
Abstract
Some studies suggest a higher risk of hypertension in people with epilepsy. Captopril, a potent and selective angiotensin-converting enzyme (ACE) inhibitor, is a well known antihypertensive drug. Besides the peripheral renin–angiotensin system (RAS), ACE inhibitors are also suggested to affect the brain RAS which might participate in the regulation of seizure susceptibility. The purpose of the current study was to evaluate the effect of captopril on the protective action of numerous antiepileptic drugs (carbamazepine [CBZ], phenytoin [PHT], valproate [VPA], phenobarbital [PB], oxcarbazepine [OXC], lamotrigine [LTG] and topiramate [TPM]) against maximal electroshock-induced seizures in mice. This study was accompanied by an evaluation of adverse effects of combined treatment with captopril and antiepileptic drugs in the passive avoidance task and chimney test. Captopril (25 and 50 mg/kg i.p.) did not influence the threshold for electroconvulsions. Among the tested antiepileptics, captopril (25 and 50 mg/kg i.p.) potentiated the antiseizure action of CBZ, decreasing its ED50 value from 12.1 to 8.9 and 8.7 mg/kg, respectively. Moreover, captopril (50 mg/kg i.p.) enhanced the anticonvulsant activity of LTG. ED50 value for LTG was lowered from 5.1 to 3.5 mg/kg. The observed interactions between captopril and CBZ or LTG were pharmacodynamic in nature as captopril did not alter plasma and total brain concentrations of these antiepileptics. The combinations of captopril with antiepileptic drugs did not lead to retention deficits in the passive avoidance task or motor impairment in the chimney test. Based on the current preclinical data, it is suggested that captopril may positively interact with CBZ and LTG in epileptic patients. The combinations of captopril with the remaining antiepileptics (PHT, VPA, PB, OXC and TPM) seem neutral.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A number of studies suggest a higher risk of hypertension in epileptic patients (Gaitatzis et al. 2004). Moreover, a history of hypertension appears to be an independent risk factor for new-onset unprovoked seizures, especially, but not only, in conjunction with a history of stroke (Ng et al. 1993). A lot of hypertensive patients suffer from increased arterial blood pressure, as a result of increased activity of renin–angiotensin system (RAS) (Laragh et al. 1980). Angiotensin-converting enzyme (ACE), a zinc metalloprotease, catalyzes the formation of angiotensin II, a potent vasoconstrictor, from circulating angiotensin I (Johnston 1990). By blocking the formation of angiotensin II in blood, ACE inhibitors significantly lower systemic vascular resistance, lower blood pressure and improve cardiac function (Thind 1990). Besides the peripheral RAS, all components (precursors and enzymes) of RAS exist in the brain (Wright and Harding 1997). In different areas of the brain, angiotensin II produces its effects acting at specific angiotensin receptors (AT1, AT2) (de Gasparo et al. 2000). The brain RAS is thought to participate in the control of the cardiovascular system, thirst, stress, memory and depression (Wright et al. 2008). In addition, this system can be involved in the regulation of seizure susceptibility as angiotensin peptides administered intracerebroventricularly (i.c.v.) appear to be anticonvulsant in experimental seizure models (Tchekalarova and Georgiev 2005).
Captopril, (D-3-mercapto-2-methylpropanoyl)-l-proline), an antihypertensive drug, is a sulfhydryl compound and a potent and selective inhibitor of ACE (Rubin et al. 1978). It can cross the blood–brain barrier and inhibit the brain RAS after systemic administration (Evered et al. 1980). Accumulating research data show that reduction in central function of angiotensin II by ACE inhibitors may lead to antidepressant and anxiolytic effects (Gard 2002). Furthermore, ACE inhibitors, such as captopril have been reported to improve learning in animals and humans (Braszko et al. 2003; Gard 2002). Interestingly, captopril has been also reported to affect convulsions in animal models. Captopril protected mice against convulsions induced by strychnine, although the drug was ineffective in bicuculline-induced seizures in mice (Minano et al. 1987). Taking into consideration, the anticonvulsant-like activity of captopril, we sought to evaluate the effect of captopril on the threshold for electroconvulsions and to assess its influence on the anticonvulsant action of numerous antiepileptic drugs (carbamazepine [CBZ], phenytoin [PHT], valproate [VPA], phenobarbital [PB], oxcarbazepine [OXC], lamotrigine [LTG] and topiramate [TPM]) against maximal electroshock-induced seizures (MES) in mice. The threshold for electroconvulsions and the MES test are regarded as experimental models of tonic–clonic seizures (Löscher et al. 1991). Among tested antiepileptics CBZ, VPA, LTG and TPM are thought to be first-line drugs, and OXC a second-line drug for generalized tonic–clonic seizures while PHT and PB can be considered in this type of seizures in adult patients (Duncan et al. 2006). These studies were accompanied by an evaluation of adverse effects of combined treatment with captopril and antiepileptic drugs. Captopril was administered intraperitoneally (i.p.) at doses enough to enter the brain and produce effective ACE inhibition (Evered et al. 1980; Minano et al. 1987).
Materials and methods
Animals
Male Swiss mice, weighing 20–26 g, were purchased from a licensed dealer (T. Górzkowska, Warsaw, Poland). The animals were housed in colony cages with free access to food (chow pellets) and tap water ad libitum. The laboratory temperature was 21 ± 1°C and the mice were kept on a 12 h light–dark cycle. The experimental groups consisting of eight animals were made up at random. Each mouse was used only once. All experimental procedures run in this study were approved by the Local Ethics Committee for Animal Experiments.
Drugs
Captopril (Captopril, Jelfa S.A., Poland), carbamazepine (Amizepin, Polpharma S.A., Poland), valproate magnesium (Dipromal, ICN Polfa S.A., Poland), phenytoin (Phenytoinum, Polfa, Poland), phenobarbital (Luminalum, Unia, Poland), oxcarbazepine (Trileptal, Novartis Pharma GmbH, Germany), lamotrigine (Lamitrin, GlaxoSmithKline, UK) and topiramate (Topamax, Janssen-Cilag International N.V., Belgium) were used in this study. VPA was directly dissolved in distilled water. Captopril, CBZ, PHT, PB, OXC, LTG and TPM were suspended in a 1% solution of Tween 80 (Sigma, St. Louis, MO, USA) in distilled water. All drugs were injected intraperitoneally (i.p.) in a volume of 5 ml/kg body weight and administered 120 min (PHT), 60 min (PB, LTG and TPM), 45 min (captopril) or 30 min (CBZ, VPA and OXC) before the tests. Control animals received injections of the vehicle. The route of systemic (i.p.) administration, doses and pretreatment time before testing of captopril was based on information about its biological activity from the literature (Minano et al. 1987).
Electroconvulsions
Electroconvulsions (50 Hz, 500 V) were produced by a Hugo Sachs generator (Rodent Shocker, Type 221, Freiburg, Germany) with the use of auricular electrodes. The stimulus duration was 0.2 s. Full-tonic extension of both hind limbs was taken as the endpoint. The convulsive threshold was evaluated as CS50, which is the current strength (mA) required to produce tonic hind limb extension in 50% of the animals tested. To calculate the convulsive threshold, at least three groups of mice were challenged with electroshocks of various intensities. An intensity–response curve was calculated with a computer based on a percentage of animals convulsing in experimental groups.
The protective activities of antiepileptic drugs were evaluated as their median effective doses (ED50 values in mg/kg) against the maximal electroshock-induced seizures. In the MES test, a fixed current intensity of 25 mA was applied. To evaluate the respective ED50 value, at least three groups of mice, after receiving progressive doses of an antiepileptic, were challenged with the maximal electroshock. A dose–response curve for each antiepileptic was subsequently constructed on the basis of a percentage of animals protected against the convulsions.
Passive avoidance test
The effect of the drugs on memory retention was assessed in the passive avoidance test (Venault et al. 1986). The mice were administered captopril alone or in combinations with antiepileptic drugs on the first day before training. The times at which mice were injected with these drugs were the same as in the MES test. The pretreated mice were placed in an illuminated box (12 × 20 × 15 cm) connected to a dark box (24 × 20 × 15 cm) that was equipped with an electric grid floor. A 4 × 7 cm doorway was located at floor level in the center of the connecting wall. Immediately, after the mouse entered the dark box, it was punished by an electric foot shock (0.6 mA for 2 s). After 24 h of the training trial, the retention test was conducted in which the same animals without any treatment were put into the illuminated box and the latency (the time) to enter the dark box was recorded for 180 s. The mice that avoided the dark compartment for 180 s were considered to remember the task.
Chimney test
The effect of the drugs on motor performance was evaluated with the chimney test of Boissier et al. (1960). Motor impairment was indicated as the inability of mice to climb backward up the plastic tube (3 cm inner diameter, 25 cm in length) within 60 s. The times during which mice were injected with captopril alone or in combinations with antiepileptics in the chimney test were the same as in the MES test.
Immunofluorescence estimation of plasma and brain concentrations of carbamazepine
The measurement of the free (non-protein bound) plasma and total brain concentrations of CBZ was undertaken at the doses of the antiepileptic drug, which corresponded to its ED50 values for the combinations with captopril (25 or 50 mg/kg) in the MES test. The mice were decapitated at times scheduled for the convulsive test and blood samples of approximately 1 ml were collected into heparinized Eppendorf tubes. Simultaneously, the whole brains of mice were removed from skulls, weighed and homogenized using Abbott buffer (1:2 weight/volume) in an Ultra-Turrax T8 homogenizer (Staufen, Germany). The homogenates were centrifuged at 10,000×g for 10 min. Blood samples were centrifuged at 5,000×g for 5 min, and plasma samples of 300 μl were pipetted into a micropartition system, MPS-1 (Amicon, Danvers, MA, USA), for separation of free from protein-bound microsolutes. Then, the MPS-1 tubes were centrifuged at 5,000×g for 10 min, and 75 μl filtrate samples or 75 μl supernatant samples were analyzed for carbamazepine content by fluorescence polarization immunoassay using a TDx analyzer and reagents exactly as described by the manufacturer (Abbott Laboratories, North Chicago, IL, USA). The free plasma and total brain concentrations of CBZ were expressed in μg/ml of plasma or brain supernatant as mean ± SD of eight determinations.
Chromatographic determination of lamotrigine plasma and brain concentrations
The quantitative determination of the plasma and total brain concentrations of LTG was undertaken at the dose of the antiepileptic drug, which corresponded to its ED50 value for the combination with captopril (50 mg/kg) in the MES test. The mice were killed by decapitation at times scheduled for the convulsive test and blood samples of approximately 1 ml were rapidly collected into heparinized Eppendorf tubes. Simultaneously, brains of mice were removed from skulls and placed into the deep freeze at −80°C. Samples of blood were centrifuged at 5,000×g for 5 min, and plasma samples of 200 ml were stocked into the deep freeze. On the next day, the brains were weighed and homogenized with a presence of Abbott buffer (1:2 weight/volume) using the Ultra-Turrax T8 homogenizer. The homogenates were centrifuged at 10,000×g for 10 min. Plasma and brain homogenate samples were prepared for analysis as follows: 200 μl of samples were pipetted into a 1.5 ml plastic tube to which was added 200 μl of 0.08 M triethylammonium phosphate buffer solution, 400 μl of acetonitrile and vortex-mixed for 1 min. After centrifugation (10,000×g for 10 min) in centrifugal filter devices (Millipore Corporation), the organic layer was removed and 20 μl of the aqueous phase was injected into HPLC system. The chromatograph (Dionex, Sunnyvale, CA, USA) was equipped with a gradient pump P580 LPG and a UV/VIS detector (UVD 340S) with a sensitivity setting of 0.1 absorbance units full scale (AUFS) and a time constant of 0.1 s. The Rheodyne 3601 injector valve with a 20 μl sample loop was used for sample injection. For HPLC, a stainless steel HP ODS column (200 × 4.6 mm) was used at an ambient temperature. The mobile phase was 40 mM triethylammonium phosphate buffer:methanol:acetonitrile (660:80:160 vol/vol/vol; Fluka, HPLC grade). The mobile phase flow rate was 1.2 ml/min, and LTG absorbance was measured at 214 nm. The peak height for LTG was linearly related to its concentrations, which ranged from 0.16 to 5.0 g/ml. Plasma levels or total brain concentrations of LTG were expressed in μg/ml of plasma or supernatant as mean ± SD of eight determinations.
Statistics
CS50 and ED50 values with their 95% confidence limits were calculated by computer log-probit analysis according to Litchfield and Wilcoxon (1949). Subsequently, the obtained 95% confidence limits were transformed to standard errors of the mean (SE) as described previously (Luszczki and Czuczwar 2005). The effect of captopril on the convulsive threshold was analyzed with one-way analysis of variance (ANOVA) and post hoc Dunett’s test for multiple comparisons. The anticonvulsant activities of tested antiepileptics injected alone or co-administered with captopril were analyzed using the log-probit method for single comparisons or one-way ANOVA followed by the post hoc Dunett’s test. A Kruskal–Wallis non-parametric ANOVA followed by Dunn’s multiple comparisons test was used to calculate results from the passive avoidance task. The data obtained in the chimney test were compared using Fisher’s exact probability test. Plasma and total brain concentrations of CBZ and LTG injected alone or in combination with captopril were analyzed using unpaired Student’s t test. Group differences were considered statistically significant at P < 0.05.
Results
Electroconvulsions
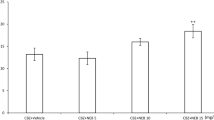
As shown in Fig. 1, captopril (25 and 50 mg/kg i.p.) did not significantly affect the threshold for electroconvulsions in mice. In the MES test, captopril at the dose of 25 and 50 mg/kg i.p. potentiated the anticonvulsant activity of CBZ, by reducing its ED50 value from 12.1 to 8.9 and 8.7 mg/kg, respectively (P < 0.01). Captopril (50 mg/kg i.p.) also potentiated the antiseizure action of LTG, lowering its ED50 value from 5.1 to 3.5 mg/kg (P < 0.05). The protective action of the remaining antiepileptics: PB, PHT, VPA, OXC and TPM were not influenced by captopril (Table 1).

Effect of captopril on the threshold for electroconvulsions in mice. Captopril (25 and 50 mg/kg i.p.) was administered 45 min before the test. The presented data are median current strengths (CS50 in mA) with SE values. Number of animals at those current strengths, whose convulsant effects ranged between 4 and 6 probit (16 and 84%) according to Litchfield and Wilcoxon (1949) and is indicated within each column. Not significant versus control group (ANOVA/Dunnett’s test)
Passive avoidance and chimney test
Captopril (50 mg/kg) did not impair memory retention in the passive avoidance task and motor performance in the chimney test. Similarly, co-administration of captopril (50 mg/kg) and the tested antiepileptics at their ED50 values did not significantly affect the performance of mice in both tests (data not shown). In combinations of captopril with PB, PHT or LTG, one mice per group failed to perform in the chimney test (results not shown).
Influence of captopril on plasma and total brain concentrations of CBZ and LTG
Combination of captopril (25 or 50 mg/kg) with CBZ did not affect either plasma or total brain concentrations of CBZ (Table 2). Captopril (50 mg/kg) did not significantly change the level of plasma and brain concentrations of LTG either (Table 3).
Discussion
The current data show that captopril, a potent inhibitor of ACE activity in the brain (Cushman et al. 1989; Evered et al. 1980), enhanced the anticonvulsant activity of CBZ and LTG against MES-induced seizures. Captopril (up to 50 mg/kg) did not affect the threshold for electroconvulsions. Captopril did not alter either plasma or total brain concentrations of CBZ and LTG; hence, the observed interactions between captopril and these antiepileptics were pharmacodynamic in nature. Moreover, captopril also showed a strong tendency to reduce the ED50 value for PHT, however, without statistical significance.
It has been earlier reported that captopril, a sulfhydryl compound, but not non-sulfhydryl ACE inhibitor enalapril, produced a dose-dependent anticonvulsant effect against strychnine-induced seizures (Minano et al. 1987). Strychnine, a highly selective and potent competitive antagonist of glycine, is a convulsant acting at strychnine-sensitive inhibitory glycine receptors in the spinal cord and brainstem (Rajendra et al. 1997; Sangiah 1985). Therefore, it was suggested that the anticonvulsant activity of captopril is mediated by an action on spinal postsynaptic glycine inhibition and/or that a sulfhydryl group of captopril may, at least in part, account for its protective action (Minano et al. 1987). The sulfhydryl group is known to covalently bind to biochemicals and alter their chemical properties and biological activity (Mojaverian et al. 1984). Further, similarly to captopril, CBZ and LTG were also protective against strychnine-evoked convulsions and CBZ was the most effective anticonvulsant among tested antiepileptics (Yamashita et al. 2004). PHT showed weaker anticonvulsant action and it was unable to prevent strychnine-induced lethality as compared to CBZ (Kubová and Mares 1994; Yamashita et al. 2004). On the other hand, the anticonvulsive potency of LTG (Borowicz et al. 2002) and classical antiepileptics, VPA, PB and PHT, was reduced by strychnine administration while the antiseizure action of CBZ was not changed by strychnine in the MES test (Czuczwar et al. 1991). Noteworthy, it has been suggested that a selective accumulation of glycine induced by CBZ in the hippocampus and brainstem may be related to the enhanced anticonvulsant effect of CBZ in the MES test (Peterson et al. 1990). Therefore, it can be speculated that the increased glycine content in the brainstem might have been responsible for the unchanged anticonvulsive potency of CBZ following strychnine injection. All the above-mentioned facts may suggest the involvement of a glycinergic component in the mechanism of combined anticonvulsant action of captopril with CBZ and LTG against MES-induced seizures. However, to confirm this hypothesis, more advanced neurochemical studies on this subject including a role of sulfhydryl group of captopril are required.
When interpreting the present results, interactions of the tested drugs with GABAergic inhibitory neurotransmission should be also considered because captopril contains a structural element proline that is a precursor of GABA (Minano et al. 1987). GABA is the major inhibitory neurotransmitter in the brain, and it is responsible for the control of seizures (Czuczwar and Patsalos 2001). It has been demonstrated that angiotensin II—GABA interactions can affect seizure susceptibility. Angiotensin II potentiated the inhibitory action of GABAergic agonists, e.g. GABA itself and muscimol, on pentylenetetrazol convulsive threshold and it increased the threshold for seizures induced by bicuculline, a competitive antagonist of GABAA receptors (Tchekalarova and Georgiev 2005). In contrast, captopril was ineffective against convulsions evoked by bicuculline and even exacerbated tonic seizures and mortality caused by this convulsant (Minano et al. 1987). In the current study, a combination of captopril and PB, a drug which exerts its pharmacological effects by allosteric activation of GABAA receptors (Kwan et al. 2001) showed no positive interaction in the MES test. The same holds true for the combination of captopril and VPA, a drug which possesses GABA-mimetic properties by increasing the content of GABA in nerve terminals (Löscher 2002). Furthermore, CBZ and LTG appear not to act via an action on GABA-mediated inhibition (Löscher 1998). Thus, the involvement of GABAergic transmission in the potentiation by captopril of anticonvulsant drugs in MES-induced seizures is rather unlikely.
In conclusion, this study showed that captopril enhanced the anticonvulsant action of CBZ and LTG in mice and these interactions were pharmacodynamic in nature. The combinations of captopril with antiepileptic drugs did not lead to memory or motor impairments. Based on the current preclinical data, it is suggested that captopril may positively interact with CBZ and LTG in epileptic patients.
References
Boissier JR, Tardy J, Diverres JC (1960) Une nouvelle methode simple pour explorer l’action ‘tranquilisante’: le test de la cheminee. Med Exp (Basel) 3:81–84
Borowicz KK, Swiader M, Zgrajka W, Sawulski C, Turski WA, Czuczwar SJ (2002) Influence of several convulsants on the protective activity of a non-competitive AMPA/kainate antagonist, LY 300164, and lamotrigine against maximal electroshock in mice. J Physiol Pharmacol 53:859–869
Braszko JJ, Karwowska-Polecka W, Halicka D, Gard PR (2003) Captopril and enalapril improve cognition and depressed mood in hypertensive patients. J Basic Clin Physiol Pharmacol 14:323–343
Cushman DW, Wang FL, Fung WC, Grover GJ, Harvey CM, Scalese RJ, Mitch SL, DeForrest JM (1989) Comparisons in vitro, ex vivo, and in vivo of the actions of seven structurally diverse inhibitors of angiotensin converting enzyme (ACE). Br J Clin Pharmacol 28(Suppl 2):115S–130S
Czuczwar SJ, Patsalos PN (2001) The new generation of GABA enhancers. CNS Drugs 15:339–350
Czuczwar SJ, Włodarczyk D, Gąsior M, Kleinrok Z (1991) Is there a glycinergic component in the mechanism of action of common antiepileptic drugs? In: Klee MR, Lux HD, Speckmann EJ (eds) Physiology, pharmacology and development of epileptogenic phenomena. Springer, Berlin, pp 255–257
de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T (2000) International Union of Pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev 52:415–472
Duncan JS, Sander JW, Sisodiya SM, Walker MC (2006) Adult epilepsy. Lancet 367:1087–1100
Evered MD, Robinson MM, Richardson MA (1980) Captopril given intracerebroventricularly, subcutaneously or by gavage inhibits angiotensin-converting enzyme activity in the rat brain. Eur J Pharmacol 68:443–449
Gaitatzis A, Carroll K, Majeed A, Sander WJ (2004) The epidemiology of the comorbidity of epilepsy in the general population. Epilepsia 45:1613–1622
Gard PR (2002) The role of angiotensin II in cognition and behaviour. Eur J Pharmacol 438:1–14
Johnston CI (1990) Biochemistry and pharmacology of the renin–angiotensin system. Drugs 39(Suppl 1):21–31
Kubová H, Mares P (1994) Effects of MK-801 (dizocilpine) and ketamine on strychnine-induced convulsions in rats: comparison with benzodiazepines and standard anticonvulsants. Physiol Res 43:313–320
Kwan P, Sills GJ, Brodie MJ (2001) The mechanisms of action of commonly used antiepileptic drugs. Pharmacol Ther 90:21–34
Laragh JH, Case DB, Atlas SA, Sealey JE (1980) Captopril compared with other antirenin system agents in hypertensive patients: its triphasic effects on blood pressure and its use to identify and treat the renin factor. Hypertension 2:586–593
Litchfield JT, Wilcoxon F (1949) A simplified method of evaluating dose–effect experiments. J Pharmacol Exp Ther 96:99–113
Löscher W (1998) New visions in the pharmacology of anticonvulsion. Eur J Pharmacol 342:1–13
Löscher W (2002) Basic pharmacology of valproate. CNS Drugs 10:669–694
Löscher W, Fassbender CP, Nolting B (1991) The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs. II. Maximal electroshock seizure models. Epilepsy Res 8:79–94
Luszczki JJ, Czuczwar SJ (2005) How significant is the difference between drug doses influencing the threshold for electroconvulsions? Pharmacol Rep 57:782–786
Minano FJ, Serrano JS, Sancibrian M, Serrano MI (1987) Effect of peptidyl-dipeptidase inhibitors in experimental convulsions in mice. Fundam Clin Pharmacol 1:77–83
Mojaverian P, Swanson BN, Ferguson RK (1984) Enalapril, a new nonsulfhydryl angiotensin converting enzyme inhibitor, does not potentiate morphine analgesia. Eur J Pharmacol 98:303–306
Ng SK, Hauser WA, Brust JC, Susser M (1993) Hypertension and the risk of new-onset unprovoked seizures. Neurology 43:425–428
Peterson SL, Trzeciakowski JP, Frye GD, Adams HR (1990) Potentiation by glycine of anticonvulsant drugs in maximal electroshock seizures in rats. Neuropharmacology 29:399–409
Rajendra S, Lynch JW, Schofield PR (1997) The glycine receptor. Pharmacol Ther 73:121–146
Rubin B, Antonaccio MJ, Horovitz ZP (1978) Captopril (SQ 14, 225) (D-3-mercapto-2-methylpropranoyl-l-proline): a novel orally active inhibitor of angiotensin-converting enzyme and antihypertensive agent. Prog Cardiovasc Dis 21:183–194
Sangiah S (1985) Effects of glycine and other inhibitory amino acid neurotransmitters on threshold in mice. Vet Hum Toxicol 27:97–99
Tchekalarova J, Georgiev V (2005) Angiotensin peptides modulatory system: how is it implicated in the control of seizure susceptibility? Life Sci 76:955–970
Thind GS (1990) Angiotensin converting enzyme inhibitors: comparative structure, pharmacokinetics, and pharmacodynamics. Cardiovasc Drugs Ther 4:199–206
Venault P, Chapouthier G, Prado de Carvalho L, Simiand J, Morre M, Dodd RH, Rossier J (1986) Benzodiazepine impairs and β-carboline enhances performance in learning and memory tasks. Nature 321:864–866
Wright JW, Harding JW (1997) Important roles for angiotensin III and IV in the brain renin-angiotensin system. Brain Res Rev 25:96–124
Wright JW, Yamamoto BJ, Harding JW (2008) Angiotensin receptor subtype mediated physiologies and behaviors: new discoveries and clinical targets. Prog Neurobiol 84:157–181
Yamashita H, Ohno K, Amada Y, Hattori H, Ozawa-Funatsu Y, Toya T, Inami H, Shishikura J, Sakamoto S, Okada M, Yamaguchi T (2004) Effects of 2-[N-(4-chlorophenyl)-N-methylamino]-4H-pyrido[3.2-e]-1, 3-thiazin-4-one (YM928), an orally active alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor antagonist, in models of generalized epileptic seizure in mice and rats. J Pharmacol Exp Ther 308:127–133
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Łukawski, K., Jakubus, T., Raszewski, G. et al. Captopril potentiates the anticonvulsant activity of carbamazepine and lamotrigine in the mouse maximal electroshock seizure model. J Neural Transm 117, 1161–1166 (2010). https://doi.org/10.1007/s00702-010-0455-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-010-0455-y