
Abstract
The importance of genomic information in intrinsic brain tumors is highlighted in the World Health Organization (WHO) 2016 classification of gliomas, which now incorporates both phenotype and genotype data to assign a diagnosis. By using genetic markers to both categorize tumors and advise patients on prognosis, this classification system has minimized the risk of tissue sampling error, improved diagnostic accuracy, and reduced inter-rater variability. In the neurosurgical community, it is critical to understand the role genetics plays in tumor biology, what certain mutations mean for the patient’s prognosis and adjuvant treatment, and how to interpret the results of sequencing data that are generated following tumor resection. In this review, we examine the critical role of genetics for diagnosis and prognosis and highlight the importance of tumor genetics for neurosurgeons caring for patients with diffuse lower grade gliomas.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genomics has revolutionized how many solid malignancies are classified and treated. Not surprisingly, the introduction of next-generation sequencing has transformed the classification of low-grade gliomas (LGGs) and high-grade gliomas from a histological definition to a molecular one (for a list of landmark papers see Table 1) [99]. This revolution is highlighted in the World Health Organization (WHO) 2016 classification of gliomas, which incorporated both phenotype and genotype data to assign a diagnosis. In addition to clarifying diagnoses, a tumor’s molecular profile can aid in prognostication, identify potentially targetable mutations, and select for appropriate clinical trial enrollment. As a result, in addition to understanding how a tumor’s size, location, and performance status influence a patient’s outcome, it is now necessary for surgeons to understand the mutational profile of a diffuse lower grade glioma, as the tumor’s individual mutation profile is critical for appropriate prognostication and patient education. In this comprehensive, non-systematic review, we discuss these relevant molecular markers for low and lower (i.e., grade II or III) grade gliomas.
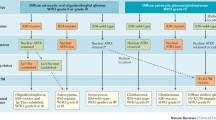
2016 World Health Organization classification
The 2016 Revised 4th edition of the WHO classification of tumors of the central nervous system represents a conceptual shift in the categorization of gliomas [54]. Diffuse gliomas (including astrocytomas, oligodendrogliomas, and glioblastoma (GBM)) are now grouped together and classified using both histological and molecular markers (see Table 2 for a list of molecular markers). These tumors are now clearly separated from more discrete astrocytic tumors, such as pilocytic astrocytoma and pleomorphic xanthoastroctyoma. This represented a significant improvement in both pathobiological understanding and diagnostic accuracy, with a reduction in interobserver variability [90]. One study showed that between 20 and 35% of oligodendrogliomas and nearly 10% of astrocytomas and GBMs were misclassified with histology alone [40]. Moreover, when interpreting clinical trials and retrospective studies that utilize histologic methods for diagnosis, readers must cautiously evaluate the findings given the high rates of tumor reclassification when genetic information is incorporated. Additionally, since tumor mutations are typically clonal, the genetic information found in a tumor mass is “volume independent,” which significantly reduces the sampling bias that used to plague small tissue samples obtained from stereotactic biopsy specimens [28, 49].
Nevertheless, a reliance on molecular diagnosis is not without limitations. The distinction between grade II and grade III tumors remained dependent of the number of mitoses seen by the neuropathologist. However, the importance of this mitotic activity on patient outcome may be less relevant for IDH-mutant gliomas, and as such using mitoses as the crucial variable for assigning a higher tumor grade may not accurately reflect the tumors’ biological behavior and/or aggressiveness [65, 74]. One group showed that IDH-mutant tumors could be better stratified by a combination of morphology and homozygous deletion of CDKN2A/2B and copy number variant status, although this genetic profile has not been incorporated in the diagnostic framework to date [83].
Relevant molecular markers in diffuse lower grade gliomas
IDH 1/2
One of the first mutations identified when the genomes of LGGs was sequenced was in the Isocitrate dehydrogenase gene (either in IDH1 or IDH2), which is involved in the tricarboxylic acid cycle. Mutations in IDH1 codon 132 or IDH2 codon 172 were found in over 70% of LGGs [97]. These mutations can be present in both astrocytic and oliogodendrocytic tumors. Importantly, when a mutation in IDH is present, other mutations that are frequently found in GBM (e.g., PTEN, EGFR, CDKN2A/2B) are absent [97]. IDH mutations also occur in a diverse range of cancers including acute myelogenous leukemia [57] and cholangiocarcinoma [7] suggesting a conserved role in oncogenesis.
IDH is involved in the canonical citric-acid cycle. Normally, IDH catalyzes isocitric acid to α-ketoglutarate and reduces NADP+ to NADPH. When there is an IDH-1 mutation present, there is neomorphic enzyme activity, which results in further conversion of α-ketoglutarate to the oncometabolite 2-hydroxyglutarate (2-HG) [26]. This inhibits DNA demethylases, specifically Ten-eleven translocation (TET) enzymes, and results in a net increase in DNA methylation, particularly at CpG islands, which is referred to as the G-CIMP hypermethylation phenotype [62, 87]. This hypermethylation of DNA methylome impairs the quaternary DNA structure and chromatin loops, making the binding of certain transcription factors challenging and brings typically disparate genes into close proximity, allowing for increased expression of certain genes and ultimately oncogenesis. Furthermore, DNA hypermethylation also interferes with the cells differentiation state.
The identification of IDH 1/2 as a gene critical for prognostication in LGG is evidenced by the inclusion of IDH mutation status in the WHO 2016 classification schema. Moreover, patients who are diagnosed with a LGG who have a wild-type IDH (i.e., a “molecular GBM”) have a poor outcome that more closely resembles the outcomes of GBM than LGG [13]. Importantly, multiple reports have suggested a huge degree of heterogeneity for patients with these IDH-wild-type (wt) lower grade gliomas, indicating more work is needed to better prognosticate these patients [2, 22].
In 2015, a review of 293 LGGs from The Cancer Genome Atlas or TCGA found that using two molecular markers, IDH status and 1p/19q status, diffuse LGGs could be stratified into two different groups [13]. Patients with 1p/19q codeleted tumors and IDH mutations had a median survival of 8 years, compared with 6.3 years for patients with IDH-mutated tumors and no codeletion, and 1.7 years for IDH-wt tumors. That same year, a population-based study of 1087 diffuse gliomas showed that grade II and grade III gliomas could be molecularly stratified into 5 subgroups with different outcomes based on the mutation status of three molecular markers: 1p/19q, IDH 1/2, and TERT (telomerase reverse transcriptase) promoter [24]. In this cohort, presence of a TERT promoter mutation was associated with a poorer prognosis.
Nearly all LGGs with IDH mutations that lack the 1p/19q codeletion are histologically astrocytomas, and the overwhelming majority have mutations in p53 (94%) and ATRX (alpha thalassemia/mental retardation syndrome X-linked, 86%). By comparison, tumors that harbor both the IDH mutation and 1p/19q codeletion are histologically oligodendrogliomas and typically also have mutations in CIC (capicua), FUBP1 (far upstream element binding protein 1), Notch1, and TERT promoter [6]. By including these additional mutations into the tumor profile, some mutational combinations appear to be predictors of outcome. For example, having an IDH mutation, MGMT methylation, and p53 mutation has been shown to increase the risk for malignant transformation of the tumor [51].
As mentioned above, about 20% of lower grade gliomas are IDH-wt, particularly grade III gliomas and those with astrocytic histology [2]. These tumors frequently contain other molecular alterations commonly seen in GBM, such as chromosome 7 gains, chromosome 10 deletions, EGFR (epidermal growth factor receptor) amplifications, TERT promoter mutations, CDKN2A deletions, and RB1 deletions. The presence or absence of some of these additional mutations plays an important role in prognosticating within the IDH-wt LGGs. For example, IDH-wt tumors that have either the EGFR amplification, H3F3A mutation, or TERT promoter mutation have a much worse prognosis compared with IDH-wt tumors without this mutations (1.2-year vs. 7.6-year overall survival) [2].
The stratification of IDH-wt LGG can get even more complex when DNA methylation patterns are incorporated. A follow-up study using the TCGA found that 3 methylation patterns could be identified for LGGs [15]. Two subgroups mimic the expression seen in the classical and mesenchymal subtypes of GBM, while a third shares similarities with pilocytic astrocytomas and, unsurprisingly, has a better prognosis. Although of great interest, this stratification is not yet in clinical use.
1p/19q codeletion
Aberrant chromosomal arrangements in glioma were noted prior to the discovery of IDH mutations. As early as 1994, Reifenberger et al. discovered that most histologically oligodendroglial tumors had heterozygous loss of chromosome 1p and 19q [73]. This was subsequently discovered to be a result of an unbalance whole arm translocation t(1p;19q) [29]. 1p/19q codeletion occurs in over 70% of tumors with oligodendroglial pathology and almost all have IDH mutations, suggesting the chromosomal aberration occurs after IDH1/2 mutation [14]. The oligodendroglial tumors that do not have fluorescence in situ hybridization (FISH) identifiable 1p/19q codeletions still harbor deletions of the 1p and 19q arms from their respective chromosomes (but not the canonical codeletion), and thus are harder to identify with FISH. Despite being discovered over two decades ago, the oncogenic pathobiology of 1p/19q codeletion remains surprisingly poorly understood. Recent work has shown that 1p/19q codeleted tumors have less ultra-long protrusions, or tumor associated microtubules (TMs), which work to interconnect tumor cells and potentially make them resistant to chemotherapy and radiotherapy by allowing them to supply damaged organelles to one another [66, 67].
Oligodendrogliomas are also associated with mutations in CIC and FUBP1, located on chromosome 19q and 1p respectively[76]. Allelic loss of 1p/19q unmasks these mutations [6], promoting cell migration, inhibiting apoptosis, and interfering with citric acid regulation in conjunction with IDH mutations [17]. 1p/19q codeletion is associated with improved survival [41] and predictive of response to chemotherapy, particularly PCV [12, 89].
The close association of 1p/19q co-deletion and oligodendroglial histology has resulted in significant alterations in diagnostic criteria. Almost all tumors previously categorized as oligoastrocytoma can now be recategorized as either astrocytoma or oligodendroglioma based on 1p/19q [13, 86]. This has resulted in the all but disappearance oligoastrocytoma, a diagnosis with poor interobserver reliability[20, 88] that is now reserved for situations where molecular testing is not possible, or extremely rare circumstances where tumors have molecular and histological features of both astrocytoma and oligodendroglioma [77].
MGMT
O6-methylguanine-DNA methyltransferase (MGMT) is a DNA repair enzyme located on 10q, and methylation of the promotor region of the MGMT is known to predict TMZ response in high-grade gliomas [34]. When the promotor region of the MGMT gene is methylated, there are lower levels of this DNA repair enzyme, and as a result, the tumor cells are unable to repair the alkylating damage caused by TMZ, leading to more abundant TMZ-induced DNA damage and ultimately more cytotoxicity within tumor cells. MGMT promotor methylation is also frequently observed in LGG (45–89%) [4, 32, 51] with the lowest frequency observed in IDH-wt low-grade gliomas [4]. Although MGMT methylation is usually reported as binary value (methylated or unmethylated), the degree of methylation is different across tumors and may influence how patients respond to TMZ therapy.
Leu et al. reported that MGMT methylation in conjunction with IDH-mut status in LGGs was associated with a favorable impact on survival [51]. However, the utility of MGMT methylation as a prognostic marker within IDH-mut LGGs may be hampered by the relatively high methylation rate of MGMT seen in this subgroup, and more work is needed to determine its utility as a prognostic marker in the subgroup of IDH-wt tumors. In terms of the predictive abilities of MGMT methylation on treatment response, some studies suggest that TMZ response rates may be better in low-grade patients harboring methylation of MGMT [25, 39, 52]. Within a cohort of patients undergoing treatment with surgery alone, MGMT promoter methylation did not appear to be a prognostic marker [32].
Some studies in high-grade glioma and GBM have suggested that overwhelming the MGMT enzyme with either dose dense TMZ or additional alkylating agents like CCNU in conjunction with TMZ may improve outcomes, but since this is also associated with added toxicity, more work is needed to determine which LGG patients could potentially benefit from this approach [27, 35]. Alternatively, PARP inhibitors, which target the PARP-mediated DNA repair pathway and appear to be effective in tumors with homologous recombination defects like BRCA mutations, may also be effective in patients with deficient DNA repair such as those with methylated MGMT.
TERT
Telomerase reverse transcriptase (TERT) is an enzyme that ensures the telomere caps at the end of chromosomes, which are necessary for cell division, do not shorten, and as a result can divide ad infinitum. In tumors, telomerase activity is much higher than what is observed in healthy, normal cells, particularly cell types that are not frequently self-renewing. Given this, it is not surprising that TERT mutations have been associated with gliomas [48].
TERT promoter (TERTp) mutations, which lead to an upregulation of TERT and are nearly mutually exclusive to ATRX mutations, have been found in gliomas [15]. TERT activation and are seen primarily in either IDH-mut 1p/19q co-del (molecular oligodendrogliomas 50–96%) or IDH-wt (diffuse astrocytoma 13–64%) LGGs [14, 15]. In IDH mut-only LGG, TERTp mutations are rare accounting for only 2.8–4% of tumors in this category [14, 15].
While some reports demonstrate TERT protein expression levels are associated with survival in LGG cohorts [24], other reports have not confirmed this [15], and protein expression can be challenging to accurately measure. Eckel-Passow et al. separated grade II/III gliomas into 5 distinct molecular groups based on the status of IDH mutation, TERTp mutation, and 1p/19 codeletion. TERTp mutation-only was associated with the worst prognosis of all 5 subgroups whereas TERTp mutation in conjunction with IDH mutation and/or 1p/19q codeletion conferred a much better prognosis [24]. Examining these molecular subgroups is essential as other reports demonstrate that only using TERTp mutation status alone to prognostic tumors misses these important subgroup differences and would suggest that having a TERTp mutation is actually associated with an improved prognosis [98]. When looking specifically at IDH-wt LGGs, TERTp mutation or gain of chr 7 or loss of 10q (+7/-10q) is thought to confer a prognosis similar to GBM. Wijnenga et al. demonstrated that 18.9% of patients with IDH-wt LGGs carried TERTp mutation but no +7/-10q pattern, and this distinct subgroup had an overall very poor prognosis, even worse than IDH-wt +7/-10q tumors. Therefore, IDH-wt TERTp mut-only tumors may constitute a distinct molecular group that closely resembles GBM [93].
ATRX
Another commonly mutated gene in adult gliomas is the ATRX gene, which was first described in the X-linked mental retardation syndrome. The ATRX protein is a chromatin remodeling protein important for maintaining genetic stability and repairing DNA damage, and its loss can result in an alternative lengthening of telomeres (ALT) which is seen in small percentage of malignancies. However, to date, the presence of an ATRX deletion or mutation has not been utilized in diagnostic or prognostic criteria by the WHO classification. Still, there is a strong association between IDH mutations and ATRX deletions or inactivating mutations, meaning the ATRX mutation is found in the majority of low-grade astrocytomas [43]. However, inactivating mutations in ATRX are rarely seen in 1p/19q mutant gliomas (i.e., oligodendrogliomas), primary GBMs, or in tumors with TERT mutations [30, 79]. ATRX mutations appear to be associated with a better prognosis for IDH-mutant astrocytomas [92]. In the future, DNA repair and chromatin structure pathways that are affected by ATRX mutations may be potential therapeutic targets for small molecule inhibitors, but no targeted therapies have progressed to clinical trials.
Histone H3 K27M
The Histone H3 gene encodes H3.3 and H3.1 proteins which are integral for DNA packing into nucleosomes and regulation in gene expression. H3 mutations were first noted in sequencing of pediatric diffuse intrinsic pontine gliomas and pediatric GBMs [81, 96]. Subsequent work lead to the definition of a new entity in the WHO classification: diffuse midline glioma, with H3 K27M-mutant. Although this entity is considered grade IV, the molecular marker is relevant to LGGs because midline tumors with this mutation will behave aggressively despite appearing low grade histologically.
How molecular markers influence patient management
Surgical management
Now that molecular features are included in the diagnosis of gliomas, there is little role for watchful waiting in patients with imaging findings concerning for a LGG as tissue is critical for prognostication and treatment. Moreover, numerous retrospective studies have shown that maximal safe surgical resection improves survival in patients with LGG [1, 36]. Additionally, a large meta-analysis showed that gross total resection is associated with improved overall survival and progression free survival at 2, 5, and 10 years compared with a subtotal resection [9].
However, less is known how the genetic mutations found in a tumor affect the benefit of extended surgical resection and there have been conflicting reports in the literature. Patel et al. found greater EOR improved overall survival in patients with grade II gliomas that were IDH-wt, but this benefit was not observed for patients with IDH-mut tumors [68]. Alternatively, a recent study by Kavouridis et al. found that increased post-operative residual tumor volume was associated with a worse overall survival, progression-free survival, and malignant progression-free survival for all molecular tumor subtypes (i.e., oligodendrogliomas, IDH-mutant astrocytomas, and IDH-wild type astrocytomas) [45]. In a retrospective review of nearly 600 adult patients with 1p/19q codeleted grade II oligodendrogliomas, Harary et al. found gross total resection, but not subtotal resection, improved overall survival compared with biopsy or watchful waiting [31]. Interestingly, Beiko et al. found that IDH-mutant tumors may be more amenable to gross total resection, possibly due to their predominantly frontal location [5].
Taken together, these findings argue for a maximal resection that minimizes the residual tumor volume for LGGs, regardless of the tumors underlying mutational profile. Future work is needed to determine the impact that a new post-operative neurological deficit has on survival and quality of life, which has been shown to worsen outcomes for patients with GBM [58]. Since LGGs are most likely to involve cortex and white matter that can retain their function, and some patients with these low-grade tumors can be expected to live for more than a decade, maximizing safety may be of even greater significance.
Defining low-grade high-risk gliomas
After the publication of RTOG 9802 [10], patients who are over 40 years old and have less than a GTR are considered “low-grade high risk” and the general consensus is these patients should be treated with adjuvant therapy following surgical resection. Nevertheless, the optimal timing and adjuvant therapy is an ongoing subject of controversy. This is an important subset of LGG patients for neurosurgeons to be mindful of, as patients should be appropriately counseled post-operatively on the role and importance of adjuvant therapy pending the extent of surgical resection.
Adjuvant therapy
After a surgical resection of low-grade glioma, no single chemotherapy regimen has proved itself to be the gold standard, and there are large discrepancies in the timing and choice of chemotherapy, radiation, and combined treatments among neuro-oncologists [21]. As a result, there are ongoing trials comparing TMZ with other regimens, but some of the highest quality evidence supports the use of PCV chemotherapy to radiotherapy [10].
Patients with IDH-mutated tumors or 1p/19q codeletions appear to benefit more from chemotherapy than those without IDH mutations [10, 61]. In fact, there is a phase III clinical trial exploring whether patients with IDH-mutant tumors should undergo adjuvant treatment or if they should be observed and wait for treatment until there is evidence of recurrence (NCT03763422).
Early radiation has been shown to extend progression-free survival, although the effect on overall survival is less clear, and doses of 45–55 Gy seem to be as effective as higher doses with less toxicity [9, 44]. Moreover, delaying radiation treatment until the time of first progression has not been shown to compromise patient survival.
Using imaging to aid in pretissue diagnosis or identifying recurrence
Recent work has focused on imaging characteristics that correlate with molecular profiles to help guide clinical decision-making. The “T2-FLAIR mismatch sign” on conventional MRI (i.e., hyperintense signal on T2W MRI with relative hypointense signal on FLAIR sequences except for a hyperintense peripheral rim) has been shown to be a highly specific marker for IDH-mutant, 1p/19q intact tumors [8, 69]. Magnetic resonance spectroscopy for α-ketogluterate was described by multiple groups in 2012 [3, 18, 71] and may be useful for distinguishing non-specific FLAIR abnormalities from low-grade tumors and to distinguish scar around resection cavities from tumor.
Other work has shown that ADC values can be used to distinguish between oligodendrogliomas and astrocytomas with higher ADC values associated with astrocytomas [46, 47]. Additionally, although PET scans are not commonly used in clinical practice, the presence of 2-HG can be identified with O-(2-[18F]fluoroethyl)-l-tyrosine (FET) PET, which in the future could provide a reliable, non-invasive measure of IDH status.
Tumor genetics is also relevant when interpreting surveillance scans for possible progression. MGMT promoter methylation and IDH mutations are associated with pseudoprogression for patients treated with TMZ for GBM [53]. Although significance of these mutations for imaging LGGs is unclear, it will be important to understand how these genetic factors affect imaging responses to treatment as more patients are living longer and receiving adjuvant therapies.
Targeting novel therapies
While the focus of this manuscript is on molecular markers and not treatment, we want to briefly cover some of the novel therapies that are under investigation for patients with diffuse LGG.
Given the seemingly major role IDH mutations play in the prognosis of patients, there has been significant interest in developing a targeted therapy against these mutations. Mechanistically, as mentioned above, the IDH mutation results in the production of 2-hydroxyglutarate (2-HG), which downstream inhibits tumor infiltrating lymphocytes and results in suppression of anti-tumor immune responses [11, 91]. As such, there is biologic plausibility for an IDH inhibitor to generate an antitumor immune effect. Alternatively, some researchers have argued against IDH inhibitors given that the epigenetic changes that take place after an IDH mutation may render any inhibition of mutant IDH ineffective, and there has been some evidence to suggest that although IDH is an early mutation in gliomagenesis, it may not be a mutation that is essential for glioma survival.
The small molecule inhibitor Ivosidenib (AG-120) is an inhibitor of IDH1 mutations and has been shown to be safe in patients. In a phase I trial of 66 glioma patients, AG-120 was shown to be safe and may have stabilized the growth of non-enhancing, LGGs [59]. There are other IDH-mutant inhibitors that inhibit both the IDH1 and IDH2 mutation and are capable of penetrating the brain. One such molecule, Vorasidenib or AG-881, is a potent oral inhibitor of both IDH1- and IDH2-mutated proteins and is currently in a phase III clinical trials. In preclinical studies, this compound has been shown to reduce 2-HG levels by over 95% and the phase I data suggests a favorable safety profile at doses less than 100 mg [60]. Unfortunately, to date, the trials have not shown any benefit for contrast enhancing recurrent tumors. More research into these exciting compounds has the potential to influence how LGG patients are managed in the future.
Beyond direct inhibitors of IDH, there are other agents that may have a therapeutic effect in IDH mutated tumors. For example, another action of 2-HG is the inhibition homologous recombination, which can make tumors with the IDH mutation sensitive to poly(ADP-ribose) polymerase (PARP) inhibition [55, 85]. This finding has led to an upcoming study investigating the role of PARP inhibitors with TMZ for recurrent IDH mutated gliomas [70].
Additionally, similar to high-grade tumors, significant research efforts have led to a series of tumor vaccines for LGGs [23, 38]. Groundbreaking work showed that the most common IDH mutation, R132H, creates a peptide epitope that is immunogenic and presented on MHC molecules to T cells to generate an immune response [80]. As such, peptide vaccination has been shown to induce immunity and control tumor growth in transgenic murine models with human MHCs. Another mutation that has been shown to function as a neoantigen for the purpose of vaccine generation is the H3K27M histone mutation [16, 63]. Moreover, some of the tumor peptides that are targeted by the peptide vaccines used in GBM have been shown to generate spontaneous immune responses in LGGs as well, arguing for the potential effectiveness of these vaccines in the low-grade setting [23]. As some of these vaccines are currently in active clinical trials for malignant gliomas, we are eagerly awaiting the results to emerge.
In addition to targeting mutations such as IDH with either small molecule inhibitors or creating tumor vaccines, the relative ease of tumor sequencing has made genomic testing for numerous mutations commonplace at major academic institutions. This creates a personalized profile for each patient’s tumor, and as a result opens up the possibility to try off-label targeted therapies such as small molecular inhibitors or enroll in specific clinical trials with strong biological plausibility for success.
Genetic risks for the development of LGG
In addition to caring for the patient, surgeons can use molecular information to help provide appropriate counseling for the patient’s family. Most of the risk for developing LGG is non-modifiable, but nevertheless the information on genetic risk can be valuable for some patients.
Hereditary cancer syndromes
Although the vast majority of LGGs come from spontaneous somatic mutations, a small percentage of patients with gliomas, approximately 5%, have germline mutations [37]. Some hereditary cancer syndromes that have been associated with gliomas include Li-Fraumeni syndrome, neurofibromatosis 1, and tuberous sclerosis [75]. Interestingly, patients with Li-Fraumeni syndrome, who harbor a germline TP53 mutation, tend to have IDH R132C mutations, which is typically an uncommon IDH mutation [33, 64].
Single nucleotide polymorphisms can increase gliomagenesis risk
The observation that patients with a family history of glioma have a higher risk of developing a glioma (5–10% of glioma patients have a family history of glioma) [56, 94], led to large genome wide association studies (GWAS) to further understand how germline variants affect glioma risk. A large GWAS trial found 19 single nucleotide polymorphisms (SNPs) that were associated with LGG formation and 5 additional SNPs associated with both LGG and GBM [50]. For instance, variants at 8q24.21 confer more than a sixfold relative risk of developing an IDH-mutant astrocytoma or oligodendroglioma [42]. Gene variants in EGFR, TERT, TP53, CCDC26, CDKN2B, PHLDB1, and RTEL1 have all been associated with an increased risk of developing glioma [72, 78, 82, 84, 95].
While there is still a relatively low rate of glioma in the general population and these SNPs are not currently included in any screening recommendations, knowledge of these high-risk alleles may be helpful when an incidental lesion is discovered for predicting how it will behavior during surveillance.
Conclusion
There is no doubt molecular characterization of diffuse LGGs has improved the diagnostic accuracy and prognostication of patients. As neurosurgeons, an understanding of the clinical course for a patient based on these molecular traits of the tumor is crucial for managing recurrences and understanding the adjuvant therapies that may be available for a particular patient. In the future, it is possible that imaging features of the tumor may be able to differentiate the tumor mutational profile and help clarify if there is true progression versus pseudoprogression, and it will be important for surgeons to take this information into consideration during operative planning. Ongoing research is investigating the potential for targeted agents against certain mutations and it remains to be seen if these may eventually play a role in patient care.
Abbreviations
- LGG:
-
Low-grade glioma
- GBM:
-
Glioblastoma
- WHO:
-
World Health Organization
- IDH:
-
Isocitrate dehydrogenase
- GWAS:
-
Genome wide association study
- IHC:
-
Immunohistochemistry
- 2-HG:
-
2-Hydroxyglutarate
References
Aghi MK, Nahed BV, Sloan AE, Ryken TC, Kalkanis SN, Olson JJ (2015) The role of surgery in the management of patients with diffuse low grade glioma: a systematic review and evidence-based clinical practice guideline. J Neuro-Oncol 125(3):503–530
Aibaidula A, Chan AKY, Shi Z et al (2017) Adult IDH wild-type lower-grade gliomas should be further stratified. Neuro-Oncology. https://doi.org/10.1093/neuonc/nox078
Andronesi OC, Kim GS, Gerstner E, Batchelor T, Tzika AA, Fantin VR, Vander Heiden MG, Sorensen AG (2012) Detection of 2-hydroxyglutarate in IDH-mutated glioma patients by in vivo spectral-editing and 2D correlation magnetic resonance spectroscopy. Sci Transl Med. https://doi.org/10.1126/scitranslmed.3002693
Baumert BG, Hegi ME, van den Bent MJ et al (2016) Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol 17(11):1521–1532
Beiko J, Suki D, Hess KR et al (2014) IDH1 mutant malignant astrocytomas are more amenable to surgical resection and have a survival benefit associated with maximal surgical resection. Neuro-Oncology. https://doi.org/10.1093/neuonc/not159
Bettegowda C, Agrawal N, Jiao Y et al (2011) Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science (80- ). https://doi.org/10.1126/science.1210557
Borger DR, Tanabe KK, Fan KC et al (2012) Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. https://doi.org/10.1634/theoncologist.2011-0386
Broen MPG, Smits M, Wijnenga MMJ, Dubbink HJ, Anten MHME, Schijns OEMG, Beckervordersandforth J, Postma AA, van den Bent MJ (2018) The T2-FLAIR mismatch sign as an imaging marker for non-enhancing IDH-mutant, 1p/19q-intact lower-grade glioma: a validation study. Neuro-Oncology 20(10):1393–1399
Brown TJ, Bota DA, van Den Bent MJ et al (2019) Management of low-grade glioma: a systematic review and meta-analysis. Neurol Pract 6(4):249–258
Buckner JC, Shaw EG, Pugh SL et al (2016) Radiation plus procarbazine, CCNU, and vincristine in low-grade glioma. N Engl J Med. https://doi.org/10.1056/NEJMoa1500925
Bunse L, Pusch S, Bunse T et al (2018) Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med. https://doi.org/10.1038/s41591-018-0095-6
Cairncross G, Wang M, Shaw E et al (2013) Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: Long-term results of RTOG 9402. J Clin Oncol. https://doi.org/10.1200/JCO.2012.43.2674
Cancer Genome Atlas Research Network, Brat DJ, Verhaak RGW et al (2015) Comprehensive, integrative genomic analysis of diffuse lower-gade gliomas. N Engl J Med. https://doi.org/10.1056/NEJMoa1402121
Cancer Genome Atlas Research Network, Brat DJ, Verhaak RGW, Aldape KD, Yung WKA, Salama SR, Cooper LAD et al (2015) Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 372(26):2481–2498
Ceccarelli M, Barthel FP, Malta TM et al (2016) Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. https://doi.org/10.1016/j.cell.2015.12.028
Chheda ZS, Kohanbash G, Okada K et al (2018) Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med. https://doi.org/10.1084/jem.20171046
Chittaranjan S, Chan S, Yang C et al (2014) Mutations in CIC and IDH1 cooperatively regulate 2-hydroxyglutarate levels and cell clonogenicity. Oncotarget. https://doi.org/10.18632/oncotarget.2401
Choi C, Ganji SK, DeBerardinis RJ et al (2012) 2-Hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat Med. https://doi.org/10.1038/nm.2682
Clark KH, Villano JL, Nikiforova MN, Hamilton RL, Horbinski C (2013) 1p/19q testing has no significance in the workup of glioblastomas. Neuropathol Appl Neurobiol 39(6):706–717
Coons SW, Johnson PC, Scheithauer BW, Yates AJ, Pearl DK (1997) Improving diagnostic accuracy and interobserver concordance in the classification and grading of primary gliomas. Cancer. https://doi.org/10.1002/(SICI)1097-0142(19970401)79:7<1381::AID-CNCR16>3.0.CO;2-W
Darlix A, Mandonnet E, Freyschlag CF et al (2019) Chemotherapy and diffuse low-grade gliomas: a survey within the European Low-Grade Glioma Network. Neuro-Oncology Pract. https://doi.org/10.1093/nop/npy051
DiCarlo DT, Duffau H, Cagnazzo F, Benedetto N, Morganti R, Perrini P (2018) IDH wild-type WHO grade II diffuse low-grade gliomas. A heterogeneous family with different outcomes. Systematic review and meta-analysis. Neurosurg Rev. https://doi.org/10.1007/s10143-018-0996-3
Dutoit V, Migliorini D, Ranzanici G et al (2018) Antigenic expression and spontaneous immune responses support the use of a selected peptide set from the IMA950 glioblastoma vaccine for immunotherapy of grade II and III glioma. Oncoimmunology. https://doi.org/10.1080/2162402X.2017.1391972
Eckel-Passow JE, Lachance DH, Molinaro AM et al (2015) Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med 372(26):2499–2508
Everhard S, Kaloshi G, Criniere E et al (2006) MGMT methylation: a marker of response to temozolomide in low-grade gliomas. Ann Neurol 60(6):740–743
Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suvà ML, Bernstein BE (2016) Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. https://doi.org/10.1038/nature16490
Gilbert MR, Wang M, Aldape KD et al (2013) Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol. https://doi.org/10.1200/JCO.2013.49.6968
Glantz MJ, Burger PC, Herndon JE, Friedman AH, Cairncross JG, Vick NA, Schold SC (2012) Influence of the type of surgery on the histologic diagnosis in patients with anaplastic gliomas. Neurology. https://doi.org/10.1212/wnl.41.11.1741
Griffin CA, Burger P, Morsberger L, Yonescu R, Swierczynski S, Weingart JD, Murphy KM (2006) Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J Neuropathol Exp Neurol. https://doi.org/10.1097/01.jnen.0000235122.98052.8f
Haase S, Garcia-Fabiani MB, Carney S, Altshuler D, Núñez FJ, Méndez FM, Núñez F, Lowenstein PR, Castro MG (2018) Mutant ATRX: uncovering a new therapeutic target for glioma. Expert Opin Ther Targets. https://doi.org/10.1080/14728222.2018.1487953
Harary M, Kavouridis VK, Torre M, Zaidi HA, Chukwueke UN, Reardon DA, Smith TR, Iorgulescu JB (2019) Predictors and early survival outcomes of maximal resection in WHO grade II 1p/19q-codeleted oligodendrogliomas. Neuro-Oncology. https://doi.org/10.1093/neuonc/noz168
Hartmann C, Hentschel B, Tatagiba M et al (2011) Molecular markers in low-grade gliomas: predictive or prognostic? Clin Cancer Res 17(13):4588–4599
Hayes J, Yu Y, Jalbert LE et al (2018) Genomic analysis of the origins and evolution of multicentric diffuse lower-grade gliomas. Neuro-Oncology. https://doi.org/10.1093/neuonc/nox205
Hegi ME, Diserens A-C, Gorlia T et al (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352(10):997–1003
Herrlinger U, Tzaridis T, Mack F et al (2019) Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA–09): a randomised, open-label, phase 3 trial. Lancet. https://doi.org/10.1016/S0140-6736(18)31791-4
Hervey-Jumper SL, Berger MS (2016) Maximizing safe resection of low- and high-grade glioma. J Neuro-Oncol. https://doi.org/10.1007/s11060-016-2110-4
Hervey-Jumper SL, van de Bent MJ, Mehta MP, Berger MS (2019) WHO II and III gliomas BT - oncology of cns tumors. In: Tonn J-C, Reardon DA, Rutka JT, Westphal M (eds) . Springer International Publishing, Cham, pp 217–236
Hilf N, Kuttruff-Coqui S, Frenzel K et al (2019) Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 565(7738):240–245
Houillier C, Wang X, Kaloshi G et al (2010) IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology 75(17):1560–1566
Iorgulescu JB, Torre M, Harary M, Smith TR, Aizer AA, Reardon DA, Barnholtz-Sloan JS, Perry A (2019) The misclassification of diffuse gliomas: rates and outcomes. Clin Cancer Res 25(8):2656 LP–2652663
Jenkins RB, Blair H, Ballman KV et al (2006) A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-06-1796
Jenkins RB, Xiao Y, Sicotte H et al (2012) A low-frequency variant at 8q24.21 is strongly associated with risk of oligodendroglial tumors and astrocytomas with IDH1 or IDH2 mutation. Nat Genet. https://doi.org/10.1038/ng.2388
Kannan K, Inagaki A, Silber J, Gorovets D, Zhang J, Kastenhuber ER, Heguy A, Petrini JH, Chan TA, Huse JT (2012) Whole exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget. https://doi.org/10.18632/oncotarget.689
Karim ABMF, Maat B, Hatlevoll R et al (1996) A randomized trial on dose-response in radiation therapy of low-grade cerebral glioma: European organization for research and treatment of cancer (EORTC) study 22844. Int J Radiat Oncol Biol Phys. https://doi.org/10.1016/S0360-3016(96)00352-5
Kavouridis VK, Boaro A, Dorr J, Cho EY, Iorgulescu JB, Reardon DA, Arnaout O, Smith TR (2019) Contemporary assessment of extent of resection in molecularly defined categories of diffuse low-grade glioma: a volumetric analysis. J Neurosurg 1(aop):1–11
Khayal IS, Nelson SJ (2009) Characterization of low-grade gliomas using RGB color maps derived from ADC histograms. J Magn Reson Imaging. https://doi.org/10.1002/jmri.21810
Khayal IS, Vandenberg SR, Smith KJ, Cloyd CP, Chang SM, Cha S, Nelson SJ, McKnight TR (2011) MRI apparent diffusion coefficient reflects histopathologic subtype, axonal disruption, and tumor fraction in diffuse-type grade II gliomas. Neuro-Oncology. https://doi.org/10.1093/neuonc/nor122
Killela PJ, Reitman ZJ, Jiao Y et al (2013) TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1303607110
Kim BYS, Jiang W, Beiko J, Prabhu SS, Demonte F, Gilbert MR, Sawaya R, Aldape KD, Cahill DP, McCutcheon IE (2014) Diagnostic discrepancies in malignant astrocytoma due to limited small pathological tumor sample can be overcome by IDH1 testing. J Neuro-Oncol. https://doi.org/10.1007/s11060-014-1451-0
Kinnersley B, Labussière M, Holroyd A et al (2015) Genome-wide association study identifies multiple susceptibility loci for glioma. Nat Commun. https://doi.org/10.1038/ncomms9559
Leu S, von Felten S, Frank S et al (2013) IDH/MGMT-driven molecular classification of low-grade glioma is a strong predictor for long-term survival. Neuro-Oncology 15(4):469–479
Levin N, Lavon I, Zelikovitsh B, Fuchs D, Bokstein F, Fellig Y, Siegal T (2006) Progressive low-grade oligodendrogliomas: response to temozolomide and correlation between genetic profile and O6-methylguanine DNA methyltransferase protein expression. Cancer 106(8):1759–1765
Li H, Li J, Cheng G, Zhang J, Li X (2016) IDH mutation and MGMT promoter methylation are associated with the pseudoprogression and improved prognosis of glioblastoma multiforme patients who have undergone concurrent and adjuvant temozolomide-based chemoradiotherapy. Clin Neurol Neurosurg. https://doi.org/10.1016/j.clineuro.2016.10.004
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. https://doi.org/10.1007/s00401-016-1545-1
Lu Y, Kwintkiewicz J, Liu Y et al (2017) Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-16-2773
Malmer B, Grönberg H, Bergenheim AT, Lenner P, Henriksson R (1999) Familial aggregation of astrocytoma in Northern Sweden: an epidemiological cohort study. Int J Cancer. https://doi.org/10.1002/(SICI)1097-0215(19990505)81:3<366::AID-IJC9>3.0.CO;2-0
Mardis ER, Ding L, Dooling DJ et al (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. https://doi.org/10.1056/NEJMoa0903840
McGirt MJ, Mukherjee D, Chaichana KL, Than KD, Weingart JD, Quinones-Hinojosa A (2009) Association of surgically acquired motor and language deficits on overall survival after resection of glioblastoma multiforme. Neurosurgery. https://doi.org/10.1227/01.NEU.0000349763.42238.E9
Mellinghoff IK, Touat M, Maher E et al (2017) ACTR-46. AG-120, A first-in-class mutant idh1 inhibitor in patients with recurrent or progressive idh1 mutant glioma: updated results from the phase 1 non-enhancing glioma population. Neuro-Oncology. https://doi.org/10.1093/neuonc/nox168.037
Mellinghoff IK, Penas-Prado M, Peters KB et al (2018) Phase 1 study of AG-881, an inhibitor of mutant IDH1/IDH2, in patients with advanced IDH-mutant solid tumors, including glioma. J Clin Oncol 36(15_suppl):2002
Nitta M, Muragaki Y, Maruyama T et al (2015) Proposed therapeutic strategy for adult low-grade glioma based on aggressive tumor resection. Neurosurg Focus. https://doi.org/10.3171/2014.10.FOCUS14651
Noushmehr H, Weisenberger DJ, Diefes K et al (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. https://doi.org/10.1016/j.ccr.2010.03.017
Ochs K, Ott M, Bunse T et al (2017) K27M-mutant histone-3 as a novel target for glioma immunotherapy. Oncoimmunology. https://doi.org/10.1080/2162402X.2017.1328340
Ohgaki H, Kleihues P (2013) The definition of primary and secondary glioblastoma. Clin Cancer Res. https://doi.org/10.1158/1078-0432.CCR-12-3002
Olar A, Wani KM, Alfaro-Munoz KD et al (2015) IDH mutation status and role of WHO grade and mitotic index in overall survival in grade II–III diffuse gliomas. Acta Neuropathol. https://doi.org/10.1007/s00401-015-1398-z
Osswald M, Jung E, Sahm F et al (2015) Brain tumour cells interconnect to a functional and resistant network. Nature. https://doi.org/10.1038/nature16071
Osswald M, Solecki G, Wick W, Winkler F (2016) A malignant cellular network in gliomas: potential clinical implications. Neuro-Oncology. https://doi.org/10.1093/neuonc/now014
Patel T, Bander ED, Venn RA et al (2017) The role of extent of resection in IDH1 wild-type or mutant low-grade gliomas. Neurosurgery. https://doi.org/10.1093/neuros/nyx265
Patel SH, Poisson LM, Brat DJ et al (2017) T2–FLAIR mismatch, an imaging biomarker for IDH and 1p/19q status in lower-grade gliomas: a TCGA/TCIA project. Clin Cancer Res. https://doi.org/10.1158/1078-0432.CCR-17-0560
Platten M, Bunse L, Riehl D, Bunse T, Ochs K, Wick W (2018) Vaccine strategies in gliomas. Curr Treat Options Neurol. https://doi.org/10.1007/s11940-018-0498-1
Pope WB, Prins RM, Thomas MA et al (2012) Non-invasive detection of 2-hydroxyglutarate and other metabolites in IDH1 mutant glioma patients using magnetic resonance spectroscopy. J Neuro-Oncol. https://doi.org/10.1007/s11060-011-0737-8
Rajaraman P, Melin BS, Wang Z et al (2012) Genome-wide association study of glioma and meta-analysis. Hum Genet. https://doi.org/10.1007/s00439-012-1212-0
Reifenberger J, Reifenberger G, Liu L, James CD, Wechsler W, Collins VP (1994) Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol
Reuss DE, Mamatjan Y, Schrimpf D et al (2015) IDH mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: a grading problem for WHO. Acta Neuropathol. https://doi.org/10.1007/s00401-015-1438-8
Rice T, Lachance DH, Molinaro AM et al (2015) Understanding inherited genetic risk of adult glioma – a review. Neuro-Oncol Pract. https://doi.org/10.1093/nop/npv026
Sahm F, Koelsche C, Meyer J, Pusch S, Lindenberg K, Mueller W, Herold-Mende C, Von Deimling A, Hartmann C (2012) CIC and FUBP1 mutations in oligodendrogliomas, oligoastrocytomas and astrocytomas. Acta Neuropathol. https://doi.org/10.1007/s00401-012-0993-5
Sahm F, Reuss D, Koelsche C et al (2014) Farewell to oligoastrocytoma: in situ molecular genetics favor classification as either oligodendroglioma or astrocytoma. Acta Neuropathol. https://doi.org/10.1007/s00401-014-1326-7
Sanson M, Hosking FJ, Shete S et al (2011) Chromosome 7p11.2 (EGFR) variation influences glioma risk. Hum Mol Genet. https://doi.org/10.1093/hmg/ddr192
Schiff D, Soffietti R, Huse J, Lawler S, Hegi M (2017) The clinical value of ATRX and TERT mutations in diffuse adult gliomas. Neuro-Oncology. https://doi.org/10.1093/neuonc/nox185
Schumacher T, Bunse L, Pusch S et al (2014) A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. https://doi.org/10.1038/nature13387
Schwartzentruber J, Korshunov A, Liu XY et al (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. https://doi.org/10.1038/nature10833
Shete S, Hosking FJ, Robertson LB et al (2009) Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet. https://doi.org/10.1038/ng.407
Shirahata M, Ono T, Stichel D et al (2018) Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. https://doi.org/10.1007/s00401-018-1849-4
Stacey SN, Sulem P, Jonasdottir A et al (2011) A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat Genet. https://doi.org/10.1038/ng.926
Sulkowski PL, Corso CD, Robinson ND et al (2017) 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aal2463
Suzuki H, Aoki K, Chiba K et al (2015) Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. https://doi.org/10.1038/ng.3273
Turcan S, Rohle D, Goenka A et al (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. https://doi.org/10.1038/nature10866
VanDenBent MJ (2010) Interobserver variation of the histopathological diagnosis in clinical trials on glioma: a clinician’s perspective. Acta Neuropathol. https://doi.org/10.1007/s00401-010-0725-7
VanDenBent MJ, Brandes AA, Taphoorn MJB et al (2013) Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: Long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol. https://doi.org/10.1200/JCO.2012.43.2229
VanDenBent MJ, Hartmann C, Preusser M et al (2013) Interlaboratory comparison of IDH mutation detection. J Neuro-Oncol. https://doi.org/10.1007/s11060-013-1056-z
Ward PS, Patel J, Wise DR et al (2010) The Common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. https://doi.org/10.1016/j.ccr.2010.01.020
Wiestler B, Capper D, Holland-Letz T, Korshunov A, Von Deimling A, Pfister SM, Platten M, Weller M, Wick W (2013) ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol. https://doi.org/10.1007/s00401-013-1156-z
Wijnenga MMJ, Dubbink HJ, French PJ, Synhaeve NE, Dinjens WNM, Atmodimedjo PN, Kros JM, Dirven CMF, Vincent AJPE, van den Bent MJ (2017) Molecular and clinical heterogeneity of adult diffuse low-grade IDH wild-type gliomas: assessment of TERT promoter mutation and chromosome 7 and 10 copy number status allows superior prognostic stratification. Acta Neuropathol 134(6):957–959
Wrensch M, Lee M, Miike R, Newman B, Barger G, Davis R, Wiencke J, Neuhaus J (1997) Familial and personal medical history of cancer and nervous system conditions among adults with glioma and controls. Am J Epidemiol. https://doi.org/10.1093/oxfordjournals.aje.a009154
Wrensch M, Jenkins RB, Chang JS et al (2009) Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat Genet. https://doi.org/10.1038/ng.408
Wu G, Broniscer A, McEachron TA et al (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. https://doi.org/10.1038/ng.1102
Yan H, Parsons DW, Jin G et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med. https://doi.org/10.1056/NEJMoa0808710
You H, Wu Y, Chang K, Shi X, Chen X-D, Yan W, Li R (2017) Paradoxical prognostic impact of TERT promoter mutations in gliomas depends on different histological and genetic backgrounds. CNS Neurosci Ther 23(10):790–797
Young JS, Prados MD, Butowski N (2018) Using genomics to guide treatment for glioblastoma. Pharmacogenomics. https://doi.org/10.2217/pgs-2018-0078
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Comments
This valuable review deals with the current molecular and histological classification of low-grade gliomas and addresses the therapeutic and prognostic implications of such recent acquisitions. References are appropriate and the timeline highlighted in Table 1, of help in summarizing the pivotal discoveries of this field. Since Bailey and Cushing’s proposal in 1926, human gliomas have been classified according to their histological features, lineage of differentiation, and putative embryological origin. In the course of the twentieth century, Bailey and Cushing’s model inspired huge achievements on glioma biology, leading to a progressive refinement of these tumors’ classification.
In recent years, the understanding of the molecular events driving gliomagenesis set the bases for novel integrated definitions of each tumor entity. This approach is endorsed by the current WHO classification of central nervous system tumors, which (for the first time in the history of glioma classification) identifies both pathological and molecular criteria for their diagnosis. Yet, for sure, this is not the end of a century long medical adventure.
Since the publication of the 2016 WHO classification, many steps forward have been taken, which will likely change the landscape of glioma diagnosis, therapy, and prognostic stratification. These novel acquisitions include (i) tumor epigenetic and methylation profiles, (ii) single cell resolution of molecular and metabolic features, (iii) the characterization of intra-tumor clonal heterogeneity, and (iv) an in-depth definition of the tumor microenvironment. These studies highlight a much more complex scenario than previously thought and pose challenging questions to the current neuro-oncology practice.
Only a tight collaboration among health professionals will succeed in the management of these complex diseases.
Domenico d’Avella and Marco Pizzi Padova, Italy
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Tumor - Glioma
Rights and permissions
About this article

Cite this article
Young, J.S., Gogos, A.J., Morshed, R.A. et al. Molecular characteristics of diffuse lower grade gliomas: what neurosurgeons need to know. Acta Neurochir 162, 1929–1939 (2020). https://doi.org/10.1007/s00701-020-04426-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00701-020-04426-2