
Abstract
The pathogenesis of Alzheimer’s disease (AD) appears to involve several different mechanisms, the most consistent of which is an impairment of cholinergic neurotransmission; however, there is controversy about its relevance at the early stage of disease. A transcranial magnetic stimulation (TMS) protocol based on coupling peripheral nerve stimulation with motor cortex TMS (short latency afferent inhibition, SAI) may give direct information about the function of some cholinergic pathways in the human motor cortex. We evaluated SAI in a group of patients with early diagnosis of AD and compared the data with that from a control group. The amount of SAI was significantly smaller in early AD patients than in controls. This study first provides physiological evidence that a central cholinergic dysfunction occurs in the earlier stages of AD. Identification of SAI abnormalities that occur early in the course of AD will allow earlier diagnosis and treatment with cholinergic drugs.
Similar content being viewed by others

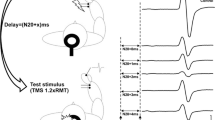
Avoid common mistakes on your manuscript.
Introduction
In animals, the cholinergic system has been implicated in a variety of activities including memory, learning and attention (Smith 1988). Although damage to multiple neurotransmitter systems have been implicated in the pathogenesis of Alzheimer’s disease (AD), impairment of cholinergic function is believed to be of particular importance (Bartus et al. 1982; Reinikainen et al. 1990; Bierer et al. 1995; Kasa et al. 1997; Mufson et al. 2000). Basal forebrain cholinergic neurons (BFCNs), which provide the major cholinergic input to hippocampal and cortical regions, are known to undergo selective and severe degeneration in AD (Davies and Maloney 1976; Whitehouse et al. 1981). In addition, decreased cholinergic neurotransmission has been demonstrated to correlate with cognitive impairment in patients with AD (Perry et al. 1978; Shinotoh et al. 2000).
Mesulam et al. (2004) demonstrated that cytopathology in cortical cholinergic pathways is a very early event in the course of the continuum that leads from advanced age to MCI (mild cognitive impairment) and AD. However, most descriptions of neurochemical and neuropathological changes in AD have been based generally on findings in brains of late-stage or terminal AD patients. Moreover, there are a few in vivo studies that indicate a significant impairment of the cholinergic system very early in AD, and they are subject to methodological limitations such as very small subject numbers (Nordberg et al. 1995; Iyo et al. 1997; Kuhl et al. 1994; Herholz et al. 2000; Rinne et al. 2003; Bohnen et al. 2007; O’Brien et al. 2007). Consequently, it remains relatively unclear how early in the course of the disease neurochemical and neuropathological alterations occur. Neurobiological changes should be examined earlier in the disease process, when presumably they are more relevant for the pathogenesis of AD. In particular, the role of the central cholinergic system in early AD needs to be further investigated in view of implications for treatment with cholinergic agents. Early detection of AD is now increasingly important as research advances with respect to therapeutic interventions. Functional neuroimaging techniques (SPECT, PET, and f-MRI) are useful for the early diagnosis of AD (Almkvist and Winblad 1999), but they are prohibitively expensive and/or require injection of radioactive tracer compounds. Accurate and inexpensive tools are clearly needed.
In vivo evaluation of some cholinergic circuits of the human brain has recently been introduced using a transcranial magnetic stimulation (TMS) protocol based on coupling electrical stimulation of peripheral nerves with TMS of the motor cortex. Peripheral nerve inputs have an inhibitory effect on motor cortex excitability at short intervals (Tokimura et al. 2000). This inhibitory phenomenon, named short latency afferent inhibition (SAI), is facilitated by cholinergic activity as demonstrated by its sensitivity to the muscarinic blockade (Di Lazzaro et al. 2000), even if it is still unknown whether different neurotransmitters such as glutamate, GABA, or dopamine are also involved in the regulation of SAI.
In this study, we evaluated SAI to explore central cholinergic activity in early AD, together with a number of other excitatory and inhibitory circuits through different TMS paradigms.
Materials and methods
Patients
We examined 17 patients (ten men and seven women, mean age 68.4 years, range 58–74 years) with a diagnosis of probable AD according to the NINCDS-ADRDA criteria (McKhann et al. 1984) and 22 age-matched neurologically healthy controls (12 men and 10 women, mean age 70.4 years, range 62–73 years).
Participants had a clinical diagnosis of probable AD in the very mild [Clinical Dementia Rating (CDR) 0.5] or mild (CDR 1) dementia stage. The CDR is a widely used dementia staging instrument; participants who had a CDR 0.5 or CDR 1 together represent early-stage AD.
There was a progress monitoring of the examined patients; all the patients showed cognitive worsening over the next 2–3 years.
Exclusion criteria were: symptoms onset more than 6 months previously, age at onset older than 75 years and presence of other major medical illness.
None of the patients were treated with anticholinergic drugs before the study. Administration of all drugs that affect motor cortex excitability in patients and control subjects was discontinued at least 2 weeks before the study.
Patients provided informed consent before participation in this study, which was performed according to the Declaration of Helsinki and approved by the institutional Ethics Committee.
Transcranial magnetic stimulation
Magnetic stimulation was performed with a High-power Magstim 200 (Magstim Co., Whitland, Dyfed, UK). A figure-of-eight coil with external loop diameters of 9 cm was held over the motor cortex at the optimum scalp position to elicit motor responses in the contralateral first dorsal interosseous (FDI) muscle. The dominant hemisphere was selected for stimulating patients and healthy subjects. Motor evoked potentials (MEPs) were recorded via two 9 mm diameter Ag–AgCl electrodes with the active electrode applied over the motor point of the muscle and the reference on the metacarpophalangeal joint of the index finger. Motor responses were amplified and filtered (bandwidth 3–3000 Hz) by D150 amplifiers (Digitimer, Welwyn Garden City, Herfordshire, UK).
SAI was studied using the recently described technique (Tokimura et al. 2000).
Conditioning stimuli were single pulse (200 μs) of electrical stimulation (with the cathode positioned proximally) applied through bipolar electrodes to the median nerve at the wrist. The intensity of the conditioning stimuli was set just above the motor threshold necessary to evoke a visible twitch of the thenar muscles. The intensity of the test cortical magnetic shock was adjusted to evoke a muscle response in relaxed FDI with an amplitude of approximately 1 mV peak-to-peak. The conditioning stimulus to the peripheral nerve preceded the test magnetic cortical stimulus. Interstimulus intervals (ISIs) were determined relative to the latency of the N20 component of the somatosensory evoked potential evoked by stimulation of the median nerve. The active electrode for recording the N20 potential was attached 3 cm posterior to C3 (10–20 system), and the reference was 3 cm posterior to C4. Five hundred responses were averaged to identify the latency of N20 peak. ISIs from the latency of the N20 component plus 2 ms to the latency of the N20 component plus 8 ms were investigated in steps of 1 ms, because at these intervals there is a clear inhibition of the MEPs and of corticospinal volleys evoked by TMS (Tokimura et al. 2000).
Five stimuli were delivered at each ISI. We calculated an average of the MEP obtained after cortical magnetic stimulation alone and of the MEP obtained by conditioning cortical magnetic stimulus with a peripheral stimulus to the median nerve at the wrist at the seven different ISIs studied. The amplitude of the conditioned MEP was expressed as percentage of the amplitude of the test MEP. The percentage inhibition of the conditioned responses at the seven different ISIs was averaged to obtain a grand mean. Subjects were given audio–visual feedback at high gain to assist in maintaining complete relaxation.
In addition to SAI, we evaluated the following TMS parameters: the resting and active motor threshold of MEP; the central motor conduction time (CMCT); the short latency intracortical inhibition (SICI) and intracortical facilitation (ICF) to paired TMS. The meaning of these measures of motor cortex excitability has been summarized in recent reviews (Hallett 2000; Ziemann 2002; Rothwell 2003).
Resting motor threshold (RMT) was defined as the minimum stimulus intensity that produced a liminal motor evoked response (about 50 μV in 50% of ten trials) at rest. Active motor threshold (AMT) was defined as the minimum stimulus intensity that produced a liminal motor evoked response of about 200 μV in 50% of ten trials during isometric contraction of the tested muscle at about 10% maximum.
CMCT was calculated by subtracting the peripheral conduction time from spinal cord to muscles from the latency of responses evoked by cortical stimulation with the formula:
(Rossini et al. 1994).
Intracortical inhibition and facilitation were studied using the technique of Kujirai et al. (1993). Using a Bistim module, two magnetic stimuli were given through the same stimulating coil over the motor cortex and the effect of the first (conditioning) stimulus on the second (test) stimulus was investigated. The intensity of the conditioning stimulus was set to 90% AMT; the second, test, shock intensity was adjusted to evoke a muscle response in relaxed FDI with an amplitude of approximately 1 mV.
The timing of the conditioning shock was altered in relation to the test shock. Inhibitory interstimulus intervals (ISIs) of 3 ms and facilitatory ISIs of 10 ms were investigated. Ten stimuli were delivered at each ISI. For these recordings, muscle relaxation is very important and the subject was given audio–visual feedback at high gain to assist in maintaining complete relaxation. The presentation of conditioned and unconditioned trials was randomised. The amplitude of the conditioned EMG responses was expressed as the percentage of the amplitude of the test EMG responses. The amplitude of the conditioned responses was averaged obtaining grand mean amplitudes of the inhibitory and of the facilitatory ISIs.
To clarify a possible spinal or peripheral contribution on the motor cortex excitability parameters, supramaximal stimulation (0.2-ms square-wave constant current pulsies) of the ulnar nerve at the wrist was used to assess spinal and peripheral motor excitability. While FDI was relaxed, the peak-to-peak amplitude of F waves (average, 20 trials) and CMAP (maximum, 3 trials) were determined.
Statistical analysis
The electrophysiological parameters of the SIVD patients were analyzed separately and compared with those of the control subjects by means of Mann–Whitney tests. The level of significance was set at 0.05.
The relation between different variables was evaluated by means of the Spearman’s r correlation coefficient.
Results
The mean amount of SAI was significantly smaller in the early AD patients (mean responses reduced to 67.4 ± 14.2% of test size) than in normal controls (mean responses reduced to 42.8 ± 13.8% of test size; P < 0.05, Mann–Whitney test, N1 = 17, N2 = 22) (Fig. 1).

Column graph showing mean values for short latency afferent inhibition (SAI) in patients with early Alzheimer’s disease (AD) and in control subjects; error bars are SD
RMT, AMT, CMCT, SICI, ICF, F-wave and CMAP were similar in both groups (P > 0.05, Mann–Whitney test, N 1 = 17, N 2 = 22).
The neurophysiological data are summarized in the Table 1.
SAI values did not correlate significantly with the CDR scores, the patient’s age and the duration of the disease (P > 0.01; Spearman’s r).
Discussion
The early discovery of acetylcholine (ACh) deficiency (Perry et al. 1978) singled out the loss of this neurotransmitter as one reason for the cognitive dysfunction in AD. Several studies support the cholinergic hypothesis (Bartus 1979; Bartus and Emerich 1999), even if little is known about the dynamic processes that underlie the cognitive dysfunction and neuropathology. Moreover, it is currently unclear whether impairment of the cholinergic system is present in AD already at an early stage.
Degeneration and loss of trophic support for the BFCNs is widely held an early and pivotal event in AD, and there is evidence for interaction with amyloid deposition and plaque formation; there is a loss of calbindin in BFCNs neurons that corresponds with the appearance of tangles before manifestation of dementia (Geula 1998), suggesting early and severe functional impairment of these neurons. Furthermore, disturbance of axonal transport in cholinergic neurons has been identified as one of the earliest signs of disease in humans and in transgenic mice (Stokin et al. 2005). Conversely, recent neuropathological studies indicate that cortical acetylcholinesterase (AChE) immunoreactivity is well preserved in mild AD (Davis et al. 1999), and BFCNs are not decreased in early AD (Gilmor et al. 1999).
A recent TMS study (Sakuma et al. 2007) reported normal SAI in patients affected by MCI, a level of cognitive functioning that reflects an intermediate state between normal age and AD. This apparent discrepancy between our results and those of Sakuma et al. may be explained by the findings of recent studies in MCI subjects. A study with functional MRI showed that short-term treatment with a AChE inhibitor appeared to enhance the activity of the frontal circuitry Saykin et al. 2004) and the presynaptic cholinergic marker choline acetyltransferase was found to be elevated in the superior frontal cortex and hippocampus (DeKosky et al. 2002). Therefore, the cholinergic system may be even upregulated in MCI individuals and some compensatory mechanisms may mark the conversion of MCI to diagnosable AD. Furthermore, SAI reflects the interaction between the sensory and motor systems and is a cortical phenomenon (Tokimura et al. 2000); if the degeneration involves the BFCNs, or if the cortical cholinergic deficiency is compensated by feed-back control, the degenerative process may not be reflected by SAI. On the other hand, amnesic MCI subjects present abnormal verbal and/or nonverbal memory for age and normal general cognitive functioning (Petersen et al. 1999), while ACh in primates seems to be more specifically involved in attentional processes than in learning and memory processes (Blokland 1996; Voytko et al. 1994).
Several studies suggest that AChE (Iyo et al. 1997; Kuhl et al. 1994; Herholz et al. 2000; Rinne et al. 2003; Bohnen et al. 2007), and presynaptic receptors (Nordberg et al. 1995; O’Brien et al. 2007) are both reduced in AD which might result in partial compensation and relatively little effect on net cholinergic activity at least at early stages of the disease. Cholinergic axons projecting to neocortical association areas appear to be significantly impaired, whereas hippocampal projections and cholinergic cell bodies in basal forebrain appear to be relatively intact at the early stages of AD (Shinotoh et al. 2003; Herholz et al. 2004).
Thus, the current evidence from post-mortem and in vivo studies on cholinergic involvement in the earliest stages of AD was inconclusive.
The present study shows that SAI, a putative marker of cholinergic cortical activity, is significantly impaired in patients with early AD compared with controls. SAI was found to be reduced in patients with AD (Di Lazzaro et al. 2002, 2004, 2005a, 2006); in contrast, SAI was found to be normal in non-cholinergic forms of dementia, such as frontotemporal dementia (Di Lazzaro et al. 2006). Drugs enhancing cholinergic transmission can increase SAI (Di Lazzaro et al. 2002, 2004, 2005a). Interestingly, changes in SAI following administration of AchE inhibitors may be related to the long-term efficacy of this treatment (Di Lazzaro et al. 2005a). SAI is also influenced by GABAergic drugs such as the benzodiazepine lorazepam in healthy subjects (Di Lazzaro et al. 2005b, c) and by dopaminergic drugs in patients with Parkinson disease (Sailer et al. 2003). However, SAI represents a non-invasive way of testing the integrity of some cholinergic cortical circuits (Ziemann 2004) while the contribution of neurotransmitters other than ACh is not well understood.
Our findings support in vivo imaging suggesting that there is early impairment of presynaptic receptors and AChE in cerebral cortex (for review, see Herholz et al. 2000), and support the concept that neocortical functional changes of the cholinergic system are an early and leading event in AD.
This method can be used as a non-invasive test for the assessment of central cholinergic pathways in patients with AD also at the early stages of illness, and may help in identifying the patients in whom cholinergic degeneration is occurred and who would be suitable for long-term treatment with cholinergic agents.
References
Almkvist O, Winblad B (1999) Early diagnosis of Alzheimer dementia based on clinical and biological factors. Eur Arch Psychiatry Clin Neurosci 249:3–9
Bartus T (1979) Physostigmine and recent memory: effects in young and aged nonhuman primates. Science 206:1085–1087
Bartus RT, Dean RL, Beer B, Lippa AS (1982) The cholinergic hypothesis of geriatric memory dysfunction. Science 217:408–417
Bartus RT, Emerich DF (1999) Cholinergic markers in Alzheimer disease. J Am Med Assoc 282:2208–2209
Bierer LM, Haroutunian V, Gabriel S, Knott PJ, Carlin LS, Purohit DP, Perl DP, Schmeidler J, Kanof P, Davis KG (1995) Neurochemical correlates of dementia severity in Alzheimer’s disease: relative importance of the cholinergic deficits. J Neurochem 64:749–760
Blokland A (1996) Acetylcholine: a neurotransmitter for learning and memory? Brain Res Rev 21:285–300
Bohnen NI, Kaufer DI, Hendrickson R, Constantine GM, Mathis CA, Moore RY (2007) Cortical cholinergic denervation is associated with depressive symptoms in Parkinson’s disease and parkinsonian dementia. J Neurol Neurosurg Psychiatry 78:641–643
Davies P, Maloney AJ (1976) Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 2(8000):1403
Davis KL, Mohs RC, Marin D, Purohit DP, Perl DP, Lantz M, Austin G, Haroutunian V (1999) Cholinergic markers in elderly patients with early signs of Alzheimer disease. JAMA 281:1401–1406
DeKosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S, Bennett DA, Cochran EJ, Kordoner JH, Mufson EJ (2002) Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol 51:145–155
Di Lazzaro V, Oliviero A, Profice P, Pennini MA, Di Giovanni S, Zito G, Tonali P, Rothwell JC (2000) Muscarinic receptor blockade has differential effects on the excitability of intracortical circuits in the human motor cortex. Exp Brain Res 135:455–461
Di Lazzaro V, Oliviero A, Tonali PA, Marra C, Daniele A, Profice P, Saturno E, Pilato F, Masullo C, Rothwll JC (2002) Noninvasive in vivo assessment of cholinergic cortical circuits in AD using transcranial magnetic stimulation. Neurology 13:392–397
Di Lazzaro V, Oliviero A, Pilato F, Saturno E, Dileone M, Marra C, Daniele A, Ghirlanda S, Gainotti G, Tonali PA (2004) Motor cortex excitability to transcranial magnetic stimulation in Alzheimer’s disease. J Neurol Neurosurg Psychiatry 75:555–559
Di Lazzaro V, Oliviero A, Pilato F, Saturno E, Dileone M, Marra C, Daniele A, Ghirlanda S, Gainotti G, Tonali PA (2005a) Neurophysiological predictors of long term response to AchE inhibitors in AD patients. J Neurol Neurosurg Psychiatry 76:1064–1069
Di Lazzaro V, Oliviero A, Saturno E, Dileone M, Pilato F, Nardone R, Ranieri F, Musumeci G, Fiorilla T, Tonali P (2005b) Effects of lorazepam on short latency afferent inhibition and short latency intracortical inhibition in humans. J Physiol 564:661–668
Di Lazzaro V, Pilato F, Dileone M, Tonali PA, Ziemann U (2005c) Dissociated effects of diazepam and lorazepam on short-latency afferent inhibition. J Physiol 569:315–323
Di Lazzaro V, Pilato F, Dileone M, Saturno E, Oliviero A, Marra C, Daniele A, Ranieri F, Gainotti G, Tonali PA (2006) In vivo cholinergic circuit evaluation in frontotemporal and Alzheimer dementias. Neurology 66:1111–1113
Geula C (1998) Abnormalities of neural circuitry in Alzheimer’s disease: hippocampus and cortical cholinergic innervation. Neurology 51:S18–S29
Gilmor ML, Erickson JD, Varoqui H, Hersh LB, Bennett DA, Cochran EJ, Mufson EJ, Levey AI (1999) Preservation of nucleus basalis neurons containing choline acetyltransferase and the vesicular acetylcholine transporter in the elderly with mild cognitive impairment and early Alzheimer’s disease. J Comp Neurol 411:693–704
Hallett M (2000) Transcranial magnetic stimulation and the human brain. Nature 406:147–150
Herholz K, Bauer B, Wienhard K, Kracht L, Mielke R, Lenz O, Strotmann T, Heiss WD (2000) In-vivo measurements of regional acetylcholine esterase activity in degenerative dementia: comparison with blood flow and glucose metabolism. J Neural Transm 12:1457–1468
Herholz K, Weisenbach S, Zundorf G, Lenz O, Schroder H, Bauer B, Kalbe E, Heiss WD (2004) In-vivo study of acetylcholine esterase in basel forebrain, amygdala, and cortex in mild to moderate Alzheimer disease. Neuroimage 21:136–143
Iyo M, Namba H, Fukushi K, Shinotoh H, Nagatsuka S, Suhara T, Sodo Y, Sukuki K, Irie T (1997) Measurement of acetylcholinesterase by positron emission tomography in the brains of healthy controls and patients with Alzheimers disease. Lancet 349:1805–1809
Kasa P, Rakonczay Z, Gulya K (1997) The cholinergic system in Alzheimer’s disease. Prog Neurobiol 52:511–535
Kuhl DE, Koeppe RA, Fessler JA, Minoshima S, Ackermann RJ, Carey JE, Gildersleeve DL, Frey KA, Wieland DM (1994) In vivo mapping of cholinergic neurons in the human brain using SPECT and IBVM. J Nucl Med 35:405–410
Kujirai T, Caramia MD, Rothwell JC, Day BL, Thompson PD, Ferbert A, Wroe S, Asselman P, Marsden CD (1993) Cortico-cortical inhibition in human motor cortex. J Physiol 471:501–520
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology 34:939–944
Mesulam M, Shaw P, Mash D, Weintraub S (2004) Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann Neurol 55(6):815–828
Mufson EJ, Ma SY, Cochran EJ, Bennett DA, Beckett LA, Jaffar S, Saragovi HU, Kordower JH (2000) Loss of nucleus basalis neurons containing trkA immunoreactivity in individuals with mild cognitive impairment and early Alzheimer’s disease. J Comp Neurol 427:9–30
Nordberg A, Lundqvist H, Hartvig P, Lilja A, Langstrom B (1995) Kinetic analysis of regional (S)(−)11C-nicotine binding in normal and Alzheimer brains—in vivo assessment using positron emission tomography. Alzheimer Dis Assoc Disord 9:21–27
O’Brien JT, Colloby SJ, Pakrasi S, Perry EK, Pimlott S, Land DJ, Wyper DJ, McKeith IG, Williams ED (2007) Alpha4beta2 nicotinic receptor status in Alzheimer’s disease using 123I–5IA–85380 single-photon-emission computed tomography. J Neurol Neurosurg Psychiatry 78:356–362
Perry EK, Tomlinson BE, Blessed G, Bergmann K, Gibson PH, Perry RH (1978) Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Br Med J 2:1457–1459
Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E (1999) Mild vognitive impairment: clinical characterization and outcome. Arch Neurol 56:303–308
Reinikainen KJ, Soininen H, Riekkinen PJ (1990) Neurotransmitter changes in Alzheimer’s disease: implications to diagnostics and therapy. J Neurosci Res 27:576–586
Rinne JO, Kaasinen V, Jarvenpaa T, Nagren K, Roivainen A, Yu M, Oikonen V, Kurki T (2003) Brain acetylcholinesterase activity in mild cognitive impairment and early Alzheimer’s disease. J Neurol Neurosurg Psychiatry 74:113–115
Rossini PM, Barker T, Berardelli A, Caramia MD, Caruso G, Cracco RQ, Dimitrijevic MR, Hallett M, Katayama Y, Lucking CH, Maertens de Noordhout AL, Marsden CD, Murray NMF, Rothwell JC, Swash M, Tomberg C (1994) Non invasive electrical and magnetic stimulation of the brain, spinal cord and roots: basic principles and procedures for routine clinical application: report of IFCN committee. Electroencephalogr clin Neurophysiol 91:79–92
Rothwell JC (2003) Techniques of transcranial magnetic stimulation. In: Boniface SJ, Ziemann U (eds) Plasticity in human nervous system. Investigations with transcranial magnetic stimulation. University Press, Cambridge, pp 26–61
Sakuma K, Murakami T, Nakashima K (2007) Short latency afferent inhibition is not impaired in mild cognitive impairment. Clin Neurophysiol 118(7):1460–1463
Sailer A, Molnar GF, Paradiso G, Gunraj CA, Lang AE, Chen R (2003) Short and long latency afferent inhibition in Parkinson’s disease. Brain 126:1883–1894
Saykin AJ, Wishart HA, Rabin LA, Flashman LA, McHugh TL, Mamourian AC (2004) Cholinergic enhancement of frontal lobe activity in mild cognitive impairment. Brain 127:1574–1583
Shinotoh H, Namba H, Fukushi K, Nagatsuka S, Tanaka N, Aotsuka A, Ota T, Tanada S, Irie T (2000) Progressive loss of cortical acetylcholinesterase activity in association with cognitive decline in Alzheimer’s disease: a positron emission tomography study. Ann Neurol 48:194–200
Shinotoh H, Fukushi K, Nagatsuka S, Tanaka N, Aotsuka A, Ota T (2003) The amygdala and Alzheimer’s disease: positron emission tomographic study of the cholinergic system. Ann N Y Acad Sci 985:411–419
Smith G (1988) Animal models of Alzheimer’s disease: experimental cholinergic denervation. Brain Res Rev 13:103–118
Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Roman R, Davies P, Masliah E, Williams DsS Goldstein LS (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 307:1282–1288
Tokimura H, Di Lazzaro V, Tokimura Y, Oliviero A, Profice P, Insola A, Mazzone P, Tonali P, Rothwell JC (2000) Short latency inhibition of human hand motor cortex by somatosensory input from the hand. J Physiol 523:503–513
Voytko ML, Olton DS, Richardson RT, Gorman LK, Tobin JR, Price DL (1994) Basal forebrain lesions in monkeys disrupt attention but not learning and memory. J Neurosci 14:167–186
Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR (1981) Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol 10:122–126
Ziemann U (2002) Assessment of motor cortex and descending motor pathways. In: Brown WF, Bolton CF, Aminoff MJ (eds) Neuromuscular function and disease. Basis, clinical and electrodiagnostic aspects aspects, vol 1. Saunders, Philadelphia, pp 189–221
Ziemann U (2004) TMS and drugs. Clin Neurophysiol 115:1717–1729
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nardone, R., Bergmann, J., Kronbichler, M. et al. Abnormal short latency afferent inhibition in early Alzheimer’s disease: a transcranial magnetic demonstration. J Neural Transm 115, 1557–1562 (2008). https://doi.org/10.1007/s00702-008-0129-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-008-0129-1