
Abstract
Parkinson’s disease (PD) is a neurodegenerative movement disorder characterized by the progressive loss of dopaminergic neurons in the substantia nigra and depletion of dopamine in the striatum, which lead to pathological and clinical abnormalities. Increasing evidence has demonstrated that inflammation is the fundamental process contributing to neuron death in PD. Neuroinflammation, which is characterized by activated microglia and infiltrating T cells at sites of neuronal injury, is a prominent contributor to the pathogenesis of progressive PD. Microglia play a critical role in forming a self-propelling cycle leading to sustained chronic neuroinflammation and driving the progressive neurodegeneration in PD. This activation depends heavily on the respiratory burst within the microglia, which in turn regulates a number of downstream pro-inflammatory activities. On the other hand, the adaptive immune responses, most notably T cells, are now emerging as important components of the inflammatory response that contribute to the pathogenesis of PD. This review paper focus on the understanding of the inflammatory etiology of PD, as well as the molecular signaling involved in this inflammatory response, with the aim to provide more effective treatments to slow down or halt the progression of chronic inflammation-induced CNS disorders, such as PD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Parkinson’s disease: a chronic inflammatory, progressive neurodegenerative disease
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after Alzheimer’s disease. It is characterized by a slow and progressive degeneration of dopaminergic (DA) neurons in the substantia nigra (SN) and degeneration of the nerve terminals in the striatum, which will eventually lead to the development of progressive movement disorders, including resting tremor, rigidity, bradykinesia, and gait disturbance (Jellinger 2001). Epidemiological studies have revealed that most (90%) PD cases are sporadic and have a late onset (Tanner 2003). About 10% of cases are characterized by early onset, and these mostly occur in familial clusters (Mizuno et al. 2001). Development of parkinsonian syndrome in such individuals has been attributed to mutations in several recently identified genes, including parkin, Leucine-rich repeat kinase 2 (LRRK2), á-synuclein, PINK-1, or DJ-1 (Polymeropoulos et al. 1997; Sun et al. 2006; Bonifati et al. 2008; Weng et al. 2007; Abou-Sleiman et al. 2003; Jiang et al. 2007). In contrast, development of idiopathic PD may represent the final outcome of a complex set of interactions among the innate vulnerabilities of the nigro-striatal DA system, potential genetic predisposition, and exposure to environmental toxins. Among the environmental toxins, infectious agents, pesticides and heavy metals have been implicated to be associated with the development and progression of PD (Mizuno et al. 2001). However, there is now a growing recognition of the central role of neuroinflammation in the pathogenesis of PD (McGeer et al. 2001; Hirsch and Hunot 2009).
While the pathogenic mechanisms that ultimately cause PD are still unclear, it is believed that the progressive nature of PD is characterized by chronic inflammation-induced DA neurodegeneration within the SN area (Gao and Hong 2008; Bartels and Leenders 2007; Dauer and Przedborski 2003). It is well documented that microglial activation results in the loss of DA neurons in patients with PD. The premise of microglial activation in PD has been supported by the analysis of post mortem brains from PD patients that provides clear evidence of microglial activation in the SN (Loeffler et al. 1994; Ghosh et al. 2007; Cicchetti et al. 2002). In the brains of patients with PD, large numbers of human leukocyte antigen (HLA-DR) and CD11b-positive microglia were found in the SN, a region in which the degeneration of DA neurons was most prominent (McGeer et al. 1988). In addition, levels of pro-inflammatory mediators, including TNFα, IL-1β, IL-6, reactive oxygen species (ROS), and eicosanoids are elevated in the brains and peripheral blood mononuclear cells (PBMC) of PD patients (McGeer et al. 1988; Nagatsu et al. 2000; Mogi et al. 1994). Nitrite (as an indicator for nitric oxide free radicals) in the cerebrospinal fluid, as well as increased expression of inducible nitric oxide synthase (iNOS) within the SN, has also been found in PD patients (Qureshi et al. 1995). Consequently, the vast majority of therapeutic attention directed at inflammation within the CNS in patients with PD has been directed toward microglial cell activity.
Peripheral inflammation may initiate or contribute to neuroinflammation and neurodegeneration in the CNS
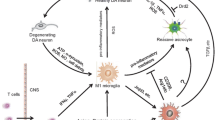
It is known that peripheral inflammatory response can affect CNS manifestations. For example, reduced locomotor activity and sickness behaviors caused by fever are attributed to the activation of the brain during peripheral inflammation (Simard and Rivest 2005). Previously, it has been shown that injection of the selective dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) results in chronic neuroinflammation and progressive neurotoxicity in human (Langston et al. 1999) and monkeys (McGeer et al. 2003). However, the mechanism underlying the progressive nature of the disease remains unclear. Recently, our group demonstrated that a single systemic administration of lipopolysaccharide (LPS) results in significant loss of DA neurons beginning at 7-months post treatment (23% loss of TH-IR neurons) and increasing in severity with time to a 47% loss at 10-months post treatment (Qin et al. 2007). This delayed and progressive loss of DA neurons in the SN (over months) recapitulate some of the cardinal features of PD. Results from our animal study is in consonant with a clinical case report documenting that a patient displayed PD-related symptoms after accidental peripheral exposure of LPS (Niehaus 2003). Since it is known that systemically injected LPS cannot readily reach the brain, further studies show that pro-inflammatory factors, such as TNFα underlie the mechanism of systemic LPS-induced DA neurotoxicity (Qin et al. 2007). TNFα produced in the periphery after systemic LPS administration is transported through BBB to reach brain through a TNFα-receptor dependent mechanism. Once TNFα reaches the brain, it will initiate a cascade event by activating TNFα receptors on the microglia, leading to the synthesis of additional TNFα and other pro-inflammatory factors, creating a persistent and self-propelling neuroinflammation that drives delayed and progressive loss of DA neurons in the SN (Fig. 1). Nguyen et al. (2004) also reported that systemic administration of LPS enhanced motor neuron degeneration in animal models of amyotrophic lateral sclerosis 6 months after LPS injection. Systemic exposure to LPS in the neonate was also shown to significantly amplify neuronal death associated with ischemic insult (Lehnardt et al. 2003). The effects of peripheral inflammation on neuron survival may depend upon several factors such as the brain region examined, length of time investigated to allow cumulative effects, age of exposure to the inflammagen, presence of systemic TNFα, and severity of the inflammatory stimuli tested. Thus, it would be interesting to determine epidemiologically whether there is a link between patients surviving septic shock and the incidence of neurodegenerative diseases, such as PD.

Peripheral inflammation induces brain inflammation and neurodegeneration: Role of TNFα in mediating brain inflammation and DA neuron death
Role of microglia in neuroinflammation-mediated neurodegeneration
Among the factors produced by inflammatory cells, the release of superoxide by microglia has been shown to be the predominant factor fueling neurodegeneration, consistent with the notion that DA neurons have an increased vulnerability to oxidative insults. This evidence lends additional support to the strong association of microglial activation with progressive PD (Block et al. 2007; Czlonkowska et al. 1996). Furthermore, we have previously found that the midbrain encompasses the SN contains 4.5 times more microglia when compared to the other brain regions (Kim et al. 2000). Understanding the progressive nature of microglia-mediated neurotoxicity, and the common characteristics/mechanism of microglial activation in response to several diverse toxins, has significant therapeutic importance for many neurodegenerative diseases, since inflammation and microglial activation is a common component of the pathogenesis for multiple neurodegenerative diseases, including Alzheimer’s disease, PD, Huntington’s disease, multiple sclerosis, and amyotrophic lateral sclerosis (Nguyen et al. 2002).
Role of NADPH oxidase and MAC1 in inflammation-induced PD
NADPH oxidase (PHOX), is the major superoxide-producing enzyme of microglia. It is a multi-component enzyme consisting of a membrane-associated cytochrome b558 (composed of 2 subunits: gp91phox and p22phox) and the cytosolic components: p47phox, p67phox, p40phox, and a small GTPase rac2 (Groemping and Rittinger 2005). Upon activation, its cytosolic subunits will translocate to cellular membrane and form a functional enzyme to generate superoxide. Increasing evidence has shown that therapy directed against the oxidative stress response can play a beneficial role in PD (Smith and Zigmond 2003; Jackson-Lewis and Smeyne 2005), as anti-inflammatory agents which function by the inhibition of the NADPH oxidase activity can be neuroprotective (Liu et al. 2003; Qian et al. 2006, 2007a). Both in vivo and in vitro studies from our laboratory using PHOX-deficient mice have clearly demonstrated reduced DA neurotoxicity induced by LPS or MPTP in PHOX−/− compared to PHOX+/+ wild-type mice (Qin et al. 2004; Gao et al. 2003. In addition, a pharmacological PHOX inhibitor diphenyliodonium (DPI), also shows potent DA-neuroprotection in vitro (Qian et al. 2007b). Moreover, PHOX activity also regulates production of pro-inflammatory cytokines such as TNFα by microglia following LPS stimulation (Qin et al. 2004), indicating that PHOX not only mediates superoxide production, but also controls the levels of other pro-inflammatory neurotoxic factors produced by activated microglia.
Further studying the up-stream signaling pathways that regulate the PHOX activity, we found that microglial MAC1 (macrophage antigen complex 1; an adhesion molecule) is closely linked with PHOX and plays an important role in microglia-mediated neuro-inflammation and neurotoxicity. We recently reported that microglial MAC1 was indispensable for the enhanced neurotoxicity induced by LPS, α-synuclein, or MPTP in neuron-glia cultures (Pei et al. 2007; Hu et al. 2008; Zhang et al. 2007), and MAC1-deficient mice show more resistance to MPTP-induced DA neurotoxicity in vivo. Furthermore, it was also reported that NADPH oxidase-generated oxygen free radicals are required for MAC1-mediated phagocytosis in neutrophils (Coxon et al. 1996). Therefore, the coupling between MAC1 and NADPH oxidase might be a central mechanism underlying the reactive microgliosis that mediates immunologic insults and oxidative damage and consequent progressive neurodegeneration.
Role of T cell in mediating neuroinflammation-induced PD
The CNS has traditionally been considered ‘‘immune privileged’’ and protected through the blood–brain barrier. However, recent findings indicate that both innate and adaptive immune systems play critical roles in the pathogenesis of PD (Benner et al. 2008; Olson and Miller 2004). Increasing evidence also demonstrates a role for an adaptive immune response in the etiology of PD. For example, T cell infiltration has been found in CNS tissues of PD patients (Miklossy et al. 2006), and adoptively transferred immune splenocytes into MPTP-treated mice results in significant infiltration into the brain and localization within the inflamed SN (Benner et al. 2008). In addition, there are reports indicated that nitrated a-synuclein activate peripheral leukocytes in draining lymphoid tissue (Benner et al. 2008), and mediated adaptive immune responses in potentiating microglial activation and exacerbating neuronal death. Th17 cells also might contribute to neurotoxicity through the release of pro-inflammatory cytokine IL-17, and secretion of granzyme B, a cytolytic enzyme (Kebir et al. 2007). However, whether this inflammation is the consequence or the cause of neuronal injury is unclear. Recently, Brochard et al. (2009) report that CD4+ T cells are cytotoxic in a MPTP-induced mouse PD model, and invading CD4+ T cell-mediated immune response contributes to DA neurodegeneration through Fas/FasL pathway. These studies implicate that the adaptive immune system, similar to the innate immune system, not only respond to, but also actively participate in the pathogenesis of PD. However, more work need to be done to determine if and how they will serve as a potential target for therapy in PD.
Strategies for anti-inflammatory therapy in PD
Emerging evidence demonstrates that numerous inflammatory mediators such as TNFα, PGE2, NO, free radicals, and potentially other products of activated immune cells can also play a role in the degeneration of nigral dopamine-producing neurons in several models of PD. Therefore, treatment with anti-inflammatory reagents directed at a number of different pro-inflammatory targets could also potentially halt or slow disease progression. Indeed, for example, steroidal anti-inflammatory drugs (SAIDS) such as dexamethasone have been reported to show neuroprotection against MPTP or LPS-induced toxicity (Kurkowska-Jastrzebska et al. 1999; Castano et al. 2002). Non-steroidal anti-inflammatory drugs (NSAIDS) such as aspirin and ibuprofen reduce inflammation by inhibition of COX activity (Sairam et al. 2003). The use of microglia inhibitor such as minocycline has shown potential neuroprotection in PD models (Wu et al. 2002; Du et al. 2001). Recently, strategies by inactivating pro-inflammatory transcription factor NF-ΚB (Zhang et al. 2010), and activating the peroxisome proliferator-activated receptor-γ (PPARγ) (Bernardo et al. 2005), have shown beneficial effects in the modulation of inflammatory responses. Other strategies including inhibiting ion channels in microglial cells (Thomas et al. 2007) are also the highly effective treatments against progressive PD in an animal study, suggesting that there are several critical loci of activation and regulation that control the complex activation pattern of microglial cells in PD. Since extensive discussion of the above-mentioned approaches is beyond the scope of this review, we chose to focus on three relatively new approaches in anti-inflammatory therapy for PD: (1) the use of endogenous anti-inflammatory cytokines IL10 or TGFβ1, as well as the use of regulatory T cells (Tregs, which are the major source of these anti-inflammatory cytokines in vivo), in the resolution of neuroinflammation; (2) the use of morphinan-related compounds; and (3) the targeting of NF-κB, the major transcriptional regulator of inflammation.
Therapies using anti-inflammatory cytokines in PD
Failure to adequately resolve acute inflammation will normally lead to the relatively uncontrolled chronic inflammation seen in neurodegenerative diseases such as PD. Endogenously, anti-inflammatory cytokines serve as negative-feedback regulators that keep potentially pathological activation of immune and immune-like cells under control (Strle et al. 2001; Moore et al. 2001). IL10 and TGFβ1, the two major anti-inflammatory cytokines produced by Tregs, show potent effects in reducing neurotoxicity induced by either LPS or MPTP in PD models. IL10 was shown to function through its inhibition of the production of TNFα, nitric oxide, and extracellular superoxide in microglia cells (Qian et al. 2006). The potent effects of IL10 in the in vitro PD models are consistent with a recent in vivo report that showed sustained administration of IL10 in a viral vector significantly protects DA neuron loss and behavior deficits induced by intra-striatally infused 6-OHDA in a rat PD model (Johnston et al. 2008). Conversely, numerous in vitro studies have shown TGFβ1 can protect neurons from cell death induced by glutamate excitotoxicity (Zhu et al. 2002), chemical hypoxia (Ruocco et al. 1999), apoptosis (Prehn et al. 1994) and oxidative injury (Prehn et al. 1994), and in vivo studies show that TGFβ suppresses the progression of EAE (Szczepanik et al. 2005). In addition, recombinant TGFβ delivered intracerebrally or via virus vectors protects animals against brain injury induced by ischemic (Unsicker and Krieglstein 2002), excitotoxic (Ruocco et al. 1999), and oxidative stress (Henrich-Noack et al. 1996). Although TGFβ has been strongly implicated as a neuroprotective factor, the molecular mechanism underlying its neuroprotection has not been clearly elucidated. Recent evidence from our laboratory has shown that the neuroprotective effects of IL10 and TGFβ1 are mainly attributed to their ability to inhibit the production of ROS from microglia during their activation or reactivation, and the molecular mechanisms appear to be through the inhibition of PHOX activity by preventing the ERK-dependent phosphorylation on p47phox in microlgia to reduce oxidase activities induced by LPS. The exact role that IL10 and TGFβ1 play in the physiological regulation of chronic CNS inflammation in PD, and how they may be used either alone or in synergy to therapeutically treat microglia-mediated neurotoxicity in PD are yet to be determined.
Therapies using regulatory T cells in PD
Another approach to the directed introduction of anti-inflammatory cytokines is the use of Treg cells in therapy for PD. Tregs, which are the primary source of IL10 and TGFβ1 in vivo, are widely recognized as being a major regulatory mechanism which controls both innate and adaptive immune responses. Consequently, they have significant potential as a therapy for inducing neuroprotection in PD. Several studies using MPTP-induced PD model indicate that induction of Tregs response inhibits microglial activation, and promote neuronal survival (Reynolds et al. 2007, 2009a, b). Recent report also demonstrates that Tregs can attenuate Th17 cell-mediated nigrostriatal DA neurodegeneration after MPTP intoxication (Reynolds et al. 2010). Tregs have been proposed to suppress immune reactivity through multiple mechanisms, such as release of IL10 and TGFβ1, induction of apoptotic tolerance, and suppression of metabolic functions in effector cells. In addition, Tregs have also been shown to promote neurotrophic factor production from astrocytes (Reynolds et al. 2007; Stone et al. 2009). Conversely, strategies have been used to induce another type of regulatory T cell, the Th2 cell, which inhibits microglia activation through the production of IL4 and IL10, and further stimulates the synthesis of GDNF from astrocyte, thereby conferring beneficial effects against MPTP-induced neuronal death. For example, copaxone, a polypeptide-based therapy approved for multiple sclerosis patients, is thought to promote the development of Th2 cells which function to dampen the CNS inflammation through the release of anti-inflammatory cytokines and neurotrophic factors (Schwartz and Kipnis 2004; Benner et al. 2004; Angelov et al. 2003. Copaxone-reactive T cells are also neuroprotective in animal models of ALS and PD (Benner et al. 2004; Angelov et al. 2003). However, the interplay between regulatory T cells, glial cells, neurons and other infiltrating leukocytes within the SN is complex and incompletely understood. Therefore, a greater understanding of the modulation of adaptive immune system, and of the infiltration and regulation of inflammation by regulatory T cells within the CNS, is necessary for the development of effective adaptive immune cell-based therapies in PD patients.
Therapies using morphinan-related anti-inflammatory compounds in PD
Morphinans are compounds with structures resembling morphine alkaloid. We have previously reported that several analogs of morphinans, such as naloxone, naltrexone or dextromethorphan are potent anti-inflammatory and neuroprotective compounds (Zhang et al. 2004). Naloxone and naltrexone are non-selective antagonists of the G-protein-coupled opioid receptors. Dextromethorphan, widely used as a cough suppressant, is a dextrorotatory morphinan and therefore, shows little affinity in binding to opioid receptors. Mechanistic studies reveal that neuroprotective effects of both naloxone and dextromethorphan are due to their anti-inflammatory properties by reducing the over-activity of brain microglia and thus dampening the degree of inflammation (Block et al. 2007).
Mechanism underlying the anti-inflammatory and neuroprotective effects of morphinans and its analogs
To search for the binding site mediating the neuroprotective effects of morphinans, we recently conducted an experiment using both levo-morphine (l-morphine) and its synthetic stereo-enantiomer, dextro-morphine (d-morphine), an ineffective opioid receptor agonist. We found that both morphine isomers are equipotent in their anti-inflammatory and neuroprotective effects against LPS or MPP+-induced DA neurotoxicity in neuron-glia cultures (Qian et al. 2007a). We also observed similar neuroprotection mediated by sinomenine, a naturally occurring dextrorotatory morphinan isomer (Qian et al. 2007c). The neuroprotective potency of morphine is also similar to opioid receptor antagonist naloxone (Liu et al. 2000a, b). These findings clearly indicate that morphinans bind to a novel site to exert anti-inflammatory and neuroprotective effects, which is independent of the conventional opioid receptor pathway. We have now identified that the catalytic subunit of PHOX activity, gp91phox is a novel binding site for morphinans (preliminary data). By binding to the gp91phox subunit, morphinans inhibit the activity of PHOX and reduce the production of superoxide and dampen the subsequent release of other pro-inflammatory factors (Qian et al. 2007a).
Recently, after screening a series of dextromethorphan analogs, and 3-hydroxy-morphinan, a metabolite of dextromethorphan, emerged as an excellent candidate for the treatment of PD. Studies using primary midbrain neuron-glia cultures showed that 3-hydroxy-morphinan is more potent in neuroprotection against LPS-induced neurotoxicity than its parent compound (Zhang et al. 2005). The higher potency of this dextromethorphan analog is attributed to its neurotrophic effect in addition to the anti-inflammatory effect shared by both dextromethorphan and 3-hydroxy-morphinan (Zhang et al. 2005). In vivo studies confirmed that 3-hydroxy-morphina is more potent than dextromethorphan in protecting DA neurons in the SN and preventing motor deficits. More significantly, 3-hydroxy-morphinan also attenuated the depletion of striatal levels of dopamine and its metabolites. It is important to point out that in this study, the neuroprotective effects of 3-hydroxy-morphinan were still observed even when this drug was given after MPTP injections (Zhang et al. 2006). This finding suggests the possibility that 3-hydroxy-morphinan can be considered not only for the treatment of PD, but also as a preventive drug.
Therapies targeting pro-inflammatory transcription factor NF-κB in PD
Nuclear transcription factor NF-κB is a family of transcription factors that play an important role in the regulation of chronic diseases and cancer through the promotion of inflammation and of cell survival/oncogenesis. Compounds that block the activation of NF-κB are capable of inhibiting the two major inflammatory pathways in microglia—activation of oxidative stress and pro-inflammatory cytokine and chemokine production (Anrather et al. 2006; Gauss et al. 2007; Kim et al. 2006), NF-κB activation is detected within the SN of PD patients and a PD animal model created by MPTP (Ghosh et al. 2007; Hunot et al. 1997), and of particular interest is marked colocalization of NF-κB-p65 with CD11b-positive activated microglia in the SN of postmortem PD brains (Ghosh et al. 2007). These findings indicate that inflammatory components that are regulated by NF-κB play a significant role in the pathogenesis of PD. Activation of NF-κB requires the activity of IκB kinase (IKK) complex, in a manner dependent mainly on the IKK-β catalytic subunit (Karin 1999). Previous reports have shown that selective inhibition of NF-κB activation using a peptide against the IKK complex suppressed microglial activation and prevented DA neuronal loss against MPTP-intoxicated PD mice (Ghosh et al. 2007). Similarly, inhibition of NF-κB activity is involved in the neuroprotection of pioglitazone (a PPARγ agonist) against LPS-induced DA neurotoxicity (Xing et al. 2007). Recently, a pharmacological inhibitor of IKK-β generated neuroprotection against LPS-induced toxicity both in vitro and in vivo (Zhang et al. 2010), and the mechanistic studies using this compound showed that its neuroprotective effects were mediated by suppressing the activity of NADPH oxidase and the NF-κB signaling pathway in microglia. Therefore, it appears as though NF-κB is also a strong potential target for anti-inflammatory therapy in the treatment of PD.
Conclusion
Neuroinflammation, which is characterized by activated microglia and infiltrating T cells at sites of neuronal injury, is a prominent contributor to the pathogenesis of progressive PD. Microglia play a critical role in forming a self-propelling cycle leading to sustained chronic neuroinflammation and driving the progressive neurodegeneration in PD. This activation depends heavily on the respiratory burst within the microglia, which in turn regulates a number of downstream pro-inflammatory activities, including NF-κB activation and pro-inflammatory mediator production. Anti-inflammatory therapies have already been developed in animal model systems of PD that demonstrate inhibition of the NADPH oxidase response by microglia is a highly potent and effective approach for inhibiting the progression of neurodegeneration induced by either a strong pro-inflammatory signal such as LPS, or even by DA neuron-specific toxins such as MPTP and 6-OHDA. These therapies, which include direct inhibition of NADPH-mediated ROS production by DPI, or inhibition of NADPH oxidase activation by anti-inflammatory cytokines, morphinan compounds, or NF-κB inhibitors, demonstrate that the oxidative stress response by microglia is a key target for future development of therapies that effectively treat PD. On the other hand, the adaptive immune responses, most notably T cells, are now emerging as important components of the inflammatory response that contribute to the pathogenesis of PD. It remains to be determined how these adaptive immune cells, including Treg, Th2, Th17, or other T cell subsets, will serve as effective targets for immune therapy in PD. A further understanding of the inflammatory etiology of PD, as well as better analysis of the molecular signaling involved in this inflammatory response, should help to identify these new targets for immune-based therapy in neurodegenerative disorders such as PD.
References
Abou-Sleiman PM, Healy DG, Quinn N, Lees AJ, Wood NW (2003) The role of pathogenic DJ-1 mutations in Parkinson’s disease. Ann Neurol 54:283–286
Angelov DN, Waibel S, Guntinas-Lichius O, Lenzen M, Neiss WF, Tomov TL, Yoles E, Kipnis J, Schori H, Reuter A, Ludolph A, Schwartz M (2003) Therapeutic vaccine for acute and chronic motor neuron diseases: implications for amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 100:4790–4795
Anrather J, Racchumi G, Iadecola C (2006) NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem 281:5657–5667
Bartels AL, Leenders KL (2007) Neuroinflammation in the pathophysiology of Parkinson’s disease: evidence from animal models to human in vivo studies with [11C]-PK11195 PET. Mov Disord 22:1852–1856
Benner EJ, Mosley RL, Destache CJ, Lewis TB, Jackson-Lewis V, Gorantla S, Nemachek C, Green SR, Przedborski S, Gendelman HE (2004) Therapeutic immunization protects dopaminergic neurons in a mouse model of Parkinson’s disease. Proc Natl Acad Sci USA 101:9435–9440
Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, Nemachek C, Ciborowski P, Przedborski S, Mosley RL, Gendelman HE (2008) Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS One 3:e1376
Bernardo A, Ajmone-Cat MA, Gasparini L, Ongini E, Minghetti L (2005) Nuclear receptor peroxisome proliferator-activated receptor-gamma is activated in rat microglial cells by the anti-inflammatory drug HCT1026, a derivative of flurbiprofen. J Neurochem 92:895–903
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69
Bonifati V, Wu-Chou YH, Schweiger D, Fonzo AD, Lu CS, Oostra B (2008) Re: LRRK2 mutation analysis in Parkinson disease families with evidence of linkage to PARK8. Neurology 70:2348–2349
Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, Duyckaerts C, Flavell RA, Hirsch EC, Hunot S (2009) Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest 119:182–192
Castano A, Herrera AJ, Cano J, Machado A (2002) The degenerative effect of a single intranigral injection of LPS on the dopaminergic system is prevented by dexamethasone, and not mimicked by rh-TNF-alpha, IL-1beta and IFN-gamma. J Neurochem 81:150–157
Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O (2002) Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur J Neurosci 15:991–998
Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, Arnaout MA, Mayadas TN (1996) A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity 5:653–666
Czlonkowska A, Kohutnicka M, Kurkowska-Jastrzebska I, Czlonkowski A (1996) Microglial reaction in MPTP (1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine) induced Parkinson’s disease mice model. Neurodegeneration 5:137–143
Dauer W, Przedborski S (2003) Parkinson’s disease: mechanisms and models. Neuron 39:889–909
Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DL, Luecke S, Phebus LA, Bymaster FP, Paul SM (2001) Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci USA 98:14669–14674
Gao HM, Hong JS (2008) Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol 29:357–365
Gao HM, Liu B, Zhang W, Hong JS (2003) Critical role of microglial NADPH oxidase-derived free radicals in the in vitro MPTP model of Parkinson’s disease. Faseb J 17:1954–1956
Gauss KA, Nelson-Overton LK, Siemsen DW, Gao Y, DeLeo FR, Quinn MT (2007) Role of NF-kappaB in transcriptional regulation of the phagocyte NADPH oxidase by tumor necrosis factor-alpha. J Leukoc Biol 82:729–741
Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ, Hartley DM, Ghosh S, Mosley RL, Gendelman HE, Pahan K (2007) Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci USA 104:18754–18759
Groemping Y, Rittinger K (2005) Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J 386:401–416
Henrich-Noack P, Prehn JH, Krieglstein J (1996) TGF-beta 1 protects hippocampal neurons against degeneration caused by transient global ischemia. Dose–response relationship and potential neuroprotective mechanisms. Stroke 27:1609–1614 (discussion 1615)
Hirsch EC, Hunot S (2009) Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol 8:382–397
Hu X, Zhang D, Pang H, Caudle WM, Li Y, Gao H, Liu Y, Qian L, Wilson B, Di Monte DA, Ali SF, Zhang J, Block ML, Hong JS (2008) Macrophage antigen complex-1 mediates reactive microgliosis and progressive dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. J Immunol 181:7194–7204
Hunot S, Brugg B, Ricard D, Michel PP, Muriel MP, Ruberg M, Faucheux BA, Agid Y, Hirsch EC (1997) Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with Parkinson disease. Proc Natl Acad Sci USA 94:7531–7536
Jackson-Lewis V, Smeyne RJ (2005) MPTP and SNpc DA neuronal vulnerability: role of dopamine, superoxide and nitric oxide in neurotoxicity. Minireview. Neurotox Res 7:193–202
Jellinger KA (2001) The pathology of Parkinson’s disease. Adv Neurol 86:55–72
Jiang H, Wu YC, Nakamura M, Liang Y, Tanaka Y, Holmes S, Dawson VL, Dawson TM, Ross CA, Smith WW (2007) Parkinson’s disease genetic mutations increase cell susceptibility to stress: mutant alpha-synuclein enhances H2O2- and Sin-1-induced cell death. Neurobiol Aging 28:1709–1717
Johnston LC, Su X, Maguire-Zeiss K, Horovitz K, Ankoudinova I, Guschin D, Hadaczek P, Federoff HJ, Bankiewicz K, Forsayeth J (2008) Human interleukin-10 gene transfer is protective in a rat model of Parkinson’s disease. Mol Ther 16:1392–1399
Karin M (1999) How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene 18:6867–6874
Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A (2007) Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med 13:1173–1175
Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS (2000) Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci 20:6309–6316
Kim HJ, Hawke N, Baldwin AS (2006) NF-kappaB and IKK as therapeutic targets in cancer. Cell Death Differ 13:738–747
Kurkowska-Jastrzebska I, Wronska A, Kohutnicka M, Czlonkowski A, Czlonkowska A (1999) The inflammatory reaction following 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine intoxication in mouse. Exp Neurol 156:50–61
Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D (1999) Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine exposure. Ann Neurol 46:598–605
Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T (2003) Activation of innate immunity in the CNS triggers neurodegeneration through a toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA 100:8514–8519
Liu B, Du L, Hong JS (2000a) Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharmacol Exp Ther 293:607–617
Liu B, Jiang JW, Wilson BC, Du L, Yang SN, Wang JY, Wu GC, Cao XD, Hong JS (2000b) Systemic infusion of naloxone reduces degeneration of rat substantia nigral dopaminergic neurons induced by intranigral injection of lipopolysaccharide. J Pharmacol Exp Ther 295:125–132
Liu Y, Qin L, Li G, Zhang W, An L, Liu B, Hong JS (2003) Dextromethorphan protects dopamanergic neurons against inflammation-mediated degeneration through inhibition of microglial activation. J Pharmacol Exp Ther 305:1–7
Loeffler DA, DeMaggio AJ, Juneau PL, Havaich MK, LeWitt PA (1994) Effects of enhanced striatal dopamine turnover in vivo on glutathione oxidation. Clin Neuropharmacol 17:370–379
McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38:1285–1291
McGeer PL, Yasojima K, McGeer EG (2001) Inflammation in Parkinson’s disease. Adv Neurol 86:83–89
McGeer PL, Schwab C, Parent A, Doudet D (2003) Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine administration. Ann Neurol 54:599–604
Miklossy J, Doudet DD, Schwab C, Yu S, McGeer EG, McGeer PL (2006) Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Exp Neurol 197:275–283
Mizuno Y, Hattori N, Kitada T, Matsumine H, Mori H, Shimura H, Kubo S, Kobayashi H, Asakawa S, Minoshima S, Shimizu N (2001) Familial Parkinson’s disease. Alpha-synuclein and parkin. Adv Neurol 86:13–21
Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T (1994) Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett 180:147–150
Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A (2001) Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 19:683–765
Nagatsu T, Mogi M, Ichinose H, Togari A (2000) Changes in cytokines and neurotrophins in Parkinson’s disease. J Neural Transm Suppl 60:277–290
Nguyen MD, Julien JP, Rivest S (2002) Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci 3:216–227
Nguyen MD, D’Aigle T, Gowing G, Julien JP, Rivest S (2004) Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis. J Neurosci 24:1340–1349
Niehaus I (2003) Lippopolysaccharides induce inflammation-mediated neurodeheneration in the substantia nigra and the cerebral cortex (a case report). AD/PD 6th International Conference, pp 1–38
Olson JK, Miller SD (2004) Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol 173:3916–3924
Pei Z, Pang H, Qian L, Yang S, Wang T, Zhang W, Wu X, Dallas S, Wilson B, Reece JM, Miller DS, Hong JS, Block ML (2007) MAC1 mediates LPS-induced production of superoxide by microglia: the role of pattern recognition receptors in dopaminergic neurotoxicity. Glia 55:1362–1373
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Prehn JH, Bindokas VP, Marcuccilli CJ, Krajewski S, Reed JC, Miller RJ (1994) Regulation of neuronal Bcl2 protein expression and calcium homeostasis by transforming growth factor type beta confers wide-ranging protection on rat hippocampal neurons. Proc Natl Acad Sci USA 91:12599–12603
Qian L, Block ML, Wei SJ, Lin CF, Reece J, Pang H, Wilson B, Hong JS, Flood PM (2006) Interleukin-10 protects lipopolysaccharide-induced neurotoxicity in primary midbrain cultures by inhibiting the function of NADPH oxidase. J Pharmacol Exp Ther 319:44–52
Qian L, Tan KS, Wei SJ, Wu HM, Xu Z, Wilson B, Lu RB, Hong JS, Flood PM (2007a) Microglia-mediated neurotoxicity is inhibited by morphine through an opioid receptor-independent reduction of NADPH oxidase activity. J Immunol 179:1198–1209
Qian L, Gao X, Pei Z, Wu X, Block M, Wilson B, Hong JS, Flood PM (2007b) NADPH oxidase inhibitor DPI is neuroprotective at femtomolar concentrations through inhibition of microglia over-activation. Parkinsonism Relat Disord 13(Suppl 3):S316–S320
Qian L, Xu Z, Zhang W, Wilson B, Hong JS, Flood PM (2007c) Sinomenine, a natural dextrorotatory morphinan analog, is anti-inflammatory and neuroprotective through inhibition of microglial NADPH oxidase. J Neuroinflammation 4:23
Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS (2004) NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem 279:1415–1421
Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT (2007) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55:453–462
Qureshi GA, Baig S, Bednar I, Sodersten P, Forsberg G, Siden A (1995) Increased cerebrospinal fluid concentration of nitrite in Parkinson’s disease. Neuroreport 6:1642–1644
Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL (2007) Neuroprotective activities of CD4+ CD25+ regulatory T cells in an animal model of Parkinson’s disease. J Leukoc Biol 82:1083–1094
Reynolds AD, Stone DK, Mosley RL, Gendelman HE (2009a) Nitrated {alpha}-synuclein-induced alterations in microglial immunity are regulated by CD4+ T cell subsets. J Immunol 182:4137–4149
Reynolds AD, Stone DK, Mosley RL, Gendelman HE (2009b) Proteomic studies of nitrated alpha-synuclein microglia regulation by CD4+ CD25+ T cells. J Proteome Res 8:3497–3511
Reynolds AD, Stone DK, Hutter JA, Benner EJ, Mosley RL, Gendelman HE (2010) Regulatory T cells attenuate th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson’s disease. J Immunol 184:2261–2271
Ruocco A, Nicole O, Docagne F, Ali C, Chazalviel L, Komesli S, Yablonsky F, Roussel S, MacKenzie ET, Vivien D, Buisson A (1999) A transforming growth factor-beta antagonist unmasks the neuroprotective role of this endogenous cytokine in excitotoxic and ischemic brain injury. J Cereb Blood Flow Metab 19:1345–1353
Sairam K, Saravanan KS, Banerjee R, Mohanakumar KP (2003) Non-steroidal anti-inflammatory drug sodium salicylate, but not diclofenac or celecoxib, protects against 1-methyl-4-phenyl pyridinium-induced dopaminergic neurotoxicity in rats. Brain Res 966:245–252
Schwartz M, Kipnis J (2004) A common vaccine for fighting neurodegenerative disorders: recharging immunity for homeostasis. Trends Pharmacol Sci 25:407–412
Simard AR, Rivest S (2005) Do pathogen exposure and innate immunity cause brain diseases? Neurol Res 27:717–725
Smith AD, Zigmond MJ (2003) Can the brain be protected through exercise? Lessons from an animal model of Parkinsonism. Exp Neurol 184:31–39
Stone DK, Reynolds AD, Mosley RL, Gendelman HE (2009) Innate and adaptive immunity for the pathobiology of Parkinson’s disease. Antioxid Redox Signal 11:2151–2166
Strle K, Zhou JH, Shen WH, Broussard SR, Johnson RW, Freund GG, Dantzer R, Kelley KW (2001) Interleukin-10 in the brain. Crit Rev Immunol 21:427–449
Sun M, Latourelle JC, Wooten GF, Lew MF, Klein C, Shill HA, Golbe LI, Mark MH, Racette BA, Perlmutter JS, Parsian A, Guttman M, Nicholson G, Xu G, Wilk JB, Saint-Hilaire MH, DeStefano AL, Prakash R, Williamson S, Suchowersky O, Labelle N, Growdon JH, Singer C, Watts RL, Goldwurm S, Pezzoli G, Baker KB, Pramstaller PP, Burn DJ, Chinnery PF, Sherman S, Vieregge P, Litvan I, Gillis T, MacDonald ME, Myers RH, Gusella JF (2006) Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: the GenePD study. Arch Neurol 63:826–832
Szczepanik M, Tutaj M, Bryniarski K, Dittel BN (2005) Epicutaneously induced TGF-beta-dependent tolerance inhibits experimental autoimmune encephalomyelitis. J Neuroimmunol 164:105–114
Tanner CM (2003) Is the cause of Parkinson’s disease environmental or hereditary? Evidence from twin studies. Adv Neurol 91:133–142
Thomas MP, Chartrand K, Reynolds A, Vitvitsky V, Banerjee R, Gendelman HE (2007) Ion channel blockade attenuates aggregated alpha synuclein induction of microglial reactive oxygen species: relevance for the pathogenesis of Parkinson’s disease. J Neurochem 100:503–519
Unsicker K, Krieglstein K (2002) TGF-betas and their roles in the regulation of neuron survival. Adv Exp Med Biol 513:353–374
Weng YH, Chou YH, Wu WS, Lin KJ, Chang HC, Yen TC, Chen RS, Wey SP, Lu CS (2007) PINK1 mutation in Taiwanese early-onset parkinsonism : clinical, genetic, and dopamine transporter studies. J Neurol 254:1347–1355
Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S (2002) Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci 22:1763–1771
Xing B, Liu M, Bing G (2007) Neuroprotection with pioglitazone against LPS insult on dopaminergic neurons may be associated with its inhibition of NF-kappaB and JNK activation and suppression of COX-2 activity. J Neuroimmunol 192:89–98
Zhang W, Hong JS, Kim HC, Block ML (2004) Morphinan neuroprotection: new insight into the therapy of neurodegeneration. Crit Rev Neurobiol 16:271–302
Zhang W, Qin L, Wang T, Wei SJ, Gao HM, Liu J, Wilson B, Liu B, Kim HC, Hong JS (2005) 3-hydroxymorphinan is neurotrophic to dopaminergic neurons and is also neuroprotective against LPS-induced neurotoxicity. Faseb J 19:395–397
Zhang W, Shin EJ, Wang T, Lee PH, Pang H, Wie MB, Kim WK, Kim SJ, Huang WH, Wang Y, Zhang W, Hong JS, Kim HC (2006) 3-Hydroxymorphinan, a metabolite of dextromethorphan, protects nigrostriatal pathway against MPTP-elicited damage both in vivo and in vitro. Faseb J 20:2496–2511
Zhang W, Dallas S, Zhang D, Guo JP, Pang H, Wilson B, Miller DS, Chen B, McGeer PL, Hong JS, Zhang J (2007) Microglial PHOX and Mac-1 are essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T mutant alpha-synuclein. Glia 55:1178–1188
Zhang F, Qian L, Flood PM, Shi JS, Hong JS, Gao HM (2010) Inhibition of I{kappa}B kinase-{beta} (IKK-{beta}) protects dopamine neurons against lipopolysaccharide-induced neurotoxicity. J Pharmacol Exp Ther 333:822–823
Zhu Y, Yang GY, Ahlemeyer B, Pang L, Che XM, Culmsee C, Klumpp S, Krieglstein J (2002) Transforming growth factor-beta 1 increases bad phosphorylation and protects neurons against damage. J Neurosci 22:3898–3909
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Qian, L., Flood, P.M. & Hong, JS. Neuroinflammation is a key player in Parkinson’s disease and a prime target for therapy. J Neural Transm 117, 971–979 (2010). https://doi.org/10.1007/s00702-010-0428-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-010-0428-1