
Abstract
Depressive disorders, including major depression, are serious and disabling, whose mechanisms are not clearly understood. Since life stressors contribute in some fashion to depression, chronic variable stress (CVS) has been used as an animal model of depression. In the present study we evaluated some parameters of oxidative stress [thiobarbituric acid reactive substances (TBARS), catalase (CAT), superoxide dismutase (SOD) and glutathione peroxidase (GPx)], and inflammatory markers (interleukin 6, C reactive protein, tumor necrosis factor-alpha and nitrites), as well as the activity of butyrylcholinesterase in blood of rats subjected to chronic stress. Homocysteine and folate levels also were measured. Stressed animals were submitted to different mild stressors for 40 days. After CVS, a reduction in weight gain was observed in the stressed group, as well as an increase in immobility time in the forced swimming test as compared with controls. Stressed animals presented a significant increase on TBARS and SOD/CAT ratio, but stress did not alter GPx activity and any inflammatory parameters studied. CVS caused a significant inhibition on serum butyrylcholinesterase activity. Stressed rats had higher plasmatic levels of homocysteine without differences in folate levels. Although it is difficult to extrapolate our findings to the human condition, the alterations observed in this work may be useful to help to understand, at least in part, the pathophysiology of depressive disorders.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Major depression is a common, severe, chronic, and often life-threatening illness. There is a growing appreciation that, far from being a disease with purely psychological manifestations, major depression is a systemic disease with deleterious effects on multiple organ systems (Charney and Manji 2004). On the other hand, stressful life events have a substantial causal association with depression, and there is now compelling evidence that even early life stress constitutes a major risk factor for the subsequent development of depression (Charney and Manji 2004). The chronic variable stress (CVS) model of depression has high validity, since a large number of recent publications have confirmed that CVS causes behavioral changes in rodents that parallel symptoms of depression (Gamaro et al. 2008; Ni et al. 2008; Wilner 2005; Katz and Hersh 1981).
Despite extensive research, the current theories on serotonergic dysfunctions do not provide sufficient explanations for the nature of depression (Maes et al. 2009). In this context, both oxidative stress and inflammatory mediators have been suggested to contribute to the neuropathology of depression (Lucinio and Wong 1999; Cumurcu et al. 2009). Oxidative stress, characterized by the imbalance between production of free radicals and the antioxidant capacity of organism, has been implied in the pathogenesis of several psychiatric disorders such as schizophrenia, bipolar disorder, and depression (Ng et al. 2008). Free radicals are molecules that play physiological roles in cellular signaling, immunological responses, and mitosis. However, being highly unstable molecules with unpaired electrons, they have differential oxidative strengths and hence the potential to damage cellular proteins, lipids, carbohydrates and nucleic acids (Halliwell 2006). Chronic stress has been shown to cause oxidative damage in the central nervous system (CNS) (Lucca et al. 2009; Madrigal et al. 2001; Olivenza et al. 2000), but there is a lack of works investigating peripheral effects of CVS.
New developments in psychiatric research have led to the hypothesis that inflammatory processes and neural-immune interactions are involved in the pathogenesis of major depression (Maes et al. 2009). Studies have demonstrated that proinflammatory parameters such as interleukins (IL-1, IL-2, IL-6, IL-8 and IL-12), interferon-γ (IFNγ), and tumor necrosis factor-α (TNFα) are increased in patients with depression (Schiepers et al. 2005). However, other studies have failed to find an association between the immune system and depression (Haack et al. 1999; Steptoe et al. 2003), indicating the need for more investigations in order to confirm the activation of immune system as cause of depressive symptoms.
Acetylcholine (ACh) is the principal vagus neurotransmitter and its action is finished by hydrolysis catalyzed by acetylcholinesterase (AChE) (E.C.3.1.1.7) and butyrylcholinesterase (BuChE) (E.C.3.1.1.8) (Darvesh et al. 2003). In humans, AChE is more abundant in the CNS, end plate of skeletal muscle, and erythrocytes membranes, while BuChE is more abundant in serum (Massoulié et al. 1993). Although the exact physiological function of BuChE is unclear, it has been shown that it can promptly hydrolyze acetylcholine and to substitute AChE in maintaining the structural and functional integrity of central cholinergic pathways (Mesulam et al. 2002). In addition, reports from the literature suggest a relationship between BuChE activity and risk factors for coronary artery disease (Alcantara et al. 2002) and that heart disease and depression are highly co-morbid (Johnson and Grippo 2006). In relation to stress, Rada et al. (2006) demonstrated that ACh levels are elevated in animals subjected to forced swimming and that this alteration is compensated by AChE activation. Otherwise, stress insults induce hyperexcitation of cholinergic circuits (Tracey 2002; Sapolsky 1996).
Another alteration that has been related to depressed patients concerns Hcy metabolism (Jendricko et al. 2009; Levine et al. 2008; Tolmunen et al. 2004). Homocysteine (Hcy) is a sulfurated amino acid derived from ingested methionine. It is directly toxic to neurons and blood vessels and can induce DNA strand breakage, oxidative stress and apoptosis (Mattson and Shea 2003; Lipton et al. 1997). On the other hand, the methionine–homocysteine metabolic pathway produces methyl groups required for the synthesis of catecholamines and DNA. This is accomplished by remethylating homocysteine—using B12 and folate as cofactors—back to methionine. A recent study demonstrated that serum homocysteine levels correlate positively with cortisol levels (Cascalheira et al. 2008). Furthermore, there are evidences supporting an association of depression with high blood homocysteine in humans (Bottiglieri et al. 2000; Tolmunen et al. 2004; Folstein et al. 2007) though the results are still controversial.
Thus, in line of the foregoing considerations, in the present study, we evaluated some parameters of oxidative stress [thiobarbituric acid reactive substances (TBARS), catalase (CAT), superoxide dismutase (SOD) and glutathione peroxidase (GPx)], and inflammatory markers [interleukin 6 (IL-6), C reactive protein (CRP), tumor necrosis factor-alpha (TNF-α) and nitrite (NO)], as well as the activity of butyrylcholinesterase in blood of rats subjected to chronic stress. Homocysteine and folate levels were also measured.
Materials and methods
Animals and reagents
Fifty-two (20 for oxidative stress measurements; 20 for inflammatory markers and BuChE; 12 for Hcy and folate assay), male Wistar rats (60 days old; 200–270 g weight) were obtained from the Central Animal House of the Department of Biochemistry of the Federal University of Rio Grande do Sul, Porto Alegre, Brazil. The experimentally naive animals were housed in groups of 4–5 in home cages made of Plexiglas material (65 × 25 × 15 cm) with the floor covered with sawdust. They were maintained under a standard dark–light cycle (lights on between 7:00 and 19:00 h) at a room temperature of 22 ± 2°C. The rats had free access to food (standard rat chow) and water, except for the stressed group during the period when the stressor applied required no water. After being randomized to assure all groups presented similar body weights, the animals were divided into two groups: control and stressed. Animal care followed the official governmental guidelines in compliance with the Federation of Brazilian Societies for Experimental Biology and was approved by the Ethical Committee of the Universidade Federal do Rio Grande do Sul, Brazil. All chemicals were purchased from Sigma Chemical Co., St Louis, MO, USA.
Stress model
The CVS protocol was applied as described by Gamaro et al. (2003) with some modifications in the stressors applied, such as inclination of the home cages instead of food deprivation and damp bedding instead of forced swimming. Control animals were handled daily. A variable-stressor paradigm was used for the animals in the stressed group. This protocol differs from other chronic stress protocols that use only one stressor in that the different stressors used diminish adaptation to stress (Marin et al. 2007). Animals were subjected to one stressor per day, at different times each day, in order to minimize predictability. The following stressors were used: (a) 24 h of water deprivation, (b) 1–3 h of restraint, as described below, (c) 1.5–2 h of restraint at 4°C, (d) flashing light during 120–210 min, (e) isolation (2–3 days), (f) inclination of the home cages at a 45° angle for 4–6 h, (g) damp bedding (300 mL water spilled onto bedding during 1.5–2 h). Restraint was carried out by placing the animal in a 25 × 7 cm plastic tube and adjusting it with plaster tape on the outside, so that the animal was unable to move. There was a 1-cm hole at the far end for breathing. Exposure to flashing light was made by placing the animal in a 50-cm high, 40 × 60 cm open field made of brown polywood with a frontal glass wall. A 40-W lamp, flashing at a frequency of 60 flashes per minute, was used. Rats were submitted to chronic variate stress for 40 days as described in Table 1.
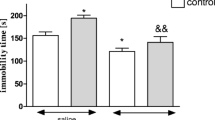
After 40 days of stress, forced swimming test was performed according to Porsolt et al. (1977), in order to confirm the ability of this stress to increase the immobility time, an indicative of depressive behavior. The test involves two individual exposures to a cylindrical tank with water in which rats cannot touch the bottom of the tank or escape. The tank is made of clear Plexiglas, 50 cm tall, 30 cm in diameter, and filled with water (22–23°C) to a depth of 30 cm. Water in the tank was changed after each rat swimming test section. For the first exposure, rats were placed in the water for 15 min (pre-test session). Twenty-four hours later, rats were placed in the water again for a 5-min session (test session), and the immobility time of rats was recorded in seconds.
Body weight was measured at different times during treatment, since several works reported that chronic stress-induced significant reduction in body weight gain (Konarska et al. 1990; Harro et al. 2001).
Erythrocyte and plasma preparation
Erythrocytes and plasma were prepared from whole blood samples obtained from rats (controls and stressed rats) after decapitation.
Whole blood was collected and transferred to heparinized tubes for erythrocyte separation. Blood samples were centrifuged at 1,000×g, plasma was removed by aspiration and frozen at −80°C until determination of TBARS, HCy, and folate levels. Erythrocytes were washed three times with cold saline solution (0.153 mol/L sodium chloride). Lysates were prepared by the addition of 1 mL of distilled water to 100 μL of washed erythrocytes and frozen at −80°C until determination of the antioxidant enzyme activities.
For antioxidant enzyme activity determination, erythrocytes were frozen and thawed three times, and centrifuged at 13,500×g for 10 min. The supernatant was diluted in order to contain approximately 0.5 mg/mL of protein.
Thiobarbituric acid reactive substances
Usually, lipid peroxidation is quantified by measuring malondialdehyde (MDA), which is formed by the degradation products of polyunsaturated fatty acid hydroperoxides (Halliwell and Gutteridge 2006). The main source of MDA in biological samples is the peroxidation of polyunsaturated fatty acids. TBARS is a widely adopted method for measuring lipid oxidation (Ferreira et al. 2010; Kunz et al. 2008; Del Rio et al. 2005; Draper and Hadley 1990); however, the TBARS assay is not specific for MDA. This way, we expressed the results in terms of the amount of thiobarbituric acid reactive substances formed per unit of time instead of the amount of malondialdehyde produced. TBARS was determined according to the method described by Ohkawa et al. (1979) for in vivo studies. TBARS measures malondialdehyde (MDA), a product of lipoperoxidation caused mainly by hydroxyls free radicals. Plasma diluted in 1.15% KCl was mixed with 20% trichloroacetic acid and 0.8% thiobarbituric acid and heated in a boiling water bath for 60 min. TBARS were determined by the absorbance at 535 nm. Calibration curve was performed using 1,1,3,3-tetramethoxypropane and each curve point was subjected to the same treatment as that of the plasmas. TBARS was calculated as nanomoles of malondialdehyde formed per milligram of protein.
Catalase assay
CAT activity was assayed by the method of Aebi (1984). H2O2 disappearance was continuously monitored with a spectrophotometer at 240 nm for 90 s. One unit of the enzyme is defined as 1 mmol of hydrogen peroxide consumed per minute and the specific activity is reported as units per mg protein.
Superoxide dismutase assay
This method for the assay of SOD activity is based on the capacity of pyrogallol to autoxidize, a process highly dependent on O −·2 , which is a substrate for SOD (Marklund 1985). The inhibition of autoxidation of this compound occurs in the presence of SOD, whose activity can be then indirectly assayed spectrophotometrically at 420 nm. A calibration curve was performed with purified SOD as standard, in order to calculate the activity of SOD present in the samples. The results were reported as units/mg protein.
Glutathione peroxidase
GSH-Px activity was measured by the method of Wendel (1981), except for the concentration of NADPH, which was adjusted to 0.1 mM after previous tests performed in our laboratory. Tert-butylhydroperoxide was used as substrate. NADPH disappearance was continuously monitored with a spectrophotometer at 340 nm for 4 min. One GSH-Px unit is defined as 1 mmol of NADPH consumed per minute and specific activity is reported as units per mg protein.
Serum preparation
After decapitation, the blood was collected and centrifuged for 10 min at 1,000×g. The serum was used for the inflammatory marker assays and enzymatic (BuChE) analyses.
Cytokines (TNF-α and IL-6) assay
TNF-α and IL-6 levels in serum were quantified by rat high-sensitivity enzyme-linked immunoabsorbent assays (ELISA) with commercially available kits (Biosource®, Camarillo, CA).
Nitrite assay (NO)
Nitrite levels were measured using the Griess reaction; 100 μL of supernatant of hippocampus and cerebral cortex was mixed with 100 μL Griess reagent (1:1 mixture of 1% sulfanilamide in 5% phosphoric acid and 0.1% naphthylethylenediamine dihydrochloride in water) and incubated in 96-well plates for 10 min at room temperature. The absorbance was measured on a microplate reader at a wavelength of 543 nm. Nitrite concentration was calculated using sodium nitrite standards (Green et al. 1982).
Acute-phase protein assay (CRP)
CRP levels in serum were determined by a colorimetric assay with commercially available kits (BioSystems® and Bioclin®, Brazil).
Butyrylcholinesterase activity assay
BuChE activity was determined by the method of Ellman et al. (1961) with some modifications. Hydrolysis rate was measured at acetylthiocholine concentration of 0.8 mM in 1 mL assay solutions with 100 mM potassium phosphate buffer pH 7.5 and 1.0 mM 5,5-dithiobis (2-nitrobenzoic acid) (DTNB). Fifty microliters of rat diluted serum was added to the reaction mixture and preincubated for 3 min. The hydrolysis was monitored by formation of the thiolate dianion of DTNB at 412 nm for 2 min (intervals of 30 s) at 25°C. All samples were run in duplicate. Specific enzyme activity was expressed as mmol acetylthiocholine hydrolyzed per hour per milligram of protein.
Homocysteine level determination
Hcy levels in plasma were determined as described by Magera et al. (1999), using liquid chromatography electrospray tandem mass spectrometry (LC–MS/MS). After reduction and deproteinization of samples, Hcy concentration was detected through the transition from the precursor to the product ion (m/z 136 to m/z 90). Homocysteine-d was added as internal standard.
Folate levels determination
For folate determination, heparinized blood was collected and plasma was separated. Plasma folate concentration was measured by an automated chemiluminescence system (ACS: 180, Siemens). The method is based on a competitive immunoassay with acridinium ester-labeled folate in solid phase.
Protein determination
Protein was measured according to Bradford (1976) for butyrylcholinesterase assay and according to Lowry et al. (1951) for all others techniques. Serum bovine albumin was used as standard.
Statistical analysis
Data were analyzed by unpaired Student’s t test. All analyses were performed using the Statistical Package for the Social Sciences (SPSS) version 15.0 in a PC-compatible computer. The results were expressed as mean ± SEM and differences were considered statistically significant if P < 0.05.
Results
The body weight of animals was evaluated before and after CVS. We verified that the control group gained more weight after 40 days than the stressed group [t(18) = 2.763; P < 0.01]. Chronic stress also increased immobility time in the forced swimming test when compared with controls [t(18) = 3.066; P < 0.01].
The effect of chronic stress upon the levels of TBARS in plasma was measured. As can be observed in Fig. 1, chronic stress increased TBARS levels [t(14) = 5.100; P < 0.001]. Figure 2a shows a significant increase in SOD/CAT ratio in the stressed group [t(18) = 3.363; P < 0.05]. To verify whether other antioxidant enzyme was compensating the imbalance verified between SOD and CAT, we determined glutathione peroxidase activity. As shown in Fig. 2b, stress did not alter GPx activity [t(18) = 0.644; P > 0.05].

Effect of chronic variable stress (CVS) on thiobarbituric acid reactive substances (TBARS) in plasma of rats. TBARS is expressed as nmol of thiobarbituric acid reactive substances per mg protein. Results are expressed as mean ± SEM for eight independent experiments performed in duplicate. ***P < 0.001 compared with control group (Student’s t test)

Effect of chronic variable stress (CVS) on superoxide dismutase/catalase ratio (a) and on glutathione peroxidase activity (b). Results are expressed as mean ± SEM for ten independent experiments performed in duplicate. *P < 0.05 compared with control group (Student’s t test). SOD superoxide dismutase, CAT catalase, GPx glutathione peroxidase
We also investigated some inflammatory parameters in serum of rats subjected to CVS. Table 2 shows that these inflammatory parameters studied were not affected in stressed group when compared with controls [IL-6: t(17) = 1.433, P > 0.05; TNF: t(18) = 1.644, P > 0.05; NO: t(18) = 3.363; P > 0.05; PCR: t(16) = 1.490; P > 0.05].
The effect of CVS on the activity of BuChE in serum of rats was also studied. Figure 3 shows that this enzyme was significantly inhibited in the stressed group [t(14) = 4.793; P < 0.001] as compared with the control group.

Effect of chronic variable stress (CVS) on butyrylcholinesterase activity in serum of rats. Results are mean ± SEM for eight independent experiments performed in duplicate. ***P < 0.001 compared with control (Student’s t test)
Plasma levels of Hcy and folate in animals submitted to CVS are demonstrated in Table 3. Results showed that the stressed group had higher plasma Hcy than the control group [t(10) = 5.433; P < 0.001], while no significant difference in folate concentration was detected between groups [t(9) = 1.078; P > 0.05].
Discussion
Major depressive disorder has traditionally been considered to have a neurochemical basis, but despite the devastating impact of the illness, little is known about its etiology or pathophysiology. However, several studies have demonstrated that although the monoaminergic neurotransmitter systems may be involved in this disease, they are limited in elucidating the pathogenesis of depression. It has been demonstrated that depression arises from the complex interaction of multiple susceptible (and likely protective) genes and environmental factors, and disease phenotypes include not only episodic and often profound mood disturbances, but also a range of cognitive, motoric, autonomic, endocrine, and sleep/wake abnormalities (Manji et al. 2001). These observations have led to the appreciation that although dysfunction within the monoaminergic neurotransmitter systems is likely to play important roles in mediating some facets of the pathophysiology of depression, there are other possible mechanisms involved (Maletic et al. 2007; Belmaker and Agam 2008).
The HPA axis and its final effector system, glucocorticoids, are essential components of an individual’s capacity to cope with stress, and in fact, a hyperactivity of the HPA axis is observed in the majority of patients with depression (Gillespie and Nemeroff 2005; Bao et al. 2008; Swaab et al. 2005). Considering that life stressors contribute in some fashion to depression and are an extension of what occurs normally, chronic stress has been used as an animal model of depression (Gamaro et al. 2008; Ni et al. 2008) since animals displayed typical changes in hedonic status. Using this model, in the present study we evaluated some parameters of oxidative stress in plasma and erythrocytes of rats. We observed a significant increase in TBARS, a method that evaluates the oxidative stress assayed for malondialdehyde, the last product of lipid breakdown caused by oxidative stress (Halliwell and Gutteridge 2006). Beyond generating pathways, in this study, we pay attention to consuming pathways of free radicals, namely SOD, CAT, and GPx, the major enzymatic system responsible for protecting cells against free radical attacks (Halliwell and Gutteridge 2006). Animals exposed to stress presented an imbalance between SOD and CAT, expressed by increased SOD/CAT ratio. When a cell has decreased activity of CAT, a large amount of H2O2 (the product of SOD action) becomes available to react with transition metals and generates the radical hydroxyl, which is the most harmful radical (Kelner et al. 1995; Matés et al. 1999). On the other hand, stress did not alter the activity of GPx; this result reinforces the view that oxidative stress responses do not always involve a coordinated regulation of all antioxidant enzymes and that their activities are regulated by different mechanisms (Röhrdanz et al. 2000; Wilson and Johnson 2000).
These data are consistent with evidences that indicate that oxidative stress is a major pathological mechanism in the maladaptation to chronic stress in rats (Lucca et al. 2009; Madrigal et al. 2001; Olivenza et al. 2000). In this line, clinical studies also demonstrate an induction of oxidative stress in serum of depressed patients (Cumurcu et al. 2009; Khanzode et al. 2003; Bilici et al. 2001). Oxidative stress induction caused by chronic stress could be explained by several pathways, for example, through over-stimulation of glucocorticoids receptors (You et al. 2009; Zafir and Banu 2009), inhibition of mitochondrial electron transport chain complexes (Tagliari et al. 2010), and alterations on homocysteine metabolism (de Souza et al. 2006).
Recent studies have demonstrated that inflammatory and neurodegenerative processes play an important role in depression and that enhanced neurodegeneration in depression may—at least partly—be caused by inflammatory processes (Maes et al. 2009; Miller et al. 2009; Dantzer 2006; Schiepers et al. 2005). Based on these studies, in the present study, we evaluated the effects of stress on some inflammatory markers such as IL-6, TNF-α, NO, and PCR. Results showed that chronic stress did not alter any of the inflammatory parameters studied. Although clinical studies showed an increase of cytokines levels in blood of depressed patients, animal models using stress as model of depression have inconsistent results. In this context, Kubera et al. (1996) demonstrated increased blood levels of IL-1 and IL-2 after 8 weeks of mild stress. On the other hand, mice exposed to a 3-week chronic mild stress had decreased expression of peripheral IL-1beta and IL-6 and an increased expression of brain IL-6 (Mormède et al. 2003). In addition, another study has also reported elevated cytokine levels in brain of mice subjected to chronic mild stress for 5 weeks (Goshen et al. 2008). These inconsistent results concerning blood interleukins may be due to different stress protocols or different periods of stress exposure.
We also measured the activity BuChE in serum of animals submitted to chronic stress. Results showed that this enzyme was inhibited in stressed animals as compared with the control group. Moreover, since the results of the present study show an imbalance between CAT and SOD activities, what could result in increased levels of H2O2, our results are in agreement with other studies demonstrating that hydrogen peroxide can inhibit serum cholinesterase (Schallreuter and Elwary 2007).
Since there are data from literature showing that Hcy metabolism can be altered in depression and/or stressed patients (Jendricko et al. 2009; Levine et al. 2008; Tolmunen et al. 2004) and that this amino acid induces oxidative stress (Matté et al. 2009; Faraci and Lentz 2004; Wyse et al. 2002), we investigate the plasma levels of homocysteine and folate in control and stressed rats. Our results showed an increase in homocysteine levels in the stressed group; however, there were no differences in folate levels between control and stressed groups. Previous studies regarding Hcy metabolism in depression have provided contradictory results. Several works suggest that Hcy levels are increased in depressed patients (Wilhelm et al. 2010; Alexopoulos et al. 2010; Resler et al. 2008; Tolmunen et al. 2004; Reif et al. 2003; Bottiglieri et al. 2000); however, the lack of correlation between Hcy and depression has been demonstrated by Kelly et al. (2004). In most cases, however, increased levels of Hcy were observed in depressed patients with vitamin B12 or folate deficiency (Kim et al. 2008; Bottiglieri et al. 2000; Refsum et al. 2006). Our results are in agreement with studies that demonstrated increased levels of Hcy in the absence of folate deficiency after restraint stress in rats (de Souza et al. 2006). In addition, Triantafyllou et al. (2008) showed that multiple sclerosis patients that present elevated Hcy levels are particularly prone to develop depressive symptomatology. Previous studies from our laboratory have shown that BuChE can be inhibited by Hcy (Scherer et al. 2007; Matté et al. 2006), and this inhibition is mediated by the generation of free radicals (Stefanello et al. 2005). Therefore, the reduced BuChE activity could be related to the altered Hcy observed in the present study. Besides, elevated levels of Hcy increase oxidative stress (Matté et al. 2009; Wyse et al. 2002).
Besides augmenting the probability of oxidative damage, increased Hcy levels in stressed animal may ultimately be related to an imbalance at the monoamine or neurotransmitter level, since the rise in Hcy levels could be ascribed to failure of methylation of Hcy to methionine. Methionine, in turn, is the precursor of S-adenosylmethionine, the methyl donor in a host of methylation reactions in the CNS involving monoamines and various neurotransmitters, amongst other cellular constituents.
In conclusion, we found in the present study that the CVS model of depression provokes an increase in oxidative stress, an inhibition of BuChE, and an increase in Hcy levels in blood of rats. Since the pathophysiology of depression still is poorly understood, if confirmed in humans, our results could be useful to explain some symptoms observed in patients.
References
Aebi H (1984) Catalase in vitro. Methods Enzymol 105:121–126
Alcantara VM, Chautard-Freire-Maia EA, Scartezini M, Cerci MS, Braun-Prado K, Picheth G (2002) Butyrylcholinesterase activity and risk factors for coronary artery disease. Scan J Clin Lab Invest 62:399–404
Alexopoulos P, Topalidis S, Irmisch G, Prehn K, Jung SU, Poppe K, Sebb H, Perneczky R, Kurz A, Bleich S, Herpertz SC (2010) Homocysteine and cognitive function in geriatric depression. Neuropsychobiology 61(2):97–104
Bao AM, Meynena G, Swaab DF (2008) The stress system in depression and neurodegeneration: focus on the human hypothalamus. Brain Res Rev 57:531–553
Belmaker RH, Agam G (2008) Major depressive disorder. N Engl J Med 358(1):55–68
Bilici M, Efe H, Köroğlu MA, Uydu HA, Bekaroğlu M, Değer O (2001) Antioxidative enzyme activities and lipid peroxidation in major depression: alterations by antidepressant treatments. J Affect Disord 64(1):43–51
Bottiglieri T, Laundy M, Crellin R, Toone BK, Carney MW, Reynolds EH (2000) Homocysteine, folate, methylation, and monoamine metabolism in depression. J Neurol Neurosurg Psychiatry 69:228–232
Bradford MM (1976) A rapid and sensitive method for the quantification of micrograms quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Cascalheira JF, Parreira MC, Viegas AN, Faria MC, Domingues FC (2008) Serum homocysteine: relationship with circulating levels of cortisol and ascorbate. Ann Nutr Metab 53(1):67–74
Charney DS, Manji HK (2004) Life stress, genes, and depression: multiple pathways lead to increased risk and new opportunities for intervention. Sci. STKE 225:1–11
Cumurcu BE, Ozyurt H, Etikan I, Demir S, Karlidag R (2009) Total antioxidant capacity and total oxidant status in patients with major depression: impact of antidepressant treatment. Psychiatry Clin Neurosci 63(5):639–645
Dantzer R (2006) Cytokine, sickness behavior and depression. Neurol Clin 24(3):441–446
Darvesh S, Hopkins DA, Geula C (2003) Neurobiology of butyrylcholinesterase. Nat Rev Neurosci 17:131–138
de Souza FG, Rodrigues MD, Tufik S, Nobrega JN, D’Almeida V (2006) Acute stressor-selective effects on homocysteine metabolism and oxidative stress parameters in female rats. Pharmacol Biochem Behav 85(2):400–407
Del Rio D, Stewart AJ, Pellegrini N (2005) A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr Metab Cardiovasc Dis 15:316–328
Draper HH, Hadley M (1990) Malondialdehyde determination as index of lipid peroxidation. Methods Enzymol 186:421–431
Ellman GL, Courtney KD, Andres V, Featherstone RM (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7:88–95
Faraci FM, Lentz SR (2004) Hyperhomocysteinemia, oxidative stress, and cerebral vascular dysfunction. Stroke 35:345–347
Ferreira AG, Lima DD, Delwing D, Mackedanz V, Tagliari B, Kolling J, Schuck PF, Wajner M, Wyse AT (2010) Proline impairs energy metabolism in cerebral cortex of young rats. Metab Brain Dis 25(2):161–168
Folstein M, Liu T, Peter I, Buel J, Arsenault L, Scott T, Qiu WW (2007) Homocysteine hypothesis of depression. Am J Psychiatry 16:861–867
Gamaro GD, Manoli LP, Torres ILS, Silveira R, Dalmaz C (2003) Effects of chronic variate stress on feeding behavior and on monoamine levels in different rat brain structures. Neurochem Int 42:107–114
Gamaro GD, Prediger ME, Bassani MG, Dalmaz C (2008) Fluoxetine alters feeding behavior and leptin levels in chronically-stressed rats. Pharmacol Biochem Behav 90:312–317
Gillespie CF, Nemeroff CB (2005) Hypercortisolemia and depression. Psychosom Med 67:26–28
Goshen I, Kreisel T, Ben-Menachem-Zidon O, Licht T, Weidenfeld J, Ben-Hur T, Yirmiya R (2008) Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry 13(7):717–728
Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR (1982) Analysis of nitrate, nitrite and [15N]nitrate in biological fluids. Anal Biochem 126:131–138
Haack M, Hinze-Selch D, Fenzel T, Kraus T, Kühn M, Schuld A, Pollmächer T (1999) Plasma levels of cytokines and soluble cytokine receptors in psychiatric patients upon hospital admission: effects of confounding factors and diagnosis. J Psychiatr Res 33:407–418
Halliwell B (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97:1634–1658
Halliwell B, Gutteridge JMC (2006) Free radicals in biology and medicine, 4th edn. Oxford University Press, Oxford
Harro J, Tõnissaar M, Eller M, Kask A, Oreland L (2001) Chronic variable stress and partial 5-HT denervation by parachloroamphetamine treatment in the rat: effects on behavior and monoamine neurochemistry. Brain Res 899(1–2):227–239
Jendricko T, Vidović A, Grubisić-Ilić M, Romić Z, Kovacić Z, Kozarić-Kovacić D (2009) Homocysteine and serum lipids concentration in male war veterans with posttraumatic stress disorder. Prog Neuropsychopharmacol Biol Psychiatry 33(1):134–140
Johnson AK, Grippo AJ (2006) Sadness and broken hearts: neurohumoral mechanisms and co-morbidity of ischemic heart disease and psychological depression. J Physiol Pharmacol 57(S11):5–29
Katz RJ, Hersh S (1981) Amitriptyline and scopolamine in an animal model of depression. Neurosci Biobehav Rev 5:265–271
Kelly CB, McDonnell AP, Johnston TG, Mulholland C, Cooper SJ, McMaster D, Evans A, Whitehead AS (2004) The MTHFR C677T polymorphism is associated with depressive episodes in patients from Northern Ireland. J. Psychopharmacol 18(4):567–571
Kelner MJ, Bagnell R, Montoya M, Estes L, Uglik SF, Cerutti P (1995) Transfection with human copper-zinc superoxide dismutase induces bidirectional alterations in other antioxidant enzymes, proteins, growth factor response, and paraquat resistance. Free Radic Biol Med 18:497–506
Khanzode SD, Dakhale GN, Khanzode SS, Saoji A, Palasodkar R (2003) Oxidative damage and major depression: the potential antioxidant action of selective serotonin re-uptake inhibitors. Redox Rep 8(6):365–370
Kim JM, Stewart R, Kim SW, Yang SJ, Shin IS, Yoon JS (2008) Predictive value of folate, vitamin B12 and homocysteine levels in latelife depression. Br J Psychiatry 192:268–274
Konarska M, Stewart RE, McCarty R (1990) Predictability of chronic intermittent stress: effects on sympathetic-adrenal medullary responses of laboratory rats. Behav Neural Biol 53:231–243
Kubera M, Symbirtsev A, Basta-Kaim A, Borycz J, Roman A, Papp M, Claesson M (1996) Effect of chronic treatment with imipramine on interleukin 1 and interleukin 2 production by splenocytes obtained from rats subjected to a chronic mild stress model of depression. Pol J Pharmacol 48(5):503–506
Kunz M, Gama CS, Andreazza AC, Salvador M, Ceresér KM, Gomes FA, Belmonte-de-Abreu PS, Berk M, Kapczinski F (2008) Elevated serum superoxide dismutase and thiobarbituric acid reactive substances in different phases of bipolar disorder and in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 32(7):1677–1681
Levine J, Timinsky I, Vishne T, Dwolatzky T, Roitman S, Kaplan Z, Kotler M, Sela BA, Spivak B (2008) Elevated serum homocysteine levels in male patients with PTSD. Depress Anxiety 25(11):E154–E157
Lipton SA, Kim WK, Choi YB, Kumar S, D’Emilia DM, Rayudu PV, Arnelle DR, Stamler JS (1997) Neurotoxicity associated with dual actions of homocysteine at the N-methyl-d-aspartate receptor. Proc Natl Acad Sci USA 94:5923–5928
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193:265–267
Lucca G, Comim CM, Valvassori SS, Réus GZ, Vuolo F, Petronilho F, Dal-Pizzol F, Gavioli EC, Quevedo J (2009) Effects of chronic mild stress on the oxidative parameters in the rat brain. Neurochem Int 54:358–362
Lucinio J, Wong ML (1999) The role of inflammatory mediators in the biology of major depression: central nervous system cytokines modulate the biological substrate of depressive symptoms, regulate stress-responsive systems, and contribute to neurotoxicity and neuroprotection. Mol Psychiatry 4:317–327
Madrigal JLM, Olivenza R, Moro MA, Lisasoain I, Lorenzo P, Rodrigo J, Leza JC (2001) Glutathione depletion, lipid peroxidation and mitochondrial dysfunction are induced by chronic stress in rat brain. Neuropsychopharmacology 24(4):420–429
Maes M, Yirmya R, Noraberg J, Brene S, Hibbeln J, Perinii G, Kubera M, Bob P, Lerer B, Maj M (2009) The inflammatory & neurodegenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression. Metab Brain Dis 24:27–53
Magera MJ, Lacey JM, Casetta B, Rinaldo P (1999) Method for the determination of total homocysteine in plasma and urine by stable isotope dilution and electrospray tandem mass spectrometry. Clin Chem 45:1517–1522
Maletic V, Robinson M, Oakes T, Iyengar S, Ball SG, Russel J (2007) Neurobiology of depression: an integrated view of key findings. Int J Clin Pract 61(12):2030–2040
Manji HK, Drevets WC, Charney DS (2001) The cellular neurobiology of depression. Nat Med 7(5):541–547
Marin MT, Cruz FC, Planeta CS (2007) Chronic restraint or variable stresses differently affect the behavior, corticosterone secretion and body weight in rats. Physiol Behav 90(1):29–35
Marklund S (1985) Handbook of methods for oxygen radical research. CRC Press, Boca Raton, pp 243–247
Massoulié J, Pezzementi L, Bon S, Krejci E, Vallette FM (1993) Molecular and cellular biology of cholinesterases. Prog Neurobiol 41(1):31–91
Matés JM, Pérez-Gomes C, De Castro IN (1999) Antioxidant enzymes and human diseases. Clin Biochem 32(8):595–603
Matté C, Durigon E, Stefanello FM, Cipriani F, Wajner M, Wyse ATS (2006) Folic acid pretreatment prevents the reduction of Na(+),K(+)-ATPase and butyrylcholinesterase activities in rats subjected to acute hyperhomocysteinemia. Int J Dev Neurosci 24(1):3–8
Matté C, Mackedanz V, Stefanello FM, Scherer EB, Andreazza AC, Zanotto C, Moro AM, Garcia SC, Gonçalves CA, Erdtmann B, Salvador M, Wyse ATS (2009) Chronic hyperhomocysteinemia alters antioxidant defenses and increases DNA damage in brain and blood of rats: protective effect of folic acid. Neurochem Int 54(1):7–13
Mattson MP, Shea TB (2003) Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci 26:137–146
Mesulam MM, Guillozet A, Shaw P, Levey A, Duysen EG, Lockridge O (2002) Acethylcolynesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyse acetylcholine. Neuroscience 110:627–639
Miller AH, Maletic V, Raison CL (2009) Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65(9):732–741
Mormède C, Castanon N, Médina C, Moze E, Lestage J, Leveu PJ, Dantzer R (2003) Chronic mild stress in mice decreases peripheral cytokine and increases central cytokine expression independently of IL-10 regulation of the cytokine network. Neuroimmunomodulation 10:359–366
Ng F, Berk M, Dean O, Bush AI (2008) Oxidative stress in psychiatric disorders: evidence base and therapeutic implications. Int J Neuropsychopharmacol 21:1–26
Ni Y, Su M, Lin J, Wang X, Qiu Y, Zhao A, Chen T, Jia W (2008) Metabolic profiling reveals disorder of amino acid metabolism in four brain regions from a rat model of chronic unpredictable mild stress. FEBS Lett 582(17):2627–2636
Ohkawa H, Ohishi N, Yagi K (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 95:351–358
Olivenza R, Moro MA, Lisasoain I, Lorenzo P, Fernandez AP, Rodrigo J, Boscá L, Leza JC (2000) Chronic stress induces the expression of inducible nitric oxide synthase in rat brain cortex. J Neurochem 74:785–791
Porsolt RD, Bertin A, Jalfre M (1977) Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther 229:327–336
Rada P, Colasante C, Skirzewski M, Hernandez L, Hoebel B (2006) Behavioral depression in the swim test causes a biphasic, long-lasting change in accumbens acetylcholine release, with partial compensation by acetylcholinesterase and muscarinic-1 receptors. Neuroscience 141:67–76
Refsum H, Nurk E, Smith AD, Ueland PM, Gjesdal CG, Bjelland I, Tverdal A, Tell GS, Nygård O, Vollset SE (2006) The Hordaland homocysteine study: a community-based study of homocysteine, its determinants, and associations with disease. J Nutr 136(6):1731S–1740S
Reif A, Schneider MF, Kamolz S, Pfuhlmann B (2003) Homocysteinemia in psychiatric disorders: association with dementia and depression, but not schizophrenia in female patients. J Neural Transm 110:1401–1411
Resler G, Lavie R, Campos J, Mata S, Urbina M, García A, Apitz R, Lima L (2008) Effect of folic acid combined with fluoxetine in patients with major depression on plasma homocysteine and vitamin B12, and serotonin levels in lymphocytes. Neuroimmunomodulation 15(3):145–152
Röhrdanz E, Obertrifter B, Ohler S, Tran-Thi Q, Kahl R (2000) Influence of Adriamycin and paraquat on antioxidant enzyme expression in primary rat hepatocytes. Arch Toxicol 74:231–237
Sapolsky RM (1996) Why stress is bad for your brain. Science 273:749–750
Schallreuter KU, Elwary S (2007) Hydrogen peroxide regulates the cholinergic signal in a concentration dependent manner. Life Sci 80(24–25):2221–2226
Scherer EB, Stefanello FM, Mattos C, Netto CA, Wyse ATS (2007) Homocysteine reduces cholinesterase activity in rat and human serum. Int J Dev Neurosci 25(4):201–205
Schiepers OJ, Wichers MC, Maes M (2005) Cytokines and major depression. Prog Neuropsychopharmacol Biol Psychiatry 29(2):201–217
Stefanello FM, Franzon R, Tagliari B, Wannmacher C, Wajner M, Wyse ATS (2005) Reduction of butyrylcholinesterase activity in rat serum subjected to hyperhomocysteinemia. Metab Brain Dis 20(2):97–103
Steptoe A, Kunz-Ebrecht SR, Owen N (2003) Lack of association between depressive symptoms and markers of immune and vascular inflammation in middle-aged men and women. Psychol Med 33:667–674
Swaab DF, Bao AM, Lucassen PJ (2005) The stress system in the human brain in depression and neurodegeneration. Ageing Res Rev 4:141–194
Tagliari B, Noschang CG, Ferreira AGK, Ferrari OA, Feksa LR, Wannmacher CMD, Dalmaz C, Wyse ATS (2010) Chronic variable stress impairs energy metabolism in brain of rats. Metab Brain Dis 25(2):169–176
Tolmunen T, Hintikka J, Voutilainen S, Ruusunen A, Alfthan G, Nyyssönen K, Viinamäki H, Kaplan GA, Salonen JT (2004) Association between depressive symptoms and serum concentrations of homocysteine in men: a population study. Am J Clin Nutr 80:1574–1578
Tracey KJ (2002) The inflammatory reflex. Nature 420:853–859
Triantafyllou N, Evangelopoulos ME, Kimiskidis VK, Kararizou E, Boufidou F, Fountoulakis KN, Siamouli M, Konstantinos N, Nikolaou C, Sfagos C, Vlaikidis N, Vassilopoulos D (2008) Increased plasma homocysteine levels in patients with multiple sclerosis and depression. Ann Gen Psychiatry 7:17–21
Wendel A (1981) Glutathione peroxidase. Methods Enzymol 77:325–332
Wilhelm J, Muller E, de Zwaan M, Fischer J, Hillemacher T, Komhuber J, Bleich S, Frieling H (2010) Elevation of homocysteine levels is only partially reversed after therapy in females with eating disorders. J Neural Transm 117:521–527
Wilner P (2005) Chronic mild stress (CMS) revisited: consistency and behavioural-neurobiological concordance in the effects of CMS. Neuropsychobiology 52:90–110
Wilson DO, Johnson P (2000) Exercise modulates antioxidant enzyme gene expression in rat myocardium and liver. J Appl Physiol 88:1791–1796
Wyse ATS, Zugno AI, Streck EL, Matté C, Calcagnotto T, Wannmacher CM, Wajner M (2002) Inhibition of Na(+),K(+)-ATPase activity in hippocampus of rats subjected to acute administration of homocysteine is prevented by vitamins E and C treatment. Neurochem Res 27(12):1685–1689
You JM, Yun SJ, Nam KN, Kang C, Won R, Lee EH (2009) Mechanism of glucocorticoid-induced oxidative stress in rat hippocampal slice cultures. Can J Physiol Pharmacol 87(6):440–447
Zafir A, Banu N (2009) Modulation of in vivo oxidative status by exogenous corticosterone and restraint stress in rats. Stress 12(2):167–177
Acknowledgments
We thank Lucas P. Mocelin for his technical assistance. This work was supported in part by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil) and by the FINEP Research Grant “Rede Instituto Brasileiro de Neurociência (IBN-Net), # 01.06.0842-00”.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tagliari, B., dos Santos, T.M., Cunha, A.A. et al. Chronic variable stress induces oxidative stress and decreases butyrylcholinesterase activity in blood of rats. J Neural Transm 117, 1067–1076 (2010). https://doi.org/10.1007/s00702-010-0445-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-010-0445-0