
Abstract
Once viewed as an isolated, immune-privileged organ, the central nervous system has undergone a conceptual change. Neuroinflammation has moved into the focus of research work regarding pathomechanisms underlying perinatal brain damage. In this review, we provide an overview of current concepts regarding perinatal brain damage and the role of inflammation in the disease pathomechanism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Injury to the developing perinatal brain is a leading cause of death and disability in children, and neurological handicap of perinatal origin has not decreased significantly in western countries within the last decades despite medical advances (Hagberg et al. 1996; Himmelmann et al. 2005; Vincer et al. 2006). The pathophysiology of perinatal brain damage has proven to be multifactorial including sensitizing/exacerbating factors such as hypoxia–ischemia, excessive glutamate release leading to excitotoxicity, oxidative stress and intracranial (sometimes secondary) inflammation. It has been acknowledged for quite some time that neuronal injury or a potent local immune stimulation can induce a cascade of immune responses within the CNS. However, until recently, the central nervous system (CNS) was viewed as an immune-privileged organ well isolated from its environment. Emerging research data expand the concept of neuroinflammation. There is growing evidence of an important cross talk between the CNS and the periphery, and the deleterious effects that systemic inflammation can have on the developing brain. It is now clear that neuroinflammation is linked to the aggravation of several diseases affecting the developing CNS. The impact and mechanisms of action of neuroinflammation is likely to differ between the developing and the adult CNS. In this review, we provide a brief overview of the major causes of perinatal brain damage and further highlight selected pathomechanisms underlying the role of neuroinflammation in perinatal brain damage. Thereby, with some conceptual bias of the authors, we summarize current concepts and ideas in the field.
Perinatal brain damage
Epidemiological point of view
The frequency of motor and/or cognitive handicaps linked to perinatal brain injury increased during the 1990s and currently seems to remain stable (Robertson et al. 2007; Vincer et al. 2006; Wilson-Costello et al. 2007). This epidemiological evidence can be explained by the progress in the field of assisted reproductive technology and intensive care that led to an increase of preterm neonate (23–32 weeks of gestation) births and survival rates of about 70% without significant improvement in the mean neurological outcome. As much as 10% of preterm neonates with birth weights <1,500 g later exhibit cerebral palsy, and about 50% of them develop cognitive and behavioral deficits (Wilson-Costello et al. 2005).
Clinical point of view
The consequences of perinatal CNS injury range among a spectrum of disorders such as mild to severe mental retardation, cerebral palsy, epilepsy, vision and hearing disorders, learning difficulties and school failure. Periventricular white matter injury, including periventricular leukomalacia, is most frequently observed in human preterm neonates (Volpe 2009). Full-term human neonates with perinatal encephalopathy generally develop gray matter damage that most frequently affects the neocortex, the basal ganglia and the hippocampus (Volpe 2001). Perinatal stroke and corresponding occlusion of cerebral arteries or veins can have sequels such as cerebral palsy, epilepsy and vision, language and behavior disorders (Kirton and deVeber 2009).
Experimental model and pathophysiology of perinatal brain damage
During the last 15 years, the etiology of perinatal brain damage has been considered by many to be multifactorial rather than simply linked to cardiovascular instability and hypoxia–ischemia (Dammann and Leviton 1997; Nelson and Grether 1999). Several prenatal, perinatal and postnatal factors have been implicated in the pathophysiology of brain lesions associated with cerebral palsy, including hypoxic–ischemic insults, maternal infection yielding excess cytokines and other pro-inflammatory agents, excess release of glutamate initiating the excitotoxic cascade (Dammann et al. 2002; Degos et al. 2008; Mesples et al. 2005; Nelson and Chang 2008), oxidative stress/hyperoxia, growth factor deficiency, exposure to medication and drugs of abuse, and maternal stress (Dammann and Leviton 1997; Dommergues et al. 2000; Follett et al. 2004, 2000; Gressens et al. 1997; Hagberg et al. 2002; Haynes et al. 2003; Inder et al. 2002; Laudenbach et al. 2001; Loeliger et al. 2003; Plaisant et al. 2003; Tahraoui et al. 2001; Volpe 2001). In addition, recent clinical studies support the existence of genetic factors for susceptibility (Harding et al. 2004). Although some of the potentially harmful factors are present in utero and are sufficient to cause permanent injury to the developing brain prior to neonatal life, several groups have hypothesized that some of these act as predisposing or sensitizing factors (“pro-damage conditions”), increasing the susceptibility to injury when a second unfavorable event occurs (Dammann et al. 2002; Dommergues et al. 2000; Eklind et al. 2001; Gressens et al. 2002; Nelson and Willoughby 2000).
Various animal models of perinatal brain damage have been established (Hagberg et al. 2002), allowing a detailed analysis of the underlying molecular and cellular mechanisms and the effect of potentially neuroprotective strategies. Perinatal brain damage is principally mimicked by infectious, inflammatory, excitotoxic or hypoxic–ischemic insults in mammals such as newborn rodents, fetal rabbits, cats and rats, as well as rabbits and sheep (Hagberg et al. 2002). Brain lesions of full-term neonates are generally mimicked by hypoxic–ischemic or excitotoxic insults in newborn rodents, rabbits, piglets and dogs (Derrick et al. 2004; Hagberg et al. 2002; Johnson et al. 1987).
Systemic inflammation and cross talk with the developing CNS
Systemic inflammation in the newborn
Epidemiological studies have shown a strong association between fetal infection secondary to systemic inflammation (chorioamnionitis) and brain damage and/or neurological handicap in survivors (Dammann and Leviton 2007). This association between systemic inflammation and neurological outcome was also observed in adult disease such as stroke and traumatic brain injury (Lim and Smith 2007; Lu et al. 2009; McColl et al. 2009). To mimic a systemic inflammatory syndrome, experimental studies classically use pro-inflammatory cytokines injection such as interleukin-1beta (IL-1β) or administration of endotoxin such as lipopolysaccharide (LPS), an essential component of Gram-negative bacteria.
Experimental studies have allowed confirming a sensitizing effect of systemic inflammation on perinatal brain lesions induced by hypoxic–ischemic or excitotoxic insults (Dommergues et al. 2000; Eklind et al. 2005). In addition, some experimental data also suggest that perinatal exposure to infectious/inflammatory factors can alter the developmental programs of the brain and thereby result in lasting neurological deficits. The relationship between this latter observation and human diseases remains to be fully demonstrated, although clinical evidence is supportive of this hypothesis (Volpe 2009).
The neuronal sensitizing effect
As perinatal brain damage is caused by a combination of several insults, experimental studies try to reproduce this clinical setting in multiple hit models including systemic infection/inflammation. Inflammation acts as a predisposing factor rendering the brain more susceptible to a second stressor (sensitization process). In this line, systemic injection of low doses of LPS to neonatal rats leaves their brain highly susceptible to a hypoxic–ischemic insult (Eklind et al. 2005). Similarly, systemic injection of IL-1β or LPS to newborn mice or rats renders their brains much more sensitive to an excitotoxic insult (Dommergues et al. 2000). In current studies, the time window during which sensitization of the brain will persist after exposure to inflammatory factors in the perinatal period is being evaluated. The sensitization mechanisms are not yet fully understood, but could include changes in gene transcription and modifications of glutamate receptor activity. The metabotropic glutamate group 1 receptors are specifically involved in the sensitizing effects in different models of perinatal excitotoxic brain damage via phospholipase C β1 (PLC β1). In vitro, the IL-1β sensitizing effect was reproduced using murine cortical neurons. After a glutamate agonist excitation, these IL-1β pre-treated neurons present a massive increase of calcium release leading eventually to apoptotic cell death (Fig. 1; V. Degos, personal communication). This sensitizing effect was also found in hypoxic–ischemic models in heterozygous G protein-coupled receptor (GPCR) kinase-2 (GRK2) knockout mice (Nijboer et al. 2008), but the exact link between GRK2 and inflammation needs further clarification.

Schematic representation of the neuronal sensitizing effect of systemic inflammation increasing the susceptibility of brain injuries
Disruption of brain development
Isolated systemic infection/inflammatory factors can also alter brain development even if they do not induce major destructive lesions. Accordingly, injection of E. coli into pregnant rabbits induces diffuse white matter cell death (Debillon et al. 2000), and injection of Ureaplasma parvum, a frequently observed pathogen in chorioamnionitis, into pregnant mice induces myelin defects and loss of interneurons in their offspring (Normann et al. 2009). Similarly, injection of LPS into pregnant rats induces transient CNS inflammation and myelination defects in their offspring (Rousset et al. 2006). Of major concern, exposure of newborn mice to low doses of systemic IL-1β, inducing a moderate and transient inflammatory response during the neonatal period, is sufficient to alter the transcription of genes implicated in oligodendrogenesis, myelin formation and axonal maturation. Furthermore, findings from animal models show a maturation-dependent vulnerability of oligodendrocyte lineage to the detriment of pre-oligodendrocytes, by several cytotoxic pathways. Pre-oligodendrocytes are the main component of white matter between 23 and 32 weeks of post-conceptional age. In vitro, TNF-α shows a cytotoxic effect on oligodendrocytes (Feldhaus et al. 2004), and IL-1β disrupts physiologic proliferative and maturational processes of the oligodendroglia lineage (Vela et al. 2002). As endothelial cells participate actively in the cross talk between systemic and intracerebral inflammation, the specific role of intracerebral inflammation in angiogenesis remains to be studied.
Perinatal blood–brain barrier and cross talk between the periphery and CNS
During brain development, there are emergence of two interfaces between blood, brain and cerebrospinal fluid (CSF). These barriers are the blood—CNS barrier (microvessels) and blood—CSF barrier (choroid plexus). Traditionally, these two types of barriers are grouped under the name of the blood–brain barrier (BBB), although histologically and developmentally these two barriers are different. The barrier concept is explained by the fact that there are intercellular tight junctions that prevent or hinder the passage of molecules between blood, brain and CSF, and in the opposite direction. However, there are different possibilities for entrance and exit based on specific transport mechanisms, which are essential for normal development and function of the brain. Recent studies consider that the choroid plexus, which secretes much of the CSF, is the main route of entry of molecules into the brain (Ghersi-Egea et al. 2006; Strazielle and Ghersi-Egea 2000). The BBB in the neonate is substantially more mature than is commonly thought. The tight junctions are present early in embryonic development (Kniesel et al. 1996), restricting the entrance of proteins into the brain, and the BBB in newborn is functional with no fenestrations (Engelhardt 2003; Ek et al. 2006, 2001). The presence of the barrier substantially affects leukocyte passage, but does not guarantee minimal leukocyte transmigration. The application of a direct inflammatory challenge through intrastriatal injections of IL-1β or TNF-α in rats did not show a linear decline of leukocyte transmigration with age (Anthony et al. 1997); in contrast, the newborn CNS proved to be more resistant to an inflammatory stressor than the juvenile brain. The reported magnitude of BBB disturbance following hypoxia–ischemia or focal stroke in neonatal rodents varies and depends on the analyzed barrier feature (Faustino et al. 2009; Svedin et al. 2007). Degradation of the extracellular matrix plays a role in neonatal ischemic injury. Excessive activation of the matrix metalloproteinase 9 (MMP-9) early after hypoxia–ischemia is deleterious to the immature brain. In the latter model, the lesion size is significantly smaller in MMP-9 knockout mice (Svedin et al. 2007) and following a pharmacological inhibition of this protease (Leonardo et al. 2008).
The precise molecular mechanisms by which circulating mediators of inflammation have a deleterious effect on perinatal brain lesions are unclear (Fig. 2; Hagberg and Mallard 2005). Circulating cytokines do not seem to cross the intact BBB easily, although this is still a mater of debate. Various alternative pathways have been proposed to link serum cytokines with brain damage (Malaeb and Dammann 2009). Firstly, circulating cytokines could alter the permeability of the BBB to inflammatory mediators and cells. Secondly, circulating cytokines could act directly on parts of the brain lacking BBB such as the circumventricular organs, meninges and choroid plexus, or, as demonstrated in the adult brain, indirectly through the activation of the vagal nerve. Thirdly, cytokine effects could be mediated by cyclooxygenase (COX) located on the BBB. In particular, cytokines could activate the inducible isoform COX-2 to enhance the local production of prostaglandin E2 (PGE2) that could have deleterious effects on the developing brain. This latter mechanism has been demonstrated in a mouse model of perinatal excitotoxic brain damage (Favrais et al. 2007). Some of these deleterious effects could involve an autocrine/paracrine loop leading to an excess production of inflammatory cytokines by brain cells.

Schematic representation of the different potential mechanisms by which intracerebral inflammation can lead to neuronal cell death
CNS inflammation
Specific effects of intracerebral cytokines and chemokines
Clinical and experimental data on the pathophysiologic role of inflammatory cytokines in perinatal term and preterm brain damage continue to emerge (Fig. 3; Bartha et al. 2004). Overall, IL-1β potentiates ischemic brain injury. Following hypoxia–ischemia or transient middle cerebral artery occlusion in neonatal rats, brain IL-1β mRNA expression is increased rapidly (Hagberg et al. 1996; Denker et al. 2007; Foster-Barber and Ferriero 2002; Grether and Nelson 1997) followed by a major rapid systemic increase of IL-1β protein (Denker et al. 2007). Brain IL-1β levels can be further amplified by concomitant infection or manipulations within the oxidant pathways (Doverhag et al. 2008; Girard et al. 2008). The pro-inflammatory shift in balance between IL-1β and IL-1ra, the physiological antagonist for the IL-1 receptor, following hypoxia–ischemia when combined with infection (modeled by endotoxin exposure) can play a role in the initiation of perinatal brain damage (Girard et al. 2008). IL-1β-induced local inflammatory reaction, chemokine expression and subsequent leukocyte attraction as well as BBB disruption greatly depend on age (Anthony et al. 1997). However, neither a genetic deletion of IL-1β or IL-1α alone nor a combined IL-1αβ knockout protected against hypoxic–ischemic injury (Hedtjarn et al. 2005). This suggests the presence of multiple and/or asynchronized pro- and anti-injurious effects of IL-1β, which are all abrogated in knockout mice. IL-18, a pro-inflammatory cytokine of the IL-1 family, also contributes to hypoxic–ischemic injury in newborn animals (Hedtjarn et al. 2002).

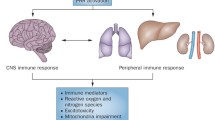
Schematic representation of the cross talk between systemic inflammation and CNS
TNF-α exhibits pleiotropic functions in the ischemic adult brain depending on the cell producing TNF-α and on the type of receptor involved. TNF-α can induce apoptosis by reacting with Fas-associated death domain (FADD) and caspase-8 with signaling through the TNF receptor 2 (TNFR2) leading to anti-inflammatory and anti-apoptotic effects. In neonatal brain injury, the pathophysiologic role of TNF-α appears to be more complex. A key role of TNF-α in injury in an excitotoxic model combined with prior inflammatory challenge (IL-1β) has been recently demonstrated (Adén et al. 2010). The TNF-α blocker etanercept did not influence brain damage when given prior to injury, but substantially ameliorated injury when given after the combined inflammatory and excitotoxic insult. The latter suggests that TNF-α can act by producing an imbalance between pro- and anti-inflammatory cytokines in the injured brain (Adén et al. 2010).
IL-10, a Th2 cytokine that is synthesized in the CNS and can act both on hematopoietic and non-hematopoietic cells, can reverse injury caused by IL-1β, TNF-α and IL-6. In a mouse model of neonatal excitotoxic brain damage, exogenously administered IL-10 affords neuroprotection (Mesples et al. 2003). Interestingly, in the same model, IL-10 knockout mice displayed brain damage comparable to wild-type mice, supporting the hypothesis that newborn pups are unable to mount an IL-10 response following perinatal brain damage (Mesples et al. 2003). This lack of anti-inflammatory response in the neonatal period could contribute to the high sensitivity of the perinatal brain to inflammatory insults.
The IL-9/IL-9 receptor pathway, which is most active in the newborn brain, has been shown to have direct anti-apoptotic action in the newborn neocortex (Fontaine et al. 2008), but at the same time to contribute to hypoxia–ischemia and excitotoxic injury in the developing brain, presumably by activating mast cells (Dommergues et al. 2000; Patkai et al. 2001).
Chemokines, small polypeptides that play a role in intracellular communication and recruitment of inflammatory cells, play also a key role in the cross talk between peripheral and CNS responses. The pathophysiologic role of the monocyte chemoattractant protein 1 (MCP-1) chemokine in neonatal brain injury is evident as its functional inactivation after an insult has proved to be protective (Galasso et al. 2000) or in mice with depleted IL-1β converting enzyme. The role of the CXC-family chemokines after neonatal brain injury is less well understood, but may be crucial for BBB regulation after an inflammatory challenge of an injured immature brain (Anthony et al. 1998). The stromal cell-derived factor 1 (SDF-1, syn. CXCL12) has been suggested to play a role in homing stem cells to regions of ischemic injury in the adult (Miller and Tran 2005). However, its role in the neonate remains to be confirmed.
Microglia-macrophage, astrocyte and mast cell activation
Microglia-macrophages of extra-cerebral origin enter the human brain early in embryonic and fetal development in two waves, first by the meninges, plexus choroid and ventricule and later through the walls of the blood vessel. They penetrate the brain with an ameboid morphology, migrate within the white matter and eventually acquire a morphology characteristic for ramified microglia (Monier et al. 2007, 2006; Rezaie and Male 1999; Rezaie et al. 1999). At mid-gestation, a cluster of activated microglia is located at the level of the axonal crossroads in the white matter where the periventricular white matter injury predominantly develops in preterm infants. The so-called resting microglial cells can be activated to functional brain macrophages, which undergo ameboid transformation and up-regulation of macrophage surface markers (Streit and Kreutzberg 1988). In the periventricular white matter of preterm infants, myelination defects associated with pre-oligodendrocyte cell death have been identified as a major component of white matter damage (Volpe 2009). Nevertheless, the specific roles of microglial-macrophages activation associated with astrogliosis in white matter damage and/or repair remain a matter of debate (C. Verney, personal communication). In various animal models of periventricular white matter injury involving excitotoxicity, inflammation and hypoxia–ischemia asphyxia, activation of microglia-macrophages was the first cellular event detected in or around the lesion (Baud et al. 2004; Mallard et al. 2003; Olivier et al. 2005; Tahraoui et al. 2001). The activation of microglia with LPS induces oligodendrocyte cell death and also greatly impairs oligodendrocyte development through cytokines and growth factor secretion modulations (Pang et al. 2010). The oligodendrocyte-microglial communication could be one of the mechanisms underlying selective white matter damage and hypomyelination in periventricular leukomalacia. Similarly, activated microglia-macrophages are seen in abundance following both neonatal hypoxia–ischemia (Ivacko et al. 1996; McRae et al. 1995) and focal stroke (Denker et al. 2007; Dingman et al. 2006), producing inflammatory cytokines, high levels of nitric oxide (NO), complement molecules and matrix metalloproteinases (MMPs). The MMPs control, by proteolytic cleavage, the components of the extracellular matrix proteins such as adhesion, membrane receptors and soluble proteins. The early post-injury macrophage population comprises predominantly resident microglia rather than invading monocytes (Denker et al. 2007; Dommergues et al. 2003). The notion that microglia contribute to rather than limit acute ischemic injury in the immature brain comes from studies illustrating the association between a reduced extent of lesion and reduced microglia activation/monocyte infiltration (Arvin et al. 2002; Dommergues et al. 2003; Fox et al. 2005). At the same time, several studies have shown that anti-inflammatory drugs, thought to protect adult brain by reducing macrophage accumulation after stroke, protect the neonatal brain without directly affecting the inflammatory mechanisms associated with microglia activation (Dingman et al. 2006; Fox et al. 2005; Tikka et al. 2001; van den Tweel et al. 2005). Distinct steps of microglial maturation and differentiation (such as expression of class II histocompatibility complex, MHC, cathepsin and other molecules) and the propensity of neurons to undergo apoptosis in the developing brain may account for this age dependence of the microglia response.
The relative contribution of pro-inflammatory mechanisms in astrocytes, as opposed to their other function in ischemic injury, is not well understood. Astrocytes express major histocompatibility complex (MHC), can up-regulate the inducible nitric oxide synthetase iNOS and increase cytokine production. Mast cells have been shown to play an injurious role in neonatal hypoxia–ischemia (Jin et al. 2007) and focal stroke (Biran et al. 2008). The injurious effects of these cells are thought to depend on the transforming growth factor β TGF-β and IL-9 (Mesples et al. 2005). Less is known about the contribution of T- and B-cell infiltration that is seen later after injury.
Conclusion
Neuroinflammation has emerged as a key pathophysiological mechanism in almost all neurological disorders affecting the adult and the developing brain. In parallel, the concept of inflammation as a sensitizing process for the developing brain has emerged opening the possibilities for the link between perinatal events and somewhat remote brain diseases. Cellular and molecular players are being identified and allow for the design of potential neuroprotective targets. The developing brain seems to be very different from the adult brain and therefore data from the adult literature cannot be extrapolated without confirmatory studies for the developing brain. Some inflammatory mediators, such as microglia, seem to be able to play deleterious or beneficial roles according to the timing after the insult and/or the cellular/molecular context. Finally, the potential side effects of some of the potential drugs targeting inflammation have to be evaluated in the context of a developing organism, and the potential benefits will need to be balanced with the potential harm.
References
Adén U, Favrais G, Plaisant F, Winerdal M, Felderhoff-Mueser UJL, Lelievre V, Gressens P (2010) Systemic inflammation sensitizes the neonatal brain to excitotoxicity through a pro-/anti-inflammatory imbalance: key role of TNF-α pathway and protection by etanercept. Brain Behav Immun (in press)
Anthony DC, Bolton SJ, Fearn S, Perry VH (1997) Age-related effects of interleukin-1 beta on polymorphonuclear neutrophil-dependent increases in blood-brain barrier permeability in rats. Brain 120(Pt 3):435–444
Anthony D, Dempster R, Fearn S, Clements J, Wells G, Perry VH, Walker K (1998) CXC chemokines generate age-related increases in neutrophil-mediated brain inflammation and blood-brain barrier breakdown. Curr Biol 8:923–926
Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM (2002) Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol 52:54–61
Bartha AI, Foster-Barber A, Miller SP, Vigneron DB, Glidden DV, Barkovich AJ, Ferriero DM (2004) Neonatal encephalopathy: association of cytokines with MR spectroscopy and outcome. Pediatr Res 56:960–966
Baud O, Daire JL, Dalmaz Y, Fontaine RH, Krueger RC, Sebag G, Evrard P, Gressens P, Verney C (2004) Gestational hypoxia induces white matter damage in neonatal rats: a new model of periventricular leukomalacia. Brain Pathol 14:1–10
Biran V, Cochois V, Karroubi A, Arrang JM, Charriaut-Marlangue C, Heron A (2008) Stroke induces histamine accumulation and mast cell degranulation in the neonatal rat brain. Brain Pathol 18:1–9
Dammann O, Leviton A (1997) Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res 42:1–8
Dammann O, Leviton A (2007) Perinatal brain damage causation. Dev Neurosci 29:280–288
Dammann O, Kuban KC, Leviton A (2002) Perinatal infection, fetal inflammatory response, white matter damage, and cognitive limitations in children born preterm. Ment Retard Dev Disabil Res Rev 8:46–50
Debillon T, Gras-Leguen C, Verielle V, Winer N, Caillon J, Roze JC, Gressens P (2000) Intrauterine infection induces programmed cell death in rabbit periventricular white matter. Pediatr Res 47:736–742
Degos V, Loron G, Mantz J, Gressens P (2008) Neuroprotective strategies for the neonatal brain. Anesth Analg 106:1670–1680
Denker SP, Ji S, Dingman A, Lee SY, Derugin N, Wendland MF, Vexler ZS (2007) Macrophages are comprised of resident brain microglia not infiltrating peripheral monocytes acutely after neonatal stroke. J Neurochem 100:893–904
Derrick M, Luo NL, Bregman JC, Jilling T, Ji X, Fisher K, Gladson CL, Beardsley DJ, Murdoch G, Back SA, Tan S (2004) Preterm fetal hypoxia–ischemia causes hypertonia and motor deficits in the neonatal rabbit: a model for human cerebral palsy? J Neurosci 24:24–34
Dingman A, Lee SY, Derugin N, Wendland MF, Vexler ZS (2006) Aminoguanidine inhibits caspase-3 and calpain activation without affecting microglial activation following neonatal transient cerebral ischemia. J Neurochem 96:1467–1479
Dommergues MA, Patkai J, Renauld JC, Evrard P, Gressens P (2000) Proinflammatory cytokines and interleukin-9 exacerbate excitotoxic lesions of the newborn murine neopallium. Ann Neurol 47:54–63
Dommergues MA, Plaisant F, Verney C, Gressens P (2003) Early microglial activation following neonatal excitotoxic brain damage in mice: a potential target for neuroprotection. Neuroscience 121:619–628
Doverhag C, Keller M, Karlsson A, Hedtjarn M, Nilsson U, Kapeller E, Sarkozy G, Klimaschewski L, Humpel C, Hagberg H, Simbruner G, Gressens P, Savman K (2008) Pharmacological and genetic inhibition of NADPH oxidase does not reduce brain damage in different models of perinatal brain injury in newborn mice. Neurobiol Dis 31:133–144
Ek CJ, Habgood MD, Dziegielewska KM, Potter A, Saunders NR (2001) Permeability and route of entry for lipid-insoluble molecules across brain barriers in developing Monodelphis domestica. J Physiol 536(Pt 3):841–853
Ek CJ, Dziegielewska KM, Stolp H, Saunders NR (2006) Functional effectiveness of the blood-brain barrier to small water-soluble molecules in developing and adult opossum (Monodelphis domestica). J Comp Neurol 496(1):13–26
Eklind S, Mallard C, Leverin AL, Gilland E, Blomgren K, Mattsby-Baltzer I, Hagberg H (2001) Bacterial endotoxin sensitizes the immature brain to hypoxic-ischaemic injury. Eur J Neurosci 13:1101–1106
Eklind S, Mallard C, Arvidsson P, Hagberg H (2005) Lipopolysaccharide induces both a primary and a secondary phase of sensitization in the developing rat brain. Pediatr Res 58:112–116
Engelhardt B (2003) Development of the blood-brain barrier. Cell Tissue Res 314:119–129
Faustino J, Liu B, Lee S, Derugin N, Wendland MF, Vexler ZS (2009) Blockade of endogenous cytokine-induced neutrophil chemoattractant protein 1 exacerbates injury after neonatal stroke Stroke meeting. San Diego
Favrais G, Schwendimann L, Gressens P, Lelievre V (2007) Cyclooxygenase-2 mediates the sensitizing effects of systemic IL-1-beta on excitotoxic brain lesions in newborn mice. Neurobiol Dis 25:496–505
Feldhaus B, Dietzel ID, Heumann R, Berger R (2004) Effects of interferon-gamma and tumor necrosis factor-alpha on survival and differentiation of oligodendrocyte progenitors. J Soc Gynecol Investig 11:89–96
Follett PL, Rosenberg PA, Volpe JJ, Jensen FE (2000) NBQX attenuates excitotoxic injury in developing white matter. J Neurosci 20:9235–9241
Follett PL, Deng W, Dai W, Talos DM, Massillon LJ, Rosenberg PA, Volpe JJ, Jensen FE (2004) Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: a protective role for topiramate. J Neurosci 24:4412–4420
Fontaine RH, Cases O, Lelievre V, Mesples B, Renauld JC, Loron G, Degos V, Dournaud P, Baud O, Gressens P (2008) IL-9/IL-9 receptor signaling selectively protects cortical neurons against developmental apoptosis. Cell Death Differ 15:1542–1552
Foster-Barber A, Ferriero DM (2002) Neonatal encephalopathy in the term infant: neuroimaging and inflammatory cytokines. Ment Retard Dev Disabil Res Rev 8:20–24
Fox C, Dingman A, Derugin N, Wendland MF, Manabat C, Ji S, Ferriero DM, Vexler ZS (2005) Minocycline confers early but transient protection in the immature brain following focal cerebral ischemia-reperfusion. J Cereb Blood Flow Metab 25:1138–1149
Galasso JM, Miller MJ, Cowell RM, Harrison JK, Warren JS, Silverstein FS (2000) Acute excitotoxic injury induces expression of monocyte chemoattractant protein-1 and its receptor, CCR2, in neonatal rat brain. Exp Neurol 165:295–305
Ghersi-Egea JF, Strazielle N, Murat A, Jouvet A, Buénerd A, Belin MF (2006) Brain protection at the blood-cerebrospinal fluid interface involves a glutathione-dependent metabolic barrier mechanism. J Cereb Blood Flow Metab 26(9):1165–1175
Girard S, Kadhim H, Larouche A, Roy M, Gobeil F, Sebire G (2008) Pro-inflammatory disequilibrium of the IL-1 beta/IL-1ra ratio in an experimental model of perinatal brain damages induced by lipopolysaccharide and hypoxia–ischemia. Cytokine 43:54–62
Gressens P, Marret S, Hill JM, Brenneman DE, Gozes I, Fridkin M, Evrard P (1997) Vasoactive intestinal peptide prevents excitotoxic cell death in the murine developing brain. J Clin Invest 100:390–397
Gressens P, Rogido M, Paindaveine B, Sola A (2002) The impact of neonatal intensive care practices on the developing brain. J Pediatr 140:646–653
Grether JK, Nelson KB (1997) Maternal infection and cerebral palsy in infants of normal birth weight. JAMA 278:207–211
Hagberg H, Mallard C (2005) Effect of inflammation on central nervous system development and vulnerability. Curr Opin Neurol 18:117–123
Hagberg H, Gilland E, Bona E, Hanson LA, Hahin-Zoric M, Blennow M, Holst M, McRae A, Soder O (1996) Enhanced expression of interleukin (IL)-1 and IL-6 messenger RNA and bioactive protein after hypoxia–ischemia in neonatal rats. Pediatr Res 40:603–609
Hagberg H, Peebles D, Mallard C (2002) Models of white matter injury: comparison of infectious, hypoxic-ischemic, and excitotoxic insults. Ment Retard Dev Disabil Res Rev 8:30–38
Harding DR, Dhamrait S, Whitelaw A, Humphries SE, Marlow N, Montgomery HE (2004) Does interleukin-6 genotype influence cerebral injury or developmental progress after preterm birth? Pediatrics 114:941–947
Haynes RL, Folkerth RD, Keefe RJ, Sung I, Swzeda LI, Rosenberg PA, Volpe JJ, Kinney HC (2003) Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol 62:441–450
Hedtjarn M, Leverin AL, Eriksson K, Blomgren K, Mallard C, Hagberg H (2002) Interleukin-18 involvement in hypoxic-ischemic brain injury. J Neurosci 22:5910–5919
Hedtjarn M, Mallard C, Iwakura Y, Hagberg H (2005) Combined deficiency of IL-1beta18, but not IL-1alphabeta, reduces susceptibility to hypoxia–ischemia in the immature brain. Dev Neurosci 27:143–148
Himmelmann K, Hagberg G, Beckung E, Hagberg B, Uvebrant P (2005) The changing panorama of cerebral palsy in Sweden. IX. Prevalence and origin in the birth-year period 1995–1998. Acta Paediatr 94:287–294
Inder T, Mocatta T, Darlow B, Spencer C, Volpe JJ, Winterbourn C (2002) Elevated free radical products in the cerebrospinal fluid of VLBW infants with cerebral white matter injury. Pediatr Res 52:213–218
Ivacko JA, Sun R, Silverstein FS (1996) Hypoxic-ischemic brain injury induces an acute microglial reaction in perinatal rats. Pediatr Res 39:39–47
Jin Y, Silverman AJ, Vannucci SJ (2007) Mast cell stabilization limits hypoxic-ischemic brain damage in the immature rat. Dev Neurosci 29:373–384
Johnson DL, Getson P, Shaer C, O’Donnell R (1987) Intraventricular hemorrhage in the newborn beagle puppy. A limited model of intraventricular hemorrhage in the premature infant. Pediatr Neurosci 13:78–83
Kirton A, deVeber G (2009) Advances in perinatal ischemic stroke. Pediatr Neurol 40:205–214
Kniesel U, Risau W, Wolburg H (1996) Development of blood-brain barrier tight junctions in the rat cortex. Brain Res Dev Brain Res 96:229–240
Laudenbach V, Calo G, Guerrini R, Lamboley G, Benoist JF, Evrard P, Gressens P (2001) Nociceptin/orphanin FQ exacerbates excitotoxic white-matter lesions in the murine neonatal brain. J Clin Invest 107:457–466
Leonardo CC, Eakin AK, Ajmo JM, Collier LA, Pennypacker KR, Strongin AY, Gottschall PE (2008) Delayed administration of a matrix metalloproteinase inhibitor limits progressive brain injury after hypoxia–ischemia in the neonatal rat. J Neuroinflammation 5:34
Lim HB, Smith M (2007) Systemic complications after head injury: a clinical review. Anaesthesia 62:474–482
Loeliger M, Watson CS, Reynolds JD, Penning DH, Harding R, Bocking AD, Rees SM (2003) Extracellular glutamate levels and neuropathology in cerebral white matter following repeated umbilical cord occlusion in the near term fetal sheep. Neuroscience 116:705–714
Lu J, Goh SJ, Tng PY, Deng YY, Ling EA, Moochhala S (2009) Systemic inflammatory response following acute traumatic brain injury. Front Biosci 14:3795–3813
Malaeb S, Dammann O (2009) Fetal inflammatory response and brain injury in the preterm newborn. J Child Neurol 24:1119–1126
Mallard C, Welin AK, Peebles D, Hagberg H, Kjellmer I (2003) White matter injury following systemic endotoxemia or asphyxia in the fetal sheep. Neurochem Res 28:215–223
McColl BW, Allan SM, Rothwell NJ (2009) Systemic infection, inflammation and acute ischemic stroke. Neuroscience 158:1049–1061
McRae A, Gilland E, Bona E, Hagberg H (1995) Microglia activation after neonatal hypoxic-ischemia. Brain Res Dev Brain Res 84:245–252
Mesples B, Plaisant F, Gressens P (2003) Effects of interleukin-10 on neonatal excitotoxic brain lesions in mice. Brain Res Dev Brain Res 141:25–32
Mesples B, Plaisant F, Fontaine RH, Gressens P (2005) Pathophysiology of neonatal brain lesions: lessons from animal models of excitotoxicity. Acta Paediatr 94:185–190
Miller RJ, Tran PB (2005) Chemokinetics. Neuron 47:621–623
Monier A, Evrard P, Gressens P, Verney C (2006) Distribution and differentiation of microglia in the human encephalon during the first two trimesters of gestation. J Comp Neurol 499:565–582
Monier A, Adle-Biassette H, Delezoide AL, Evrard P, Gressens P, Verney C (2007) Entry and distribution of microglial cells in human embryonic and fetal cerebral cortex. J Neuropathol Exp Neurol 66:372–382
Nelson KB, Chang T (2008) Is cerebral palsy preventable? Curr Opin Neurol 21:129–135
Nelson KB, Grether JK (1999) Causes of cerebral palsy. Curr Opin Pediatr 11:487–491
Nelson KB, Willoughby RE (2000) Infection, inflammation and the risk of cerebral palsy. Curr Opin Neurol 13:133–139
Nijboer CH, Kavelaars A, Vroon A, Groenendaal F, van Bel F, Heijnen CJ (2008) Low endogenous G-protein-coupled receptor kinase 2 sensitizes the immature brain to hypoxia–ischemia-induced gray and white matter damage. J Neurosci 28:3324–3332
Normann E, Lacaze-Masmonteil T, Eaton F, Schwendimann L, Gressens P, Thebaud B (2009) A novel mouse model of ureaplasma-induced perinatal inflammation: effects on lung and brain injury. Pediatr Res 65:430–436
Olivier P, Baud O, Evrard P, Gressens P, Verney C (2005) Prenatal ischemia and white matter damage in rats. J Neuropathol Exp Neurol 64:998–1006
Pang Y, Campbell L, Zheng B, Fan L, Cai Z, Rhodes P (2010) Lipopolysaccharide-activated microglia induce death of oligodendrocyte progenitor cells and impede their development. Neuroscience 166(2):464–475
Patkai J, Mesples B, Dommergues MA, Fromont G, Thornton EM, Renauld JC, Evrard P, Gressens P (2001) Deleterious effects of IL-9-activated mast cells and neuroprotection by antihistamine drugs in the developing mouse brain. Pediatr Res 50:222–230
Plaisant F, Dommergues MA, Spedding M, Cecchelli R, Brillault J, Kato G, Munoz C, Gressens P (2003) Neuroprotective properties of tianeptine: interactions with cytokines. Neuropharmacology 44:801–809
Rezaie P, Male D (1999) Colonisation of the developing human brain and spinal cord by microglia: a review. Microsc Res Tech 45:359–382
Rezaie P, Patel K, Male DK (1999) Microglia in the human fetal spinal cord—patterns of distribution, morphology and phenotype. Brain Res Dev Brain Res 115:71–81
Robertson CM, Watt MJ, Yasui Y (2007) Changes in the prevalence of cerebral palsy for children born very prematurely within a population-based program over 30 years. JAMA 297:2733–2740
Rousset CI, Chalon S, Cantagrel S, Bodard S, Andres C, Gressens P, Saliba E (2006) Maternal exposure to LPS induces hypomyelination in the internal capsule and programmed cell death in the deep gray matter in newborn rats. Pediatr Res 59:428–433
Strazielle N, Ghersi-Egea JF (2000) Choroid plexus in the central nervous system: biology and physiopathology. J Neuropathol Exp Neurol 59(7):561–574
Streit WJ, Kreutzberg GW (1988) Response of endogenous glial cells to motor neuron degeneration induced by toxic ricin. J Comp Neurol 268:248–263
Svedin P, Hagberg H, Savman K, Zhu C, Mallard C (2007) Matrix metalloproteinase-9 gene knock-out protects the immature brain after cerebral hypoxia–ischemia. J Neurosci 27:1511–1518
Tahraoui SL, Marret S, Bodenant C, Leroux P, Dommergues MA, Evrard P, Gressens P (2001) Central role of microglia in neonatal excitotoxic lesions of the murine periventricular white matter. Brain Pathol 11:56–71
Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J (2001) Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci 21:2580–2588
van den Tweel ER, Nijboer C, Kavelaars A, Heijnen CJ, Groenendaal F, van Bel F (2005) Expression of nitric oxide synthase isoforms and nitrotyrosine formation after hypoxia–ischemia in the neonatal rat brain. J Neuroimmunol 167:64–71
Vela JM, Molina-Holgado E, Arevalo-Martin A, Almazan G, Guaza C (2002) Interleukin-1 regulates proliferation and differentiation of oligodendrocyte progenitor cells. Mol Cell Neurosci 20:489–502
Vincer MJ, Allen AC, Joseph KS, Stinson DA, Scott H, Wood E (2006) Increasing prevalence of cerebral palsy among very preterm infants: a population-based study. Pediatrics 118:e1621–e1626
Volpe JJ (2001) Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res 50:553–562
Volpe JJ (2009) The encephalopathy of prematurity-brain injury and impaired brain development inextricably intertwined. Semin Pediatr Neurol 16:167–178
Wilson-Costello D, Friedman H, Minich N, Fanaroff AA, Hack M (2005) Improved survival rates with increased neurodevelopmental disability for extremely low birth weight infants in the 1990s. Pediatrics 115:997–1003
Wilson-Costello D, Friedman H, Minich N, Siner B, Taylor G, Schluchter M, Hack M (2007) Improved neurodevelopmental outcomes for extremely low birth weight infants in 2000–2002. Pediatrics 119:37–45
Acknowledgments
Our research work is supported by Inserm, Université Paris 7, PremUP, the Sixth Framework Program of the European Commission, the Fondation des Gueules Cassées, the Fondation Motrice, the ELA Foundation, the Fondation Grace de Monaco, the Institut pour la Recherche sur la Moelle épinière et l’Encéphale (IRME), the Medical Research Council, the Charité, the German Research Foundation (DFG), the German Federal Ministry of Education and Research, the Sonnenfeld Stiftung and the Sanitätsrat Dr.-Emil-Alexander-Huebner-und-Gemahlin-Stiftung.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Degos, V., Favrais, G., Kaindl, A.M. et al. Inflammation processes in perinatal brain damage. J Neural Transm 117, 1009–1017 (2010). https://doi.org/10.1007/s00702-010-0411-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-010-0411-x