 7–12: n = 9, week 13–15: n = 8; data are means + SEM (a–e) or medians and interquartile range (f); *P < 0.05, **P < 0.01, ***P < 0.001; tg transgenic, wt wild-type, KYNA kynurenic acid, SRS stereotypy rating scale)
We also tested the exploratory behaviour of the same three groups of mice. We found that, whereas the N171-82Q tg mice reared significantly less than the wt mice (by ~65%; P = 0.000062, Fischer’s LSD post hoc test), the KYNA analogue did not change this behaviour significantly, although the difference between the treated and the non-treated mice did become larger by the end of the third 3 weeks (Fig. 3b). In the independent control experiments, the KYNA analogue itself did not influence the exploratory activity of the wt mice (Fig. 3d).
The results of the elevated plus maze test at 15 weeks of age in the wt animals showed that there was no significant difference between the KYNA analogue-treated and untreated groups as concerns the percentage of time spent in the closed arms (Fig. 3e) or the number of closed arm entries (data not shown). The results on the SRS demonstrated that the treatment with the KYNA analogue did not induce the stereotypic behaviour in the wt mice (Fig. 3f).
Body weight
The average body weight of the wt control animals increased continuously until around 14 weeks of age, whereas no increase in body weight was detected in case of the N171-82Q tg mice after the age of 7 weeks, and hence the weights of the two groups were significantly different (P < 0.000001, repeated measures of ANOVA; Fig. 5a). Our results revealed that the treatment increased the body weight of the N171-82Q tg mice significantly (P = 0.00022; repeated measures of ANOVA).
Neuroanatomical changes
With regard to the CNS pathology, the brain weight of 16-week-old N171-82Q tg animals (432.2 ± 10.0 mg) was significantly lower, by 7.9%, than that of the wt mice (469.2 ± 7.6 mg; P = 0.012, independent t test), but that of the treated N171-82Q tg animals (446.8 ± 12.7 mg) was not significantly different (Fig. 5b). The stereologically measured striatal volume was also significantly lower (by 12.6%; P = 0.012, Mann–Whitney U test) in the N171-82Q tg animals (17.4 mm3, interquartile range: 14.6–17.7 mm3) than in the wt mice (18.9 mm3, interquartile range: 18.3–19.0 mm3). When the N171-82Q tg mice were treated with the KYNA analogue, their striatal volume was no longer different from that of the wt mice (19.0 mm3, interquartile range: 17.3–19.4 mm3; Fig. 5c).
Unbiased stereological methods did not indicate any significant difference in the number of neurons in the striatum of the control wt mice, the N171-82Q tg mice or the KYNA analogue-treated N171-82Q tg mice (Fig. 4a, c; Fig. 5d). However, we found that the average size of the neurons in the striatum of the N171-82Q tg animals (663.4 μm3, interquartile range: 605.6–681.0 μm3) was lower by 14% (P = 0.0022, Mann–Whitney U test) than that for the wt mice (739.0 μm3, interquartile range: 734.2–802.1 μm3). The treatment with the KYNA analogue completely prevented the development of atrophy of the neurons in the N171-82Q tg mice (744.5 μm3, interquartile range: 716.2–787.4 μm3), and the average size of these neurons was significantly different from that in the control non-treated N171-82Q tg animals (P = 0.012, Mann–Whitney U test; Fig. 5e).

Low and high-magnification photomicrographs of the striata from 16-week-old wt and N171-82Q tg mice. Such representative light micrographs do not show any obvious difference in the number of anti-NeuN-stained neurons between the wt (a) and N171-82Q tg animals (c). As concerns the EM48+ immunoreactivity, no labelled nucleus was observed in the wt mice (b), whereas cells could readily be observed in the N171-82Q tg animals (d). Stereological investigations at high magnification clearly revealed the contours of anti-NeuN-stained cells (e) and EM48+ nuclei (f). (tg transgenic, wt wild-type; scale bar a–d: 100 μm, e–f: 10 μm)

Effects of treatment with the KYNA analogue on the anatomical features of the mice. a A significant difference emerged between the body weights of wt and N171-82Q tg animals with aging (n = 7; P < 0.000001, repeated measures of ANOVA), which was significantly ameliorated by the treatment (P < 0.001, repeated measures of ANOVA). The treatment abolished the significant differences in brain weight (b) and striatal volume (c) between the wt and N171-82Q tg animals. Although the number of striatal neurons did not decrease significantly by the 16-weeks of age (d), obvious neuronal atrophy could be observed in the N171-82Q tg animals, which was completely prevented by the treatment with the KYNA analogue (e). (f) As concerns the aggregation pathology, the treatment did not change the percentage of EM48+ neurons in the striatum significantly. (data are means + SEM (a, b, f) or medians and interquartile range (c, d, e); *P < 0.05, **P < 0.01; tg transgenic, wt wild-type, KYNA kynurenic acid)
The treatment with the KYNA analogue did not induce a significant change in the percentage of anti-huntingtin immunoreactive (EM48+) neurons in the striatum of the N171-82Q tg animals (17.5% of the total neuron population) relative to the control N171-82Q tg animals (24.1% of the total neuron population; Fig. 5f).
Discussion
Although HD is caused by expansion of the cytosine–adenine–guanine (CAG) repeat in the gene coding for the N-terminal region of the huntingtin protein, the deleterious effect on neurons can not be explained by the expression pattern of the gene itself in the CNS (Fusco et al. 1999). However, the expression of the mutant huntingtin protein can sensitise the NR2B subunit containing NMDA receptors (Chen et al. 1999). Indeed, there is accumulating evidence that glutamate-induced excitotoxicity is mainly mediated by these NMDA receptor channels (Liu et al. 2007; Heng et al. 2009), which are abundantly expressed by the most vulnerable metenkephalinergic-GABAergic medium-sized spiny neurons (MSNs). The specific impairment of the MSNs may be due to the massive glutamatergic input from the cortex and thalamus (Fonnum et al. 1981; reviewed by Smith et al. 2004), or because the NMDA receptors have an especially high density on these neurons (Landwehrmeyer et al. 1995). QUIN, a possible culprit endogenous excitotoxin in HD, may also exert its deleterious effects via direct activation of the above-mentioned NMDA receptors (Stone and Perkins 1981; de Carvalho et al. 1996). Furthermore, QUIN can facilitate the release of glutamate or can inhibit its uptake (Connick and Stone 1988; Tavares et al. 2002).
From clinical aspects, glycine and polyamine site agents, NR2B subunit specific antagonists and ion channel blockers with lower affinity may come into consideration as NMDA receptor antagonists, as they exert acceptable side-effects (reviewed by Muir 2006). KYNA would theoretically be a good candidate, because it is a broad-spectrum endogenous antagonist on ionotropic excitatory amino acid receptors (Perkins and Stone 1982; Birch et al. 1988). KYNA can inhibit NMDA receptors at the glycine-binding sites (Kessler et al. 1989) and it can non-competitively inhibit α7-nicotinic acetylcholine receptors (Hilmas et al. 2001). Blockade of these nicotinic receptors can also mediate the inhibition of glutamate release in the striatum (Marchi et al. 2002). However, from pharmacological considerations, KYNA itself has several disadvantages, which rule out its systemic use in the treatment of HD. Accordingly, several new KYNA analogues or prodrugs have been designed (reviewed by Schwarcz 2004). An important group of these compounds are the KYNA amides (including our novel agent), which are excellent candidates (reviewed by Fülöp et al. 2009). A test group of these analogues showed selective inhibition of the NR2B subunit containing NMDA receptors, too, serving a possible additional target of drug action (Borza et al. 2007). In preliminary collaborative studies, using mass spectrometric analyses, we managed to detect our novel KYNA amide in the rat cerebrospinal fluid after a single intraperitoneal injection (data are not shown). Furthermore, the analogue showed a relatively prolonged clearance from the mouse serum with a detectable presence in the serum after an observation period of 6 h. Nevertheless, the detailed pharmacological characterisation, involving the thorough examination of penetrance through the blood–brain barrier and brain pharmacokinetics and pharmacodynamics, needs further experiments and after completing those studies, the results will be published separately in the near future.
In this study, we focused on testing our novel KYNA amide in the N171-82Q tg mouse model of HD. This analogue increased the survival time of the N171-82Q tg mice significantly (by ~31%). This elevation is noteworthy: the most effective agent in this model, probenecid, lengthened the survival by ~35% (Vámos et al. 2009a), while valproate did so by 31% (Zádori et al. 2009b), tiagabine by 26% (Masuda et al. 2008) and phenylbutyrate by 23% (Gárdián et al. 2005). It is important to mention that our treatment did not simply shift the survival curve of the N171-82Q tg animals, but altered the slope of the curve, i.e., the longer the treatment period, the more pronounced the prolongation of survival duration was. So our compound seems to slow down the progress of neurodegeneration instead of only delaying the onset of mortality (like in case of valproate or phenylbutyrate treatment).
The beneficial effect of this KYNA amide is supported by our findings in the open-field tests. The KYNA analogue treatment ameliorated the hypolocomotion of the N171-82Q tg mice, because the locomotor activity of the N171-82Q tg mice had improved to the wt level by the end of the 9-week treatment period. As the KYNA analogue did not influence the locomotor or exploratory activity of the wt mice and did not induce anxiolysis or stereotypy at the dose administered in the wt mice, the change in hypolocomotion is not due to any behavioural side-effect of the KYNA amide treatment.
The body weight data also support the above findings, for the treatment significantly mitigated the characteristic phenomenon that the body weight of the N171-82Q tg mice does not increase further after 2 months of age. Furthermore, our brain pathology data are in accordance with the findings of Schilling et al. (1999; the first characterization of N171-82Q tg mice), who reported that the brains of tg animals are slightly smaller (by 7.9% in our experiment). However, precise unbiased design-based stereological procedures were necessary to investigate the number of neurons and their size in the striatum, in order to reveal the possible pathomechanism of HD in these N171-82Q tg animals at the cellular level. In parallel with the difference in brain weights, the volume of the striatum was lower (by 12.6%) in the N171-82Q tg animals. This phenomenon can be well explained by the 14% lower neuronal volume. This suggests that neuronal atrophy plays a more important role in the pathomechanism in the N171-82Q tg mouse model of HD than the possible final decrease in the number of neurons. This observation is in accordance with the original characterization of these N171-82Q tg mice, which did not indicate a severe loss of neurons in the striatum. Indeed, it has been demonstrated that some drugs have beneficial effects on neuronal atrophy (e.g., Ferrante et al. 2002; Gárdián et al. 2005; Stack et al. 2010), it therefore seems to be a good parameter via which to test neuroprotective effects of drugs. In our experiments with the novel KYNA amide, the neuronal atrophy was nearly completely mitigated. As concerns the aggregation pathology, we investigated the percentage of EM48+ nuclei in the striatum. This revealed that the treatment led to a slight, but not significant improvement.
In summary, administration of the novel KYNA analogue can prevent or alleviate the deleterious effects of HD in the striatum in the N171-82Q tg mouse model. We did not detect any side-effect of this analogue at the neuroprotective dose applied, and this compound may therefore be considered a promising candidate for clinical trials.
References
Bahn A, Ljubojevic M, Lorenz H, Schultz C, Ghebremedhin E, Ugele B, Sabolic I, Burckhardt G, Hagos Y (2005) Murine renal organic anion transporters mOAT1 and mOAT3 facilitate the transport of neuroactive tryptophan metabolites. Am J Physiol Cell Physiol 289:C1075–C1084
Beal MF, Matson WR, Swartz KJ, Gamache PH, Bird ED (1990) Kynurenine pathway measurements in Huntington’s disease striatum: evidence for reduced formation of kynurenic acid. J Neurochem 55:1327–1339
Birch PJ, Grossman CJ, Hayes AG (1988) Kynurenate and FG9041 have both competitive and non-competitive antagonist actions at excitatory amino acid receptors. Eur J Pharmacol 151:313–315
Borza I, Kolok S, Galgóczy K, Gere A, Horváth C, Farkas S, Greiner I, Domány G (2007) Kynurenic acid amides as novel NR2B selective NMDA receptor antagonists. Bioorg Med Chem Lett 17:406–409
Chen N, Luo T, Wellington C, Metzler M, McCutcheon K, Hayden MR, Raymond LA (1999) Subtype-specific enhancement of NMDA receptor currents by mutant huntingtin. J Neurochem 72:1890–1898
Connick JH, Stone TW (1988) Quinolinic acid effects on amino acid release from the rat cerebral cortex in vitro and in vivo. Br J Pharmacol 93:868–876
Coyle JT, Schwarcz R (1976) Lesion of striatal neurons with kainic acid provides a model for Huntington’s chorea. Nature 263:244–246
Csillik A, Knyihár E, Okuno E, Krisztin-Péva B, Csillik B, Vécsei L (2002) Effect of 3-nitropropionic acid on kynurenine aminotransferase in rat brain. Exp Neurol 177:233–241
de Carvalho LP, Bochet P, Rossier J (1996) The endogenous agonist quinolinic acid and the non endogenous homoquinolinic acid discriminate between NMDAR2 receptor subunits. Neurochem Int 28:445–452
DiFiglia M (1990) Excitotoxic injury of the neostriatum: a model for Huntington’s disease. Trends Neurosci 13:286–289
Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF (2002) Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington’s disease. J Neurosci 22:1592–1599
Fonnum F, Storm-Mathisen J, Divac I (1981) Biochemical evidence for glutamate as neurotransmitter in corticostriatal and corticothalamic fibres in rat brain. Neuroscience 6:863–873
Fukui S, Schwarcz R, Rapoport SI, Takada Y, Smith OR (1991) Blood-brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J Neurochem 56:2007–2017
Fülöp F, Szatmári I, Vámos E, Zádori D, Toldi J, Vécsei L (2009) Synthesis, transformations and pharmaceutical applications of kynurenic acid derivatives. Curr Med Chem 16:4828–4842
Fusco FR, Chen Q, Lamoreaux WJ, Figueredo-Cardenas G, Jiao Y, Coffman JA, Surmeier DJ, Honig MG, Carlock LR, Reiner A (1999) Cellular localization of huntingtin in striatal and cortical neurons in rats: lack of correlation with neuronal vulnerability in Huntington’s disease. J Neurosci 19:1189–1202
Gárdián G, Vécsei L (2004) Huntington’s disease: pathomechanism and therapeutic perspectives. J Neural Transm 111:1485–1494
Gárdián G, Browne SE, Choi DK, Klivényi P, Gregorio J, Kubilus JK, Ryu H, Langley B, Ratan RR, Ferrante RJ, Beal MF (2005) Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J Biol Chem 280:556–563
Guidetti P, Luthi-Carter RE, Augood SJ, Schwarcz R (2004) Neostriatal and cortical quinolinate levels are increased in early grade Huntington’s disease. Neurobiol Dis 17:455–461
Guidetti P, Bates GP, Graham RK, Hayden MR, Leavitt BR, MacDonald ME, Slow EJ, Wheeler VC, Woodman B, Schwarcz R (2006) Elevated brain 3-hydroxykynurenine and quinolinate levels in Huntington disease mice. Neurobiol Dis 23:190–197
Harris CA, Miranda AF, Tanguay JJ, Boegman RJ, Beninger RJ, Jhamandas K (1998) Modulation of striatal quinolinate neurotoxicity by elevation of endogenous brain kynurenic acid. Br J Pharmacol 124:391–399
Heng MY, Detloff PJ, Wang PL, Tsien JZ, Albin RL (2009) In vivo evidence for NMDA receptor-mediated excitotoxicity in a murine genetic model of Huntington disease. J Neurosci 29:3200–3205
Hilmas C, Pereira EF, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX (2001) The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci 21:7463–7473
Jauch D, Urbańska EM, Guidetti P, Bird ED, Vonsattel JP, Whetsell WO Jr, Schwarcz R (1995) Dysfunction of brain kynurenic acid metabolism in Huntington’s disease: focus on kynurenine aminotransferases. J Neurol Sci 130:39–47
Kessler M, Terramani T, Lynch G, Baudry M (1989) A glycine site associated with N-methyl-d-aspartic acid receptors: characterization and identification of a new class of antagonists. J Neurochem 52:1319–1328
Klivényi P, Bende Z, Hartai Z, Penke Z, Németh H, Toldi J, Vécsei L (2006) Behaviour changes in a transgenic model of Huntington’s disease. Behav Brain Res 169:137–141
Landwehrmeyer GB, Standaert DG, Testa CM, Penney JB Jr, Young AB (1995) NMDA receptor subunit mRNA expression by projection neurons and interneurons in rat striatum. J Neurosci 15:5297–5307
Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT (2007) NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci 27:2846–2857
Marchi M, Risso F, Viola C, Cavazzani P, Raiteri M (2002) Direct evidence that release-stimulating alpha7* nicotinic cholinergic receptors are localized on human and rat brain glutamatergic axon terminals. J Neurochem 80:1071–1078
Masuda N, Peng Q, Li Q, Jiang M, Liang Y, Wang X, Zhao M, Wang W, Ross CA, Duan W (2008) Tiagabine is neuroprotective in the N171-82Q and R6/2 mouse models of Huntington’s disease. Neurobiol Dis 30:293–302
McGeer EG, McGeer PL (1976) Duplication of biochemical changes of Huntington’s chorea by intrastriatal injections of glutamic and kainic acids. Nature 263:517–519
McNamara FN, Clifford JJ, Tighe O, Kinsella A, Drago J, Croke DT, Waddington JL (2003) Congenic D1A dopamine receptor mutants: ethologically based resolution of behavioural topography indicates genetic background as a determinant of knockout phenotype. Neuropsychopharmacology 28:86–99
Muir KW (2006) Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr Opin Pharmacol 6:53–60
Perkins MN, Stone TW (1982) An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res 247:184–187
Sapko MT, Guidetti P, Yu P, Tagle DA, Pellicciari R, Schwarcz R (2006) Endogenous kynurenate controls the vulnerability of striatal neurons to quinolinate: implications for Huntington’s disease. Exp Neurol 197:31–40
Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR (1999) Intranuclear inclusions and neuritic aggregates in transgenic mice expressing mutant N-terminal fragment of huntingtin. Hum Mol Gen 8:397–407
Schmitt ML, Graeff FG, Carobrez AP (1990) Anxiolytic effect of kynurenic acid microinjected into the dorsal periaqueductal gray matter of rats placed in the elevated plus-maze test. Braz J Med Biol Res 23:677–679
Schmitz C, Schuster D, Niessen P, Korr H (1999) No difference between estimated mean nuclear volumes of various types of neurons in the mouse brain obtained on either isotropic uniform random sections or conventional frontal or sagittal sections. J Neurosci Methods 88:71–82
Schwarcz R (2004) The kynurenine pathway of tryptophan degradation as a drug target. Curr Opin Pharmacol 4:12–17
Schwarcz R, Guidetti P, Sathyasaikumar KV, Muchowski PJ (2010) Of mice, rats and men: revisiting the quinolinic acid hypothesis of Huntington’s disease. Prog Neurobiol 90:230–245
Smith Y, Raju DV, Pare JF, Sidibe M (2004) The thalamostriatal system: a highly specific network of the basal ganglia circuitry. Trends Neurosci 27:520–527
Stack C, Ho D, Wille E, Calingasan NY, Williams C, Liby K, Sporn M, Dumont M, Beal MF (2010) Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington’s disease. Free Radic Biol Med 49:147–158
Stone TW, Perkins MN (1981) Quinolinic acid: a potent endogenous excitant at amino acid receptors in CNS. Eur J Pharmacol 72:411–412
Tavares RG, Tasca CI, Santos CE, Alves LB, Porciúncula LO, Emanuelli T, Souza DO (2002) Quinolinic acid stimulates synaptosomal glutamate release and inhibits glutamate uptake into astrocytes. Neurochem Int 40:621–627
Thompson PD, Berardelli A, Rothwell JC, Day BL, Dick JP, Benecke R, Marsden CD (1988) The coexistence of bradykinesia and chorea in Huntington’s disease and its implications for theories of basal ganglia control of movement. Brain 111:223–244
Vámos E, Vörös K, Zádori D, Vécsei L, Klivényi P (2009a) Neuroprotective effects of probenecid in a transgenic animal model of Huntington’s disease. J Neural Transm 116:1079–1086
Vámos E, Párdutz Á, Varga H, Bohár Z, Tajti J, Fülöp F, Toldi J, Vécsei L (2009b) l-kynurenine combined with probenecid and the novel synthetic kynurenic acid derivative attenuate nitroglycerin-induced nNOS in the rat caudal trigeminal nucleus. Neuropharmacology 57:425–429
Vámos E, Fejes A, Koch J, Tajti J, Fülöp F, Toldi J, Párdutz Á, Vécsei L (2010) Kynurenate derivative attenuates the nitroglycerin-induced CamKIIalpha and CGRP expression changes. Headache 50:834–843
van Vugt JP, van Hilten BJ, Roos RA (1996) Hypokinesia in Huntington’s disease. Mov Disord 4:384–388
Vécsei L, Beal MF (1991) Comparative behavioral and pharmacological studies with centrally administered kynurenine and kynurenic acid in rats. Eur J Pharmacol 196:239–246
Walker FO (2007) Huntington’s disease. Semin Neurol 27:143–150
West NJ (2002) Design-based stereological methods for counting neurons. Prog Brain Res 135:43–51
Zádori D, Klivényi P, Vámos E, Fülöp F, Toldi J, Vécsei L (2009a) Kynurenines in chronic neurodegenerative disorders: future therapeutic strategies. J Neural Transm 116:1403–1409
Zádori D, Geisz A, Vámos E, Vécsei L, Klivényi P (2009b) Valproate ameliorates the survival and the motor performance in a transgenic mouse model of Huntington’s disease. Pharmacol Biochem Behav 94:148–153
Acknowledgments
This work was supported by grants ETT 026-04, NAP-BIO-06-BAYBIOSZ, TÁMOP-4.2.2-08/1/2008-0002, NIH (DA09158, NS030549) and OTKA (K75628). Gábor Nyiri was supported by János Bolyai Research Fellowship. We thank Andrea Varga and Gábor Patkovics for their technical assistance.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zádori, D., Nyiri, G., Szőnyi, A. et al. Neuroprotective effects of a novel kynurenic acid analogue in a transgenic mouse model of Huntington’s disease. J Neural Transm 118, 865–875 (2011). https://doi.org/10.1007/s00702-010-0573-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-010-0573-6